Dynamic Gene Network Analysis of Caco-2 Cell Response to Shiga Toxin-Producing Escherichia coli-Associated Hemolytic–Uremic Syndrome

 , ,
, ,
Abstract
1. Introduction
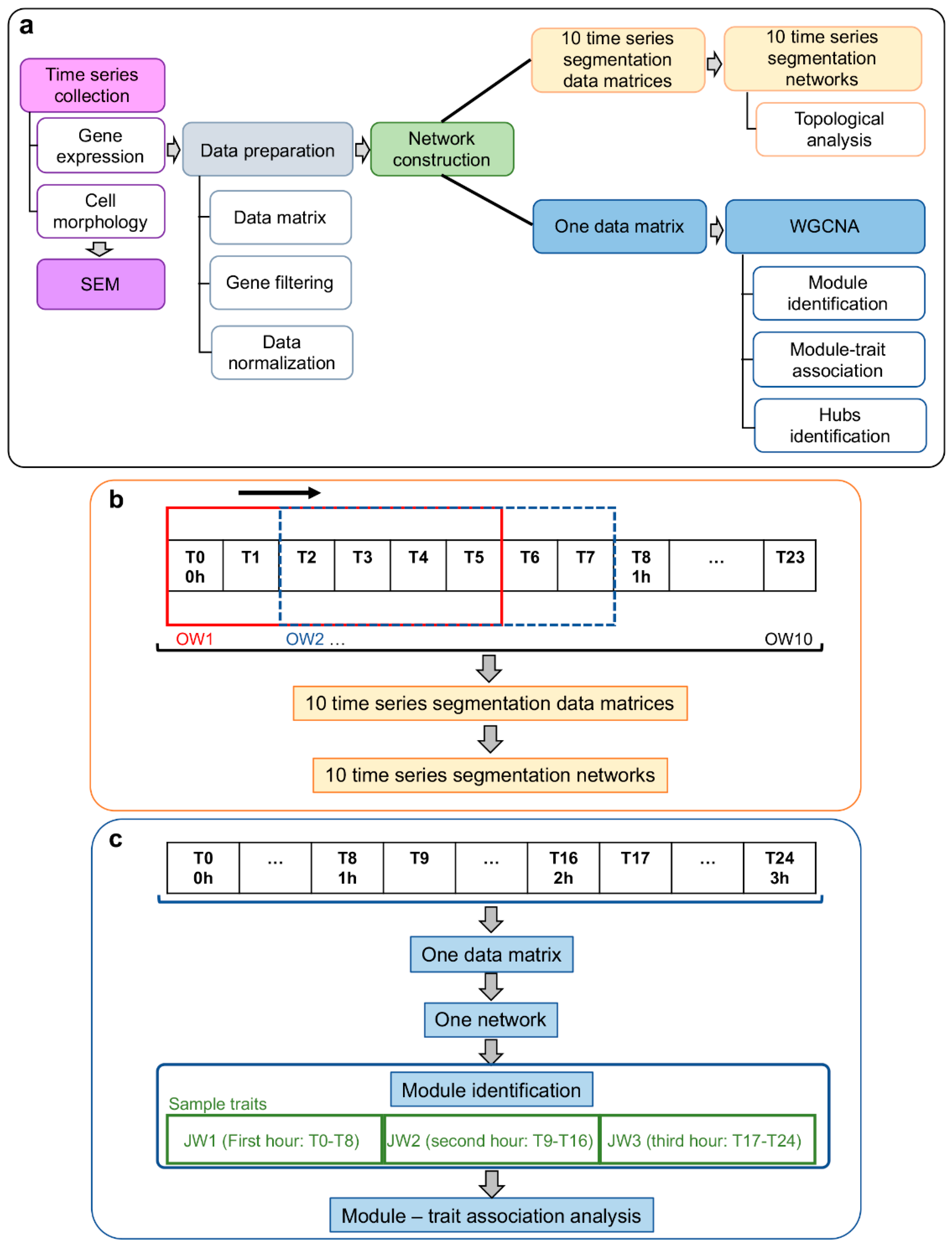
2. Materials and Methods
2.1. Bacterial Strains
2.2. Cell Culture Procedures and Temporal Shiga toxin (Stx)-producing Escherichia coli (STEC)-Enterocyte Interaction Assays
2.3. RNA Extraction
2.4. Microarray Hybridization
2.5. Systems Biology Approach
2.6. Gene Co-Expression Network (GCN) Topological Analysis
2.7. Weighted Gene Co-Expression Network Analysis (WGCNA): Module–Trait Association
2.7.1. Module–Trait Association
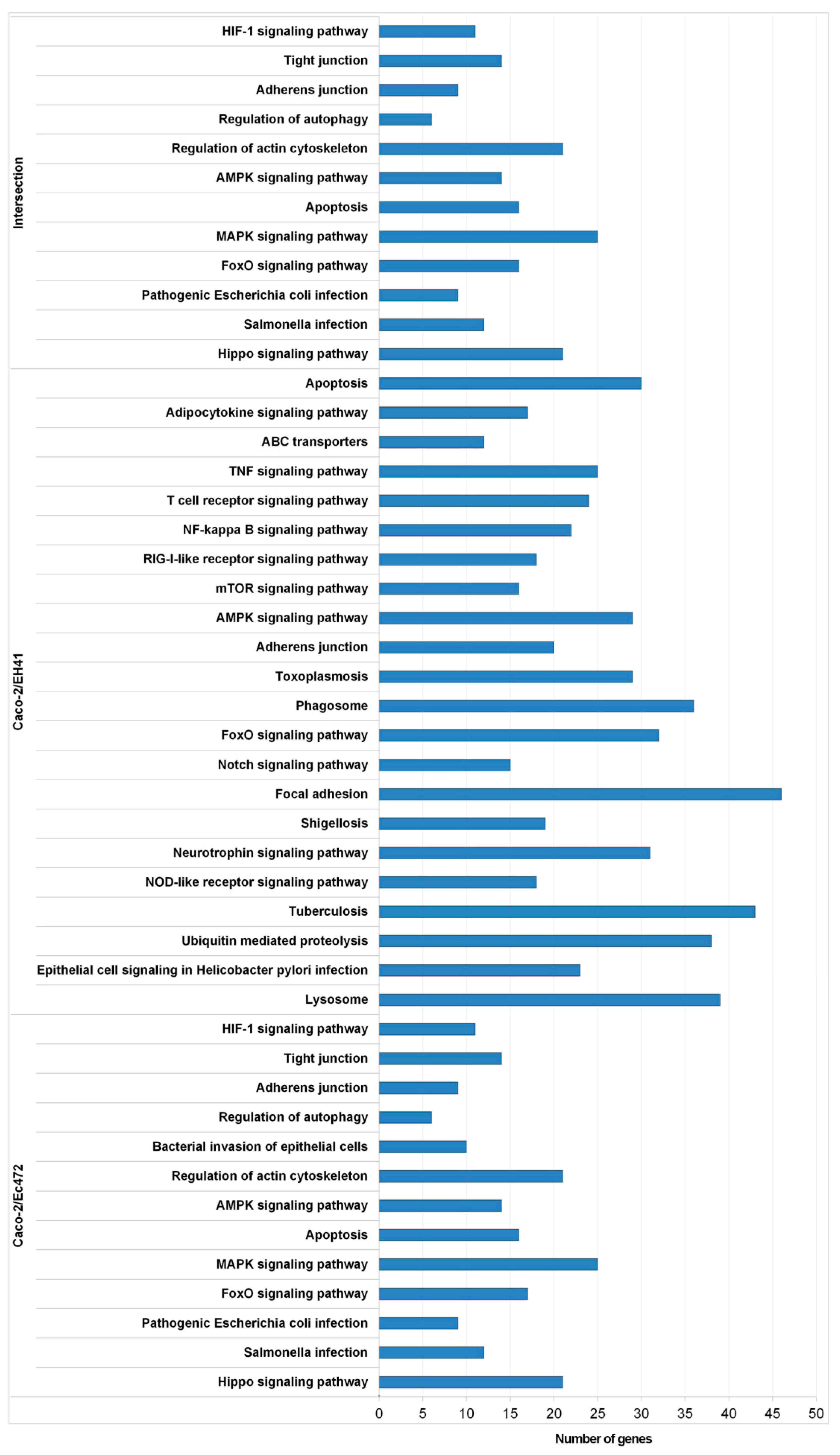
2.7.2. Functional Enrichment Analysis of Network Module Genes
2.7.3. Intramodular Analysis for Hub Selection
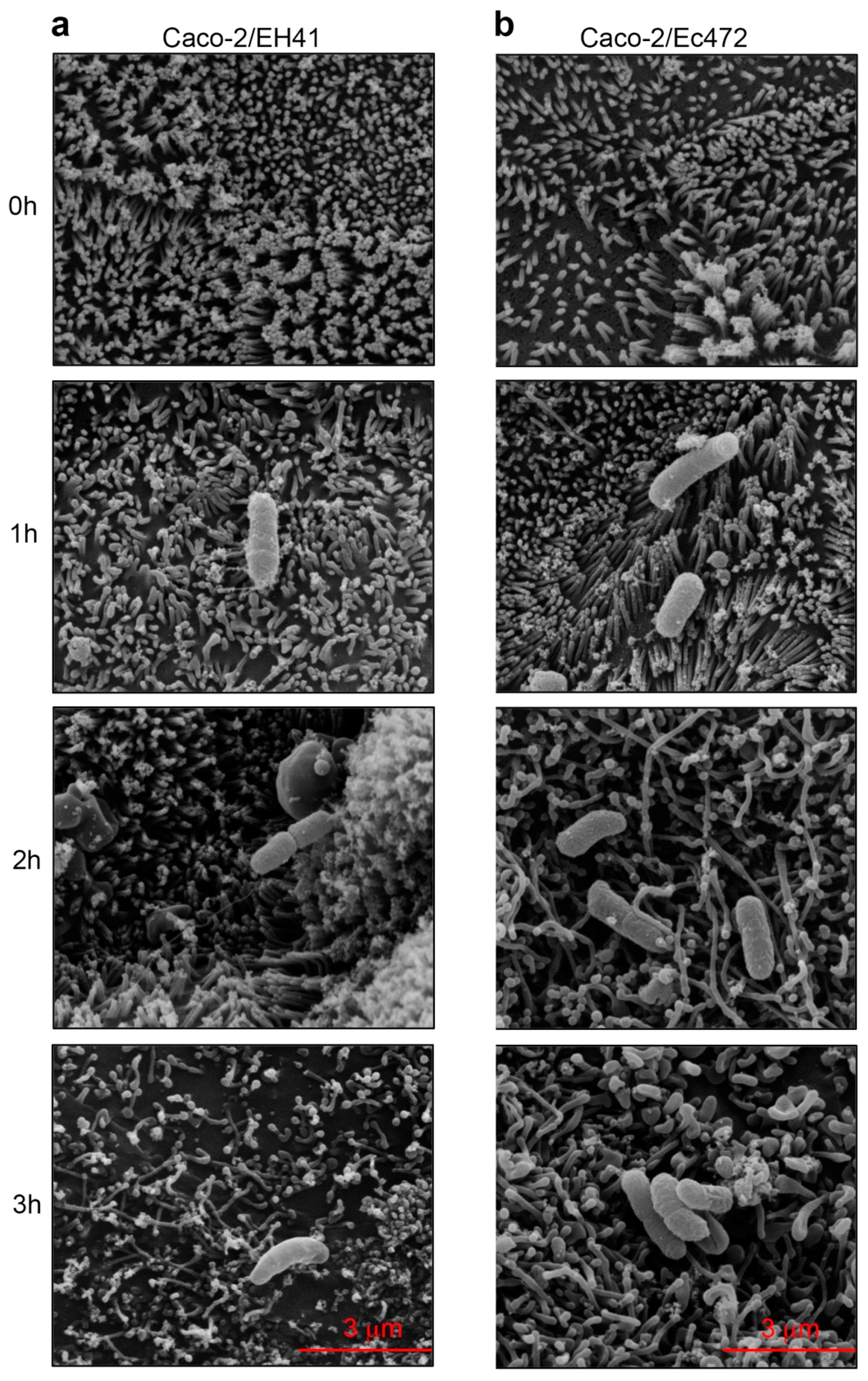
2.8. Scanning Electron Microscopy (SEM)
2.9. Validation of Microarray Data by RT-qPCR
3. Results
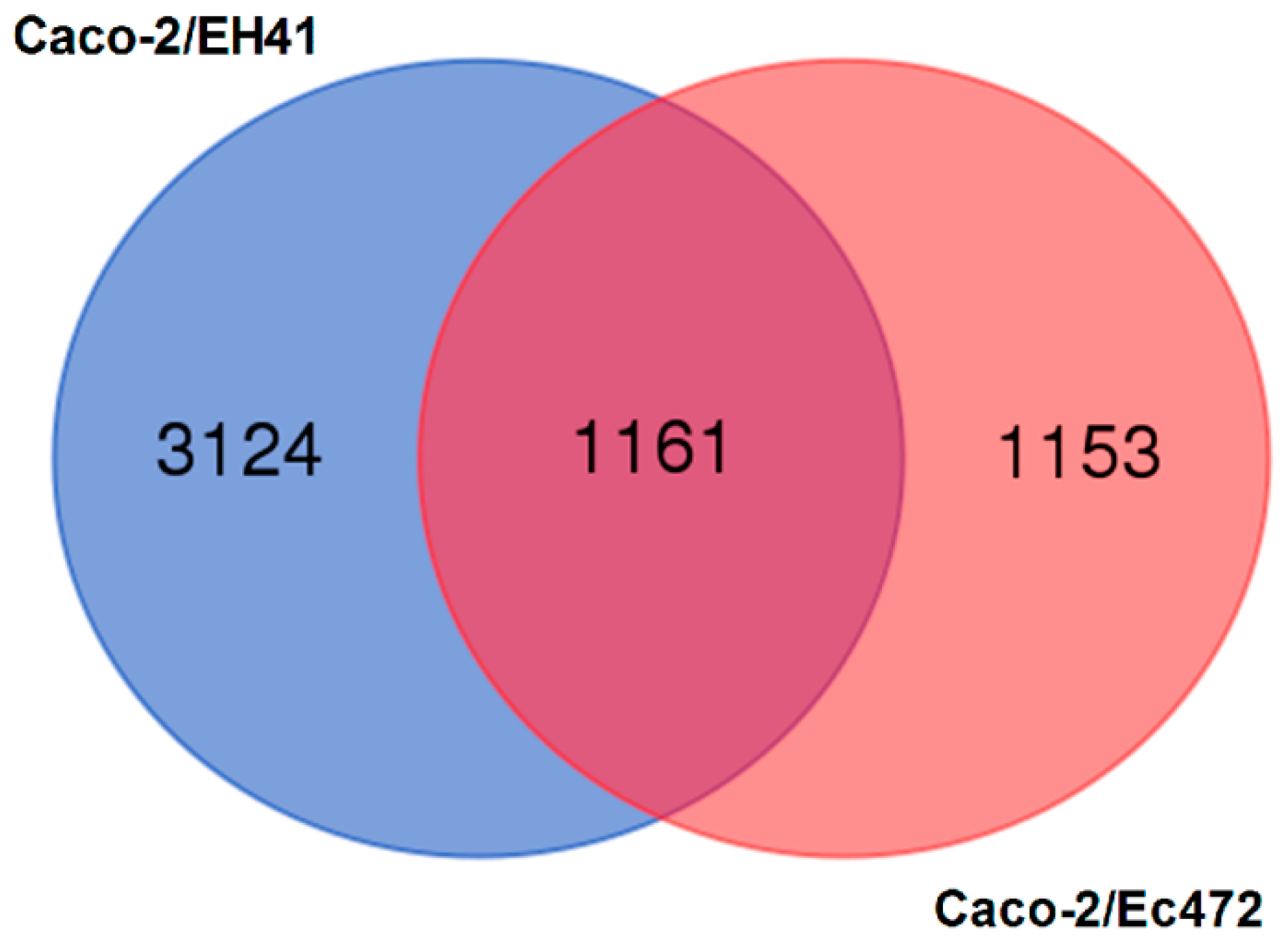
3.1. Transcriptional and Network Topology Analysis
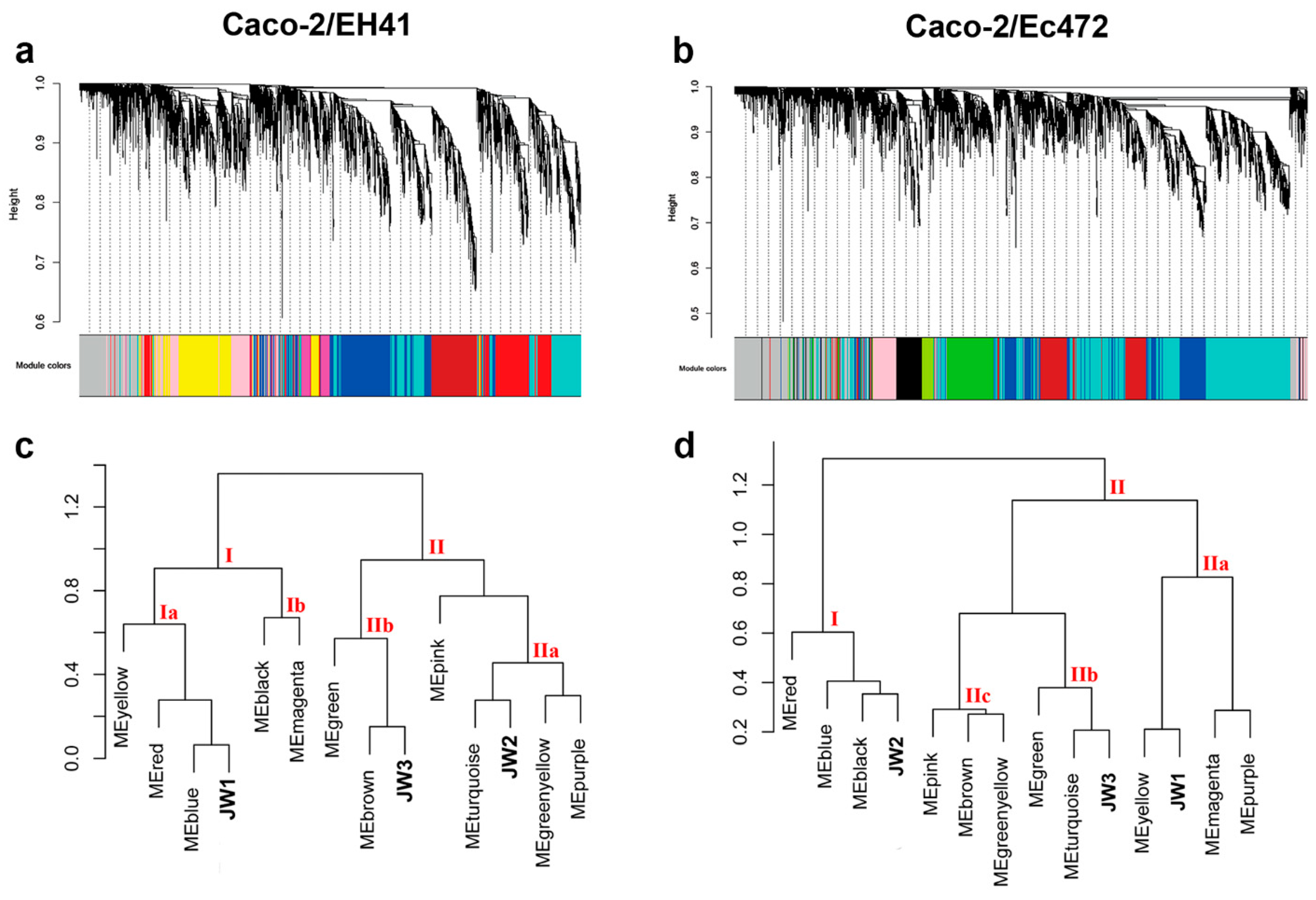
3.2. Weighted Gene Co-Expression Network Analysis (WGCNA)
3.2.1. Caco-2/EH41 Network
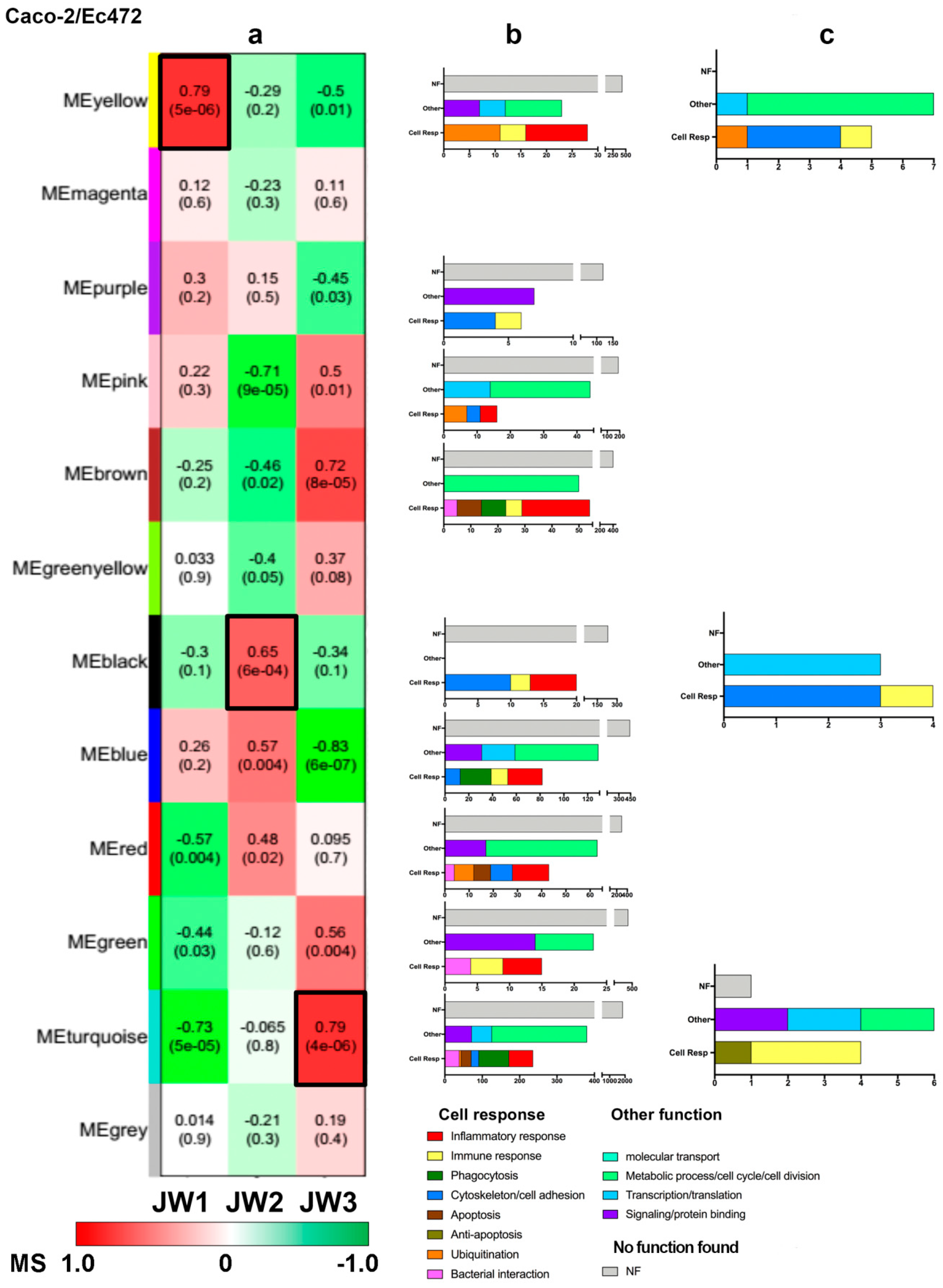
3.2.2. Caco-2/Ec472 Network
3.3. SEM
3.4. RT-qPCR Technical Validation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Petruzziello-Pellegrini, T.N.; Marsden, P.A. Shiga toxin-associated hemolytic uremic syndrome: Advances in pathogenesis and therapeutics. Curr. Opin. Nephrol. Hypertens. 2012, 21, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Karmali, M.A.; Petric, M.; Lim, C.; Fleming, P.C.; Arbus, G.S.; Lior, H. The association between idiopathic hemolytic uremic syndrome and infection by verotoxin-producing Escherichia coli. J. Infect. Dis. 1985, 151, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Paton, A.W.; Woodrow, M.C.; Doyle, R.M.; Lanser, J.A.; Paton, J.C. Molecular characterization of a shiga toxigenic Escherichia coli O113: H21 strain lacking eae responsible for a cluster of cases of hemolytic-uremic syndrome. J. Clin. Microbiol. 1999, 37, 3357–3361. [Google Scholar] [PubMed]
- Monaghan, A.M.; Byrne, B.; McDowell, D.; Carroll, A.M.; McNamara, E.B.; Bolton, D.J. Characterization of farm, food, and clinical Shiga toxin-producing Escherichia coli (STEC) O113. Foodborne Pathog. Dis. 2012, 9, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Bielaszewska, M.; Friedrich, A.W.; Aldick, T.; Schürk-Bulgrin, R.; Karch, H. Shiga toxin activatable by intestinal mucus in Escherichia coli isolated from humans: Predictor for a severe clinical outcome. Clin. Infect. Dis. 2006, 43, 1160–1167. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Delannoy, S.; Lacher, D.W.; Bosilevac, J.M.; Fach, P. Characterization and Virulence Potential of Serogroup O113 Shiga Toxin-Producing Escherichia coli Strains Isolated from Beef and Cattle in the United States. J. Food Prot. 2017, 80, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Bando, S.Y.; Iamashita, P.; Guth, B.E.; Dos Santos, L.F.; Fujita, A.; Abe, C.M.; Ferreira, L.R.; Moreira-Filho, C.A. A hemolytic-uremic syndrome-associated strain O113:H21 Shiga toxin-producing Escherichia coli specifically expresses a transcriptional module containing dicA and is related to gene network dysregulation in Caco-2 cells. PLoS ONE 2017, 12, e0189613. [Google Scholar] [CrossRef] [PubMed]
- dos Santos, L.F.; Irino, K.; Vaz, T.M.; Guth, B.E. Set of virulence genes and genetic relatedness of O113: H21 Escherichia coli strains isolated from the animal reservoir and human infections in Brazil. J. Med. Microbiol. 2010, 59, 634–640. [Google Scholar] [CrossRef]
- Gonzalez, A.G.; Cerqueira, A.M.; Guth, B.E.; Coutinho, C.A.; Liberal, M.H.; Souza, R.M.; Andrade, J.R. Serotypes, virulence markers and cell invasion ability of Shiga toxin-producing Escherichia coli strains isolated from healthy dairy cattle. J. Appl. Microbiol. 2016, 121, 1130–1143. [Google Scholar] [CrossRef]
- Lea, T. Caco-2 Cell Line. In The Impact of Food Bioactives on Health: In Vitro and Ex Vivo Models; Verhoeckx, K., Cotter, P., López-Expósito, I., Kleiveland, C., Lea, T., Mackie, A., Requena, T., Swiatecka, D., Wichers, H., Eds.; Springer: Cham, Switzerland, 2015; pp. 103–111. [Google Scholar]
- Doughty, S.; Sloan, J.; Bennett-Wood, V.; Robertson, M.; Robins-Browne, R.M.; Hartland, E.L. Identification of a novel fimbrial gene cluster related to long polar fimbriae in locus of enterocyte effacement-negative strains of enterohemorrhagic Escherichia coli. Infect. Immun. 2002, 70, 6761–6769. [Google Scholar] [CrossRef]
- Feng, P.C.H.; Delannoy, S.; Lacher, D.W.; dos Santos, L.F.; Beutin, L.; Fach, P.; Rivas, M.; Hartland, E.L.; Paton, A.W.; Guth, B.E. Genetic Diversity and Virulence Potential of Shiga Toxin-Producing Escherichia coli O113:H21 Strains Isolated from Clinical, Environmental, and Food Sources. Appl. Environ. Microbiol. 2014, 80, 4757–4763. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing: Vienna, Austria. Available online: http://www.R-project.org/ (accessed on 13 May 2018).
- Smyth, G.K. Limma: Linear models for microarray data. Bioinform. Comput. Biol. Solut. Using R Bioconductor 2005, 397–420. [Google Scholar]
- Bando, S.Y.; Silva, F.N.; Costa La, F.; Silva, A.V.; Pimentel-Silva, L.R.; Castro, L.H.; Wen, H.-T.; Amaro, E., Jr.; Moreira-Filho, C.A. Complex network analysis of CA3 transcriptome reveals pathogenic and compensatory pathways in refractory temporal lobe epilepsy. PLoS ONE 2013, 8, e79913. [Google Scholar] [CrossRef] [PubMed]
- Silva, F.N.; da F. Costa, L. Networks 3D Visualization Tools for Mac OS X. Available online: http://cyvision.ifsc.usp.br/software/networks3d (accessed on 14 May 2018).
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; McDermott, A.L.M.G.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Larionov, A.; Krause, A.; Miller, W. A standard curve based method for relative real time PCR data processing. BMC Bioinform. 2005, 6, 62. [Google Scholar] [CrossRef] [PubMed]
- Jandhyala, D.M.; Ahluwalia, A.; Schimmel, J.J.; Rogers, A.B.; Leong, J.M.; Thorpe, C.M. Activation of the Classical Mitogen-Activated Protein Kinases Is Part of the Shiga Toxin-Induced Ribotoxic Stress Response and May Contribute to Shiga Toxin-Induced Inflammation. Infect. Immun. 2015, 84, 138–148. [Google Scholar] [CrossRef]
- Bouwmeester, T.; Bauch, A.; Ruffner, H.; Angrand, P.O.; Bergamini, G.; Croughton, K.; Cruciat, C.; Eberhard, D.; Gagneur, J.; Ghidelli, S.; et al. A physical and functional map of the human TNF-alpha/NF-kappa B signal transduction pathway. Nat. Cell Biol. 2004, 6, 97–105. [Google Scholar] [CrossRef]
- Weidberg, H.; Elazar, Z. TBK1 mediates crosstalk between the innate immune response and autophagy. Sci. Signal. 2011, 4, pe39. [Google Scholar] [CrossRef]
- Lombardo, M.F.; De Angelis, F.; Bova, L.; Bartolini, B.; Bertuzzi, F.; Nano, R.; Capuani, B.; Lauro, R.; Federici, M.; Lauro, D.; et al. Human placental lactogen (hPL-A) activates signaling pathways linked to cell survival and improves insulin secretion in human pancreatic islets. Islets 2011, 3, 250–258. [Google Scholar] [CrossRef] [PubMed]
- Vareille, M.; Rannou, F.; Thélier, N.; Glasser, A.L.; de Sablet, T.; Martin, C.; Gobert, A.P. Heme oxygenase-1 is a critical regulator of nitric oxide production in enterohemorrhagic Escherichia coli-infected human enterocytes. J. Immunol. 2008, 180, 5720–5726. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Shang, J.; Zhang, Y.; Liu, S.; Peng, Y.; Zhou, Z.; Pan, H.; Wang, X.; Chen, L.; Zhao, Q. MEG3 is a prognostic factor for CRC and promotes chemosensitivity by enhancing oxaliplatin-induced cell apoptosis. Oncol. Rep. 2017, 38, 1383–1392. [Google Scholar] [CrossRef] [PubMed]
- Mysorekar, I.U.; Mulvey, M.A.; Hultgren, S.J.; Gordon, J.I. Molecular regulation of urothelial renewal and host defenses during infection with uropathogenic Escherichia coli. J. Biol. Chem. 2002, 277, 7412–7419. [Google Scholar] [CrossRef] [PubMed]
- Giogha, C.; Lung, T.W.; Pearson, J.S.; Hartland, E.L. Inhibition of death receptor signaling by bacterial gut pathogens. Cytokine Growth Factor Rev. 2014, 25, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Dagenais, M.; Dupaul-Chicoine, J.; Champagne, C.; Skeldon, A.; Morizot, A.; Saleh, M. A critical role for cellular inhibitor of protein 2 (cIAP2) in colitis-associated colorectal cancer and intestinal homeostasis mediated by the inflammasome and survival pathways. Mucosal Immunol. 2016, 9, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Im, E.; Chung, H.K.; Pothoulakis, C.; Rhee, S.H. TRIF mediates Toll-like receptor 5-induced signaling in intestinal epithelial cells. J. Biol. Chem. 2010, 285, 37570–37578. [Google Scholar] [CrossRef]
- Oshima, S.; Nakamura, T.; Namiki, S.; Okada, E.; Tsuchiya, K.; Okamoto, R.; Yamazaki, M.; Yokota, T.; Aida, M.; Yamaguchi, Y.; et al. Interferon regulatory factor 1 (IRF-1) and IRF-2 distinctively up-regulate gene expression and production of interleukin-7 in human intestinal epithelial cells. Mol. Cell Biol. 2004, 24, 6298–6310. [Google Scholar] [CrossRef]
- Qiu, W.; Wu, B.; Wang, X.; Buchanan, M.E.; Regueiro, M.D.; Hartman, D.J.; Schoen, R.E.; Yu, J.; Zhang, L. PUMA-mediated intestinal epithelial apoptosis contributes to ulcerative colitis in humans and mice. J. Clin. Investig. 2011, 121, 1722–1732. [Google Scholar] [CrossRef]
- Yu, T.; Shan, T.D.; Li, J.Y.; Huang, C.Z.; Wang, S.Y.; Ouyang, H.; Lu, X.J.; Xu, J.H.; Zhong, W.; Chen, Q.K. Knockdown of linc-UFC1 suppresses proliferation and induces apoptosis of colorectal cancer. Cell Death Dis. 2016, 7, e2228. [Google Scholar] [CrossRef]
- Celis-Gutierrez, J.; Boyron, M.; Walzer, T.; Pandolfi, P.P.; Jonjić, S.; Olive, D.; Dalod, M.; Vivier, E.; Nunès, J.A. Dok1 and Dok2 proteins regulate natural killer cell development and function. EMBO J. 2014, 33, 1928–1940. [Google Scholar] [CrossRef] [PubMed]
- Borlido, J.; D’Angelo, M.A. Nup62: A novel regulator of centrosome integrity and function. Cell Cycle 2014, 13, 14. [Google Scholar] [CrossRef] [PubMed]
- Saita, S.; Nolte, H.; Fiedler, K.U.; Kashkar, H.; Venne, A.S.; Zahedi, R.P.; Krüger, M.; Langer, T. PARL mediates Smac proteolytic maturation in mitochondria to promote apoptosis. Nat. Cell Biol. 2017, 19, 318–328. [Google Scholar] [CrossRef] [PubMed]
- Hartwig, J.H.; Thelen, M.; Rosen, A.; Janmey, P.A.; Nairn, A.C.; Aderem, A. MARCKS is an actin filament crosslinking protein regulated by protein kinase C and calcium-calmodulin. Nature 1992, 356, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.; Suk, K.; Lee, W.H. Myristoylated alanine-rich C kinase substrate (MARCKS) regulates the expression of proinflammatory cytokines in macrophages through activation of p38/JNK MAPK and NF-κB. Cell Immunol. 2015, 296, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Tollin, M.; Bergman, P.; Svenberg, T.; Jörnvall, H.; Gudmundsson, G.H.; Agerberth, B. Antimicrobial peptides in the first line defence of human colon mucosa. Peptides 2003, 24, 523–530. [Google Scholar] [CrossRef]
- Chen, Q.; Wang, L.; Jiang, M.; Huang, J.; Jiang, Z.; Feng, H.; Ji, Z. E2F1 interactive with BRCA1 pathway induces HCC two different small molecule metabolism or cell cycle regulation via mitochondrion or CD4+T to cytosol. J. Cell Physiol. 2018, 233, 1213–1221. [Google Scholar] [CrossRef]
- Crottès, D.; Félix, R.; Meley, D.; Chadet, S.; Herr, F.; Audiger, C.; Soriani, O.; Vandier, C.; Roger, S.; Angoulvant, D.; et al. Immature human dendritic cells enhance their migration through KCa3.1 channel activation. Cell Calcium. 2016, 59, 198–207. [Google Scholar] [CrossRef] [PubMed]
- Zundler, S.; Caioni, M.; Müller, M.; Strauch, U.; Kunst, C.; Woelfel, G. K+ Channel Inhibition Differentially Regulates Migration of Intestinal Epithelial Cells in Inflamed vs. Non-Inflamed Conditions in a PI3K/Akt-Mediated Manner. PLoS ONE 2016, 11, e0147736. [Google Scholar] [CrossRef]
- Yu, Z.; Liu, J.; Deng, W.M.; Jiao, R. Histone chaperone CAF-1: Essential roles in multi-cellular organism development. Cell Mol. Life Sci. 2015, 72, 327–337. [Google Scholar] [CrossRef]
- Bhattacharya, S.; Srinivasan, K.; Abdisalaam, S.; Su, F.; Raj, P.; Dozmorov, I.; Mishra, R.; Wakeland, E.K.; Ghose, S.; Mukherjee, S.; et al. RAD51 interconnects between DNA replication, DNA repair and immunity. Nucleic Acids Res. 2017, 45, 4590–4605. [Google Scholar] [CrossRef] [PubMed]
- Kelsall, I.R.; Langenick, J.; MacKay, C.; Patel, K.J.; Alpi, A.F. The Fanconi anaemia components UBE2T and FANCM are functionally linked to nucleotide excision repair. PLoS ONE 2012, 7, e36970. [Google Scholar] [CrossRef] [PubMed]
- Bielaszewska, M.; Rüter, C.; Bauwens, A.; Greune, L.; Jarosch, K.A.; Steil, D.; Zhang, W.; He, X.; Lloubes, R.; Fruth, A.; et al. Host cell interactions of outer membrane vesicle-associated virulence factors of enterohemorrhagic Escherichia coli O157: Intracellular delivery, trafficking and mechanisms of cell injury. PLoS Pathog. 2017, 13, e1006159. [Google Scholar] [CrossRef] [PubMed]
- Chono, E.; Kurokawa, T.; Oda, C.; Kawasaki, K.; Yamamoto, T.; Ishibashi, S. Expression of rac1 protein in the crypt-villus axis of rat small intestine: In reference to insulin action. Biochem. Biophys. Res. Commun. 1997, 233, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Radtke, F.; Fasnacht, N.; Macdonald, H.R. Notch signaling in the immune system. Immunity 2010, 32, 14–27. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.L.; Cai, W.F.; Wang, L.; Liu, G.S.; Paul, C.; Jiang, L.; Wang, B.; Gao, X.; Wang, Y.; Wu, S.Z. Identification of the Functional Autophagy-Regulatory Domain in HCLS1-Associated Protein X-1 That Resists Against Oxidative Stress. DNA Cell Biol. 2018, 37, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Mao, R.; Yang, R.; Chen, X.; Harhaj, E.W.; Wang, X.; Fan, Y. Regnase-1, a rapid response ribonuclease regulating inflammation and stress responses. Cell Mol. Immunol. 2017, 14, 412–422. [Google Scholar] [CrossRef] [PubMed]
- Sanada, T.; Takaesu, G.; Mashima, R.; Yoshida, R.; Kobayashi, T.; Yoshimura, A. FLN29 deficiency reveals its negative regulatory role in the Toll-like receptor (TLR) and retinoic acid-inducible gene I (RIG-I)-like helicase signaling pathway. J. Biol. Chem. 2008, 283, 33858–33864. [Google Scholar] [CrossRef]
- Hooker, E.; Baldwin, C.; Lemay, S. New insights into Dok-4 PTB domain structure and function. Biochem. Biophys. Res. Commun. 2012, 427, 67–72. [Google Scholar] [CrossRef]
- Grizzi, F.; Franceschini, B.; Di Biccari, S.; Musardo, S.; Pedretti, E.; Chiriva-Internati, M.; Osipov, V.; Fernández-Aceñero, M.J. Sperm protein 17 and AKAP-associated sperm protein cancer/testis antigens are expressed in ciliated hepatic foregut cysts. Histopathology 2015, 67, 398–403. [Google Scholar] [CrossRef]
- Naishiro, Y.; Yamada, T.; Idogawa, M.; Honda, K.; Takada, M.; Kondo, T.; Imai, K.; Hirohashi, S. Morphological and transcriptional responses of untransformed intestinal epithelial cells to an oncogenic beta-catenin protein. Oncogene 2005, 24, 3141–3153. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Morales, S.A.; Mareninov, S.; Coulam, P.; Wadehra, M.; Goodglick, L.; Braun, J.; Gordon, L.K. Functional consequences of interactions between FAK and epithelial membrane protein 2 (EMP2). Invest. Ophthalmol. Vis. Sci. 2009, 50, 4949–4956. [Google Scholar] [CrossRef] [PubMed]
- Loukil, A.; Cheung, C.T.; Bendris, N.; Lemmers, B.; Peter, M.; Blanchard, J.M. Cyclin A2: At the crossroads of cell cycle and cell invasion. World J. Biol. Chem. 2015, 6, 346–350. [Google Scholar] [CrossRef] [PubMed]
- Chu, C. Time series segmentation: A sliding window approach. Inf. Sci. 1995, 147–173. [Google Scholar] [CrossRef]
- Delbart, C.; Valdes, D.; Barbecot, F.; Tognelli, A.; Richon, P.; Couchoux, L. Temporal variability of karst aquifer response time established by the sliding-windows cross-correlation method. J. Hydrol. 2014, 580–588. [Google Scholar] [CrossRef]
- Barabási, A.L.; Oltvai, Z.N. Network biology: Understanding the cell’s functional organization. Nat. Rev. Genet. 2004, 5, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Sieberts, S.K.; Schadt, E.E. Moving toward a system genetics view of disease. Mamm. Genome. 2007, 18, 389–401. [Google Scholar] [CrossRef] [PubMed]
- Sahni, N.; Yi, S.; Taipale, M.; Fuxman Bass, J.I.; Coulombe-Huntington, J.; Yang, F.; Peng, J.; Weile, J.; Karras, G.I.; Wang, Y.; et al. Widespread macromolecular interaction perturbations in human genetic disorders. Cell 2015, 161, 647–660. [Google Scholar] [CrossRef]
- Liu, Y.; Koyutürk, M.; Barnholtz-Sloan, J.S.; Chance, M.R. Gene interaction enrichment and network analysis to identify dysregulated pathways and their interactions in complex diseases. BMC Syst. Biol. 2012, 6, 65. [Google Scholar] [CrossRef]
- Newman, M.; Girvan, M. Finding and evaluating community structure in networks. Phys. Rev. E. Stat. Nonlin. Soft Matter Phys. 2004, 69. [Google Scholar] [CrossRef]
- Newman, M.E.J. Networks: An Introduction; Oxford University Press: Oxford, UK, 2010. [Google Scholar]
- Rosenkrantz, J.T.; Aarts, H.; Abee, T.; Rolfe, M.D.; Knudsen, G.M.; Nielsen, M.B.; Thomsen, L.E.; Zwietering, M.H.; Olsen, J.E.; Pin, C. Non-essential genes form the hubs of genome scale protein function and environmental gene expression networks in Salmonella enterica serovar Typhimurium. BMC Microbiol. 2013, 13, 294. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Kuroda, M.; Sakashita, N.; Konishi, M.; Kaneda, H.; Igarashi, N.; Yamahana, J.; Taneichi, H.; Kanegane, H.; Ito, M.; et al. Cytokine profiles of patients with enterohemorrhagic Escherichia coli O111-induced hemolytic-uremic syndrome. Cytokine 2012, 60, 694–700. [Google Scholar] [CrossRef] [PubMed]
- Honda, T.; Ogata, S.; Mineo, E.; Nagamori, Y.; Nakamura, S.; Bando, Y.; Ishii, M. A novel strategy for hemolytic uremic syndrome: Successful treatment with thrombomodulin α. Pediatrics 2013, 131, e928–e933. [Google Scholar] [CrossRef] [PubMed]
- Bennett, K.M.; Walker, S.L.; Lo, D.D. Epithelial microvilli establish an electrostatic barrier to microbial adhesion. Infect. Immun. 2014, 82, 2860–2871. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Module | Gene Ontology | Functional Description |
|---|---|---|---|
| MEG3 | Blue | Long non-coding RNA | apoptosis |
| ROPN1L | epithelial cilium movement | epithelial cilium movement | |
| CSH1 | JAK-STAT signaling pathway; PI3K-Akt signaling pathway | inflammatory response | |
| ADRA1A | positive regulation of ERK1 and ERK2 cascade | inflammatory response | |
| TBKBP1 | RIG-I-like receptor signaling pathway | inflammatory response | |
| NEGR1 | protein binding | protein binding | |
| P2RX2 | positive regulation of calcium-mediated signaling | signaling | |
| FLJ40434 | ND | unknown | |
| LOC157740 | ND | unknown | |
| BBC3 | Turquoise | Hippo signaling pathway | apoptosis |
| UFC1 | response to endoplasmic reticulum stress | apoptosis | |
| YY1AP1 | regulation of cell cycle | cell cycle | |
| DOK1 | Ras protein signal transduction | immune response | |
| IRF1 | positive regulation of interferon-beta production | inflammatory response | |
| TICAM1 | positive regulation of I-kappaB kinase/NF-kappaB signaling; Toll-like receptor signaling pathway | inflammatory response | |
| BCL3 | I-kappaB kinase/NF-kappaB signaling; TNF signaling pathway | inflammatory response | |
| TNFRSF1A | positive regulation of I-kappaB kinase/NF-kappaB signaling; TNF signaling pathway | inflammatory response | |
| BIRC3 | positive regulation of I-kappaB kinase/NF-kappaB signaling; TNF signaling pathway | inflammatory response | |
| MAFF | transcription from RNA polymerase II promoter | transcription | |
| RG9MTD1 | positive regulation of mitochondrial translation | transcription | |
| NUP62 | Brown | negative regulation of apoptotic process and programmed cell death | anti-apoptosis |
| PARL | negative regulation of intrinsic apoptotic signaling pathway | anti-apoptosis | |
| NASP | DNA replication-dependent nucleosome assembly | cell division | |
| HIST1H1C | chromatin DNA binding | cell division | |
| C1orf96 | regulation of mitotic spindle assembly | cell division | |
| HIST1H2BH | chromatin organization | chromatin organization | |
| SAFB | chromatin organization | chromatin organization | |
| MARCKS | actin filament bundle assembly | cytoskeleton | |
| HIST1H2BJ | LPS binding | immune response | |
| SLC25A22 | L-glutamate transmembrane transport; mitochondrial transport | metabolic process | |
| HIST1H3H | chromatin organization | transcription | |
| HIST1H4K | telomere organization | transcription | |
| HIST2H3A | chromatin organization | transcription | |
| NUP153 | protein sumoylation | transcription | |
| DNMT1 | transcription | transcription | |
| UBE2Q1 | protein ubiquitination | ubiquitination |
| Gene | Module | Gene Ontology | Functional Description |
|---|---|---|---|
| CENPN | Yellow | mitotic cell cycle | cell cycle |
| CHAF1A | DNA repair | cell cycle | |
| ZWINT | RHO GTPase Effectors | cell division | |
| CDCA5 | mitotic cell cycle | cell division | |
| RAD51 | DNA repair | cell division | |
| WDR62 | RNA splicing | cell division | |
| CCNA2 | Ras protein signal transduction | cytoskeleton | |
| EMP2 | actin filament organization; cell adhesion | cytoskeleton | |
| PODXL | regulation of microvillus assembly | cytoskeleton | |
| KCNN4 | positive regulation of T cell receptor signaling pathway | immune response | |
| E2F2 | transcription factor | transcription | |
| UBE2T | DNA repair; protein ubiquitination | ubiquitination | |
| PRIMA1 | Black | cell junction | cell adhesion |
| ITGA9 | cell adhesion; PI3K-Akt signaling pathway | cell adhesion; immune response | |
| KRT9 | structural constituent of cytoskeleton | cytoskeleton | |
| RAC2 | actin cytoskeleton organization; Chemokine signaling pathway | cytoskeleton; inflammatory response | |
| KRTAP10-10 | keratin filament | keratinization | |
| C8orf73 | alias MROH6; lipid transport | lipid transport | |
| AQP5 | microvillus; Aquaporins | molecular transport | |
| ANKRD13B | endosome | protein transport | |
| HAX1 | Turquoise | interleukin-1 binding; regulation of apoptotic process | anti-apoptosis |
| SSNA1 | G2/M transition of mitotic cell cycle | cell cycle | |
| UPF1 | DNA repair; cellular response to LPS/interleukin-1 | immune response | |
| TRAFD1 | negative regulation of innate immune response | immune response | |
| DOK4 | protein binding; negative regulator of T cells | immune response | |
| ACOT8 | acyl-CoA hydrolase activity | metabolic process | |
| STX5 | SNARE binding; vesicle-mediated transport | protein transport | |
| COPZ1 | vesicle-mediated transport | protein transport | |
| WBP1 | WW domain binding | protein binding | |
| NOTCH2NL | Notch signaling pathway | signaling | |
| TMEM116 | transmembrane protein | unknown |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bando, S.Y.; Iamashita, P.; Silva, F.N.; Costa, L.d.F.; Abe, C.M.; Bertonha, F.B.; Guth, B.E.C.; Fujita, A.; Moreira-Filho, C.A. Dynamic Gene Network Analysis of Caco-2 Cell Response to Shiga Toxin-Producing Escherichia coli-Associated Hemolytic–Uremic Syndrome. Microorganisms 2019, 7, 195. https://doi.org/10.3390/microorganisms7070195
Bando SY, Iamashita P, Silva FN, Costa LdF, Abe CM, Bertonha FB, Guth BEC, Fujita A, Moreira-Filho CA. Dynamic Gene Network Analysis of Caco-2 Cell Response to Shiga Toxin-Producing Escherichia coli-Associated Hemolytic–Uremic Syndrome. Microorganisms. 2019; 7(7):195. https://doi.org/10.3390/microorganisms7070195
Chicago/Turabian StyleBando, Silvia Y., Priscila Iamashita, Filipi N. Silva, Luciano da F. Costa, Cecilia M. Abe, Fernanda B. Bertonha, Beatriz E. C. Guth, André Fujita, and Carlos A. Moreira-Filho. 2019. "Dynamic Gene Network Analysis of Caco-2 Cell Response to Shiga Toxin-Producing Escherichia coli-Associated Hemolytic–Uremic Syndrome" Microorganisms 7, no. 7: 195. https://doi.org/10.3390/microorganisms7070195
APA StyleBando, S. Y., Iamashita, P., Silva, F. N., Costa, L. d. F., Abe, C. M., Bertonha, F. B., Guth, B. E. C., Fujita, A., & Moreira-Filho, C. A. (2019). Dynamic Gene Network Analysis of Caco-2 Cell Response to Shiga Toxin-Producing Escherichia coli-Associated Hemolytic–Uremic Syndrome. Microorganisms, 7(7), 195. https://doi.org/10.3390/microorganisms7070195