Genomic Analyses of Bifidobacterium moukalabense Reveal Adaptations to Frugivore/Folivore Feeding Behavior

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation and Identification of Bifidobacteria
2.2. Genome Library Construction and Illumina Sequencing
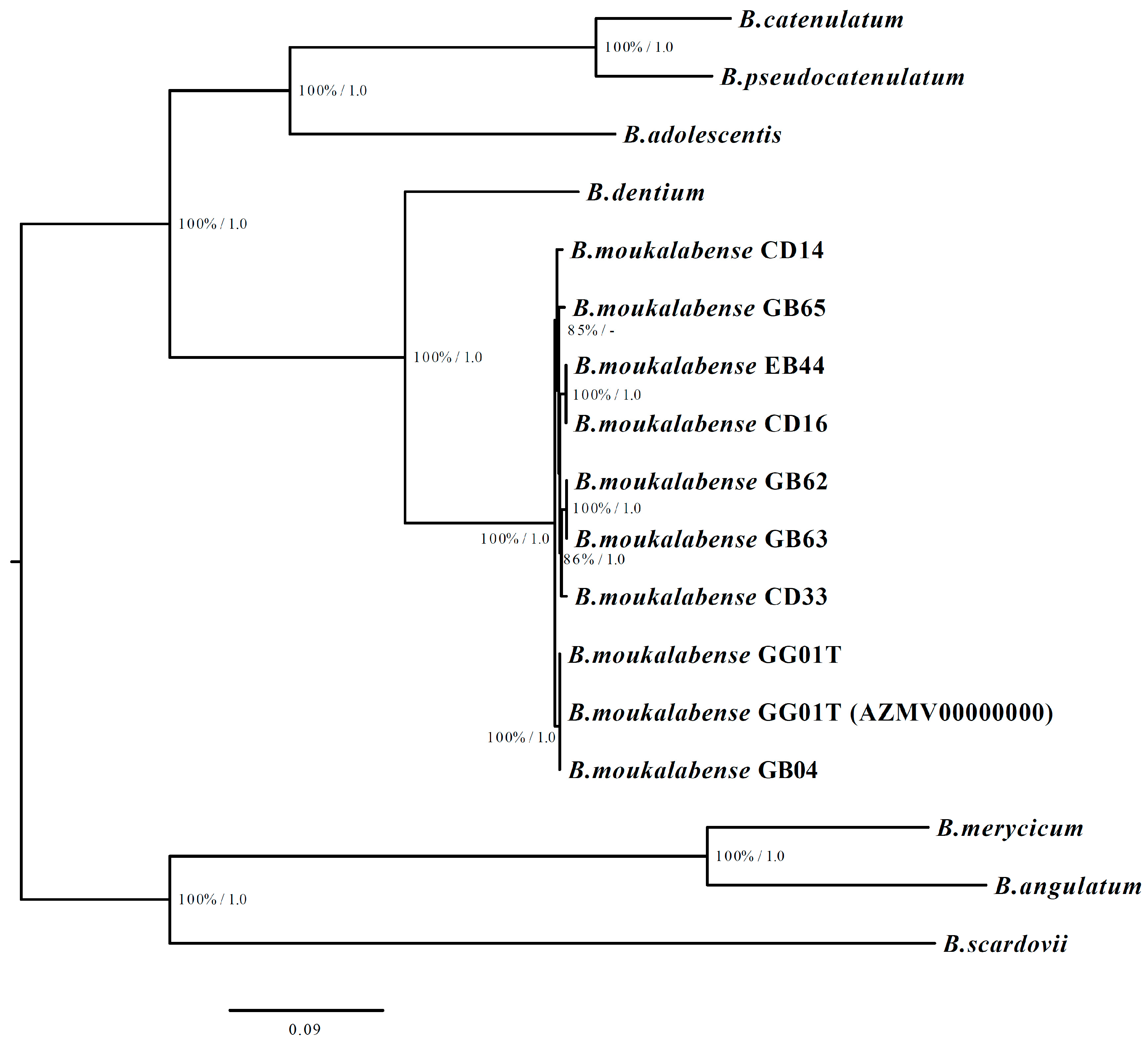
2.3. Phylogenetic Tree Construction Based on Core-Genome
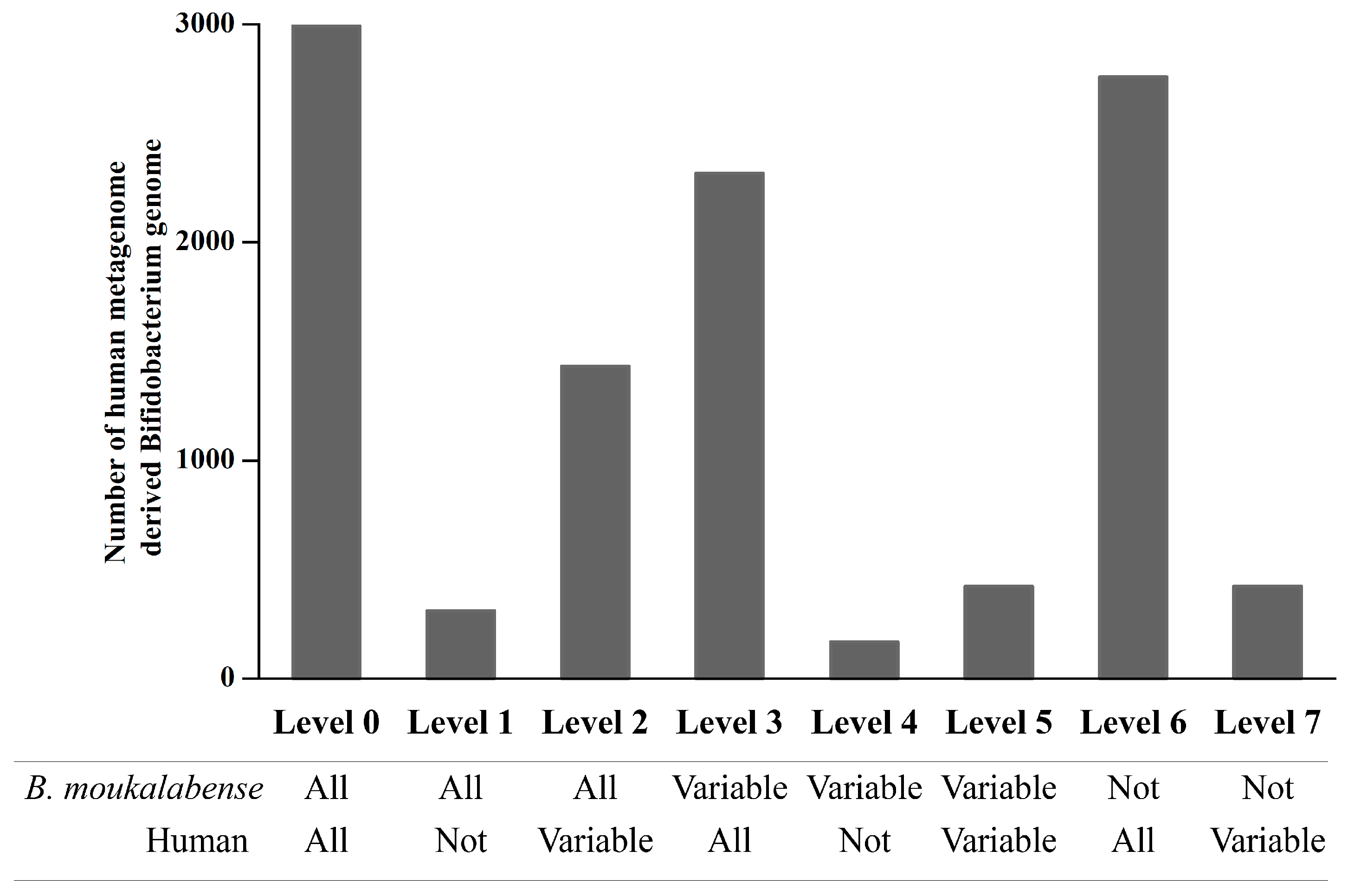
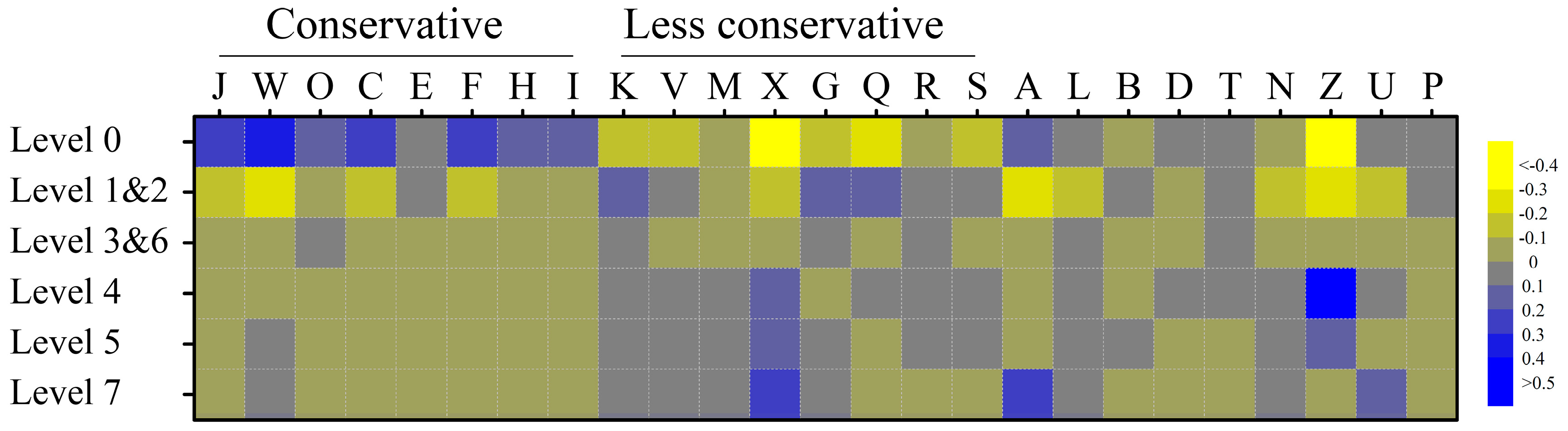
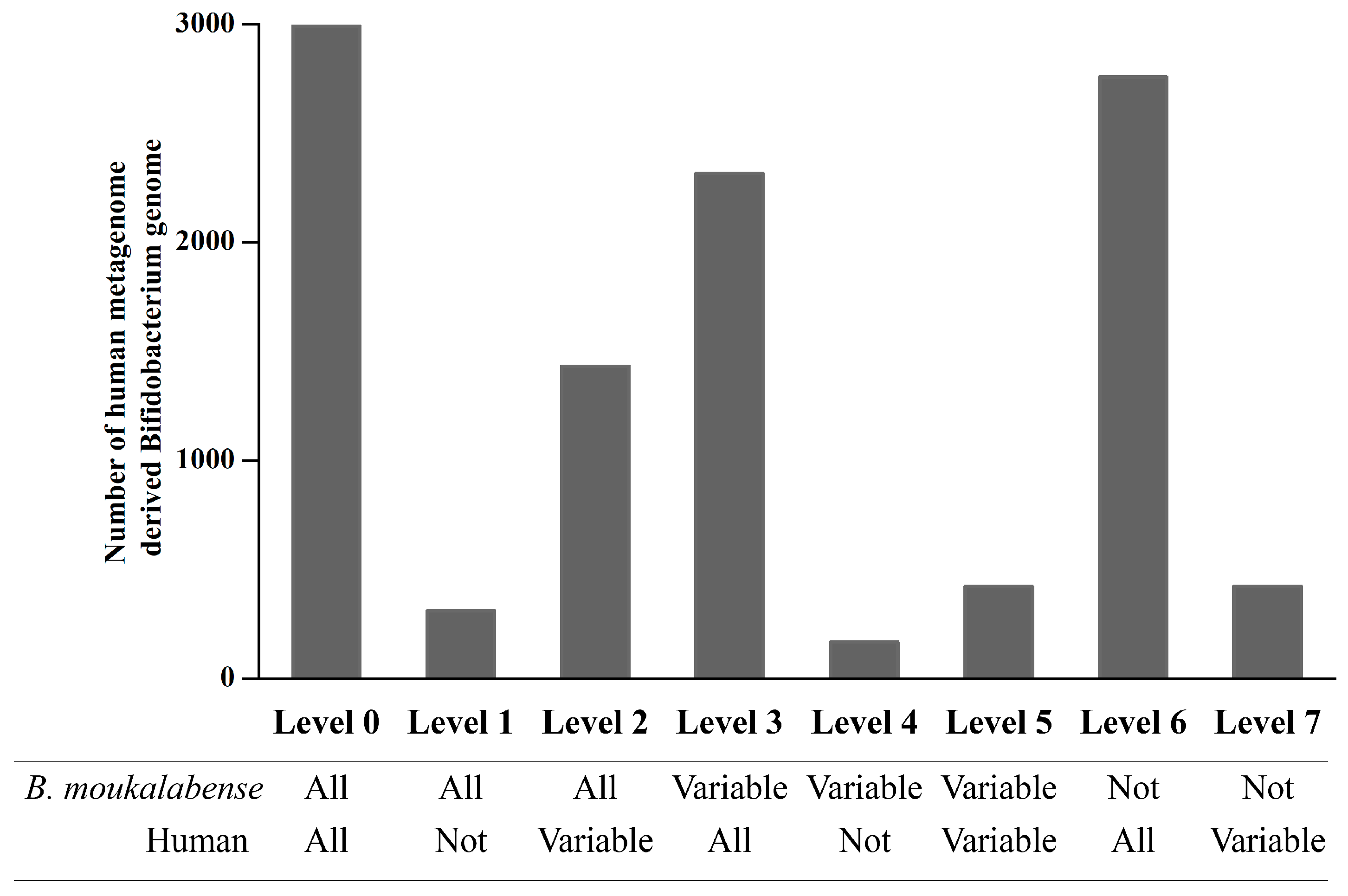
2.4. Orthologous Abundance Comparison of the Human Metagenome-Derived Bifidobacterium Genomes
2.5. High Throughput Sequencing of 16S rRNA Genes on Feces Samples
2.6. Ethics
3. Results
4. Discussion
Data Accessibility
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.-M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; Edney, R.J.; Whelan, K. Systematic review: Faecal microbiota transplantation in the management of inflammatory bowel disease. Aliment. Pharmacol. Ther. 2012, 36, 503–516. [Google Scholar] [CrossRef]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; de Roos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.-M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Belcheva, A.; Irrazabal, T.; Robertson, S.J.; Streutker, C.; Maughan, H.; Rubino, S.; Moriyama, E.H.; Copeland, J.K.; Surendra, A.; Kumar, S.; et al. Gut Microbial Metabolism Drives Transformation of Msh2-Deficient Colon Epithelial Cells. Cell 2014, 158, 288–299. [Google Scholar] [CrossRef]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, Receptor for Niacin and the Commensal Metabolite Butyrate, Suppresses Colonic Inflammation and Carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly-y, M.; Glickman, J.N.; Garrett, W.S. The Microbial Metabolites, Short-Chain Fatty Acids, Regulate Colonic T reg Cell Homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef]
- Ashraf, R.; Shah, N.P. Immune System Stimulation by Probiotic Microorganisms. Crit. Rev. Food Sci. Nutr. 2014, 54, 938–956. [Google Scholar] [CrossRef]
- Langkamp-Henken, B.; Rowe, C.C.; Ford, A.L.; Christman, M.C.; Nieves, C.; Khouri, L.; Specht, G.J.; Girard, S.-A.; Spaiser, S.J.; Dahl, W.J. Bifidobacterium bifidum R0071 results in a greater proportion of healthy days and a lower percentage of academically stressed students reporting a day of cold/flu: A randomised, double-blind, placebo-controlled study. Br. J. Nutr. 2015, 113, 426–434. [Google Scholar] [CrossRef] [PubMed]
- Arboleya, S.; Watkins, C.; Stanton, C.; Ross, R.P. Gut Bifidobacteria Populations in Human Health and Aging. Front. Microbiol. 2016, 7, 1204. [Google Scholar] [CrossRef] [PubMed]
- O’Callaghan, A.; van Sinderen, D. Bifidobacteria and Their Role as Members of the Human Gut Microbiota. Front. Microbiol. 2016, 7, 925. [Google Scholar] [CrossRef]
- Turroni, F.; Berry, D.; Ventura, M. Editorial: Bifidobacteria and Their Role in the Human Gut Microbiota. Front. Microbiol. 2017, 7, 2148. [Google Scholar] [CrossRef]
- Holzapfel, W.H.; Wood, B.J.B. Introduction to the LAB. In Lactic Acid Bacteria; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2014; pp. 1–12. [Google Scholar] [CrossRef]
- Mitsuoka, T.; Kaneuchi, C. Ecology of the bifidobacteria. Am. J. Clin. Nutr. 1977, 30, 1799–1810. [Google Scholar] [CrossRef]
- Bottacini, F.; Ventura, M.; van Sinderen, D.; O’Connell Motherway, M. Diversity, ecology and intestinal function of bifidobacteria. Microb. Cell Fact. 2014, 13, S4. [Google Scholar] [CrossRef]
- Ventura, M.; Turroni, F.; van Sinderen, D. Bifidobacteria of the Human Gut: Our Special Friends A2—Tuohy, Kieran. In Diet-Microbe Interactions in the Gut; Rio, D.D., Ed.; Academic Press: San Diego, CA, USA, 2015; Chapter 4; pp. 41–51. [Google Scholar] [CrossRef]
- Schell, M.A.; Karmirantzou, M.; Snel, B.; Vilanova, D.; Berger, B.; Pessi, G.; Zwahlen, M.-C.; Desiere, F.; Bork, P.; Delley, M.; et al. The genome sequence of Bifidobacterium longum reflects its adaptation to the human gastrointestinal tract. Proc. Natl. Acad. Sci. USA 2002, 99, 14422–14427. [Google Scholar] [CrossRef]
- Sela, D.A.; Chapman, J.; Adeuya, A.; Kim, J.H.; Chen, F.; Whitehead, T.R.; Lapidus, A.; Rokhsar, D.S.; Lebrilla, C.B.; German, J.B.; et al. The genome sequence of Bifidobacterium longum subsp. infantis reveals adaptations for milk utilization within the infant microbiome. Proc. Natl. Acad. Sci. USA 2008, 105, 18964–18969. [Google Scholar]
- Turroni, F.; Bottacini, F.; Foroni, E.; Mulder, I.; Kim, J.-H.; Zomer, A.; Sánchez, B.; Bidossi, A.; Ferrarini, A.; Giubellini, V.; et al. Genome analysis of Bifidobacterium bifidum PRL2010 reveals metabolic pathways for host-derived glycan foraging. Proc. Natl. Acad. Sci. USA 2010, 107, 19514–19519. [Google Scholar] [CrossRef]
- Barrangou, R.; Briczinski, E.P.; Traeger, L.L.; Loquasto, J.R.; Richards, M.; Horvath, P.; Coûté-Monvoisin, A.-C.; Leyer, G.; Rendulic, S.; Steele, J.L.; et al. Comparison of the Complete Genome Sequences of Bifidobacterium animalis subsp. lactis DSM 10140 and Bl-04. J. Bacteriol. 2009, 191, 4144–4151. [Google Scholar] [CrossRef]
- Andriantsoanirina, V.; Allano, S.; Butel, M.J.; Aires, J. Tolerance of Bifidobacterium human isolates to bile, acid and oxygen. Anaerobe 2013, 21, 39–42. [Google Scholar] [CrossRef]
- Ruiz, L.; Margolles, A.; Sánchez, B. Bile resistance mechanisms in Lactobacillus and Bifidobacterium. Front. Microbiol. 2013, 4, 396. [Google Scholar] [CrossRef]
- Tsuchida, S.; Takahashi, S.; Nguema, P.P.M.; Fujita, S.; Kitahara, M.; Yamagiwa, J.; Ngomanda, A.; Ohkuma, M.; Ushida, K. Bifidobacterium moukalabense sp. nov., isolated from the faeces of wild west lowland gorilla (Gorilla gorilla gorilla). Int. J. Syst. Evol. Microbiol. 2014, 64, 449–455. [Google Scholar] [CrossRef]
- Lugli, G.A.; Duranti, S.; Milani, C.; Turroni, F.; Viappiani, A.; Mangifesta, M.; van Sinderen, D.; Ventura, M. The Genome Sequence of Bifidobacterium moukalabense DSM 27321 Highlights the Close Phylogenetic Relatedness with the Bifidobacterium dentium Taxon. Genome Announc. 2014, 2, e00048-00014. [Google Scholar] [CrossRef]
- Kitahara, M.; Sakamoto, M.; Ike, M.; Sakata, S.; Benno, Y. Bacteroides plebeius sp. nov. and Bacteroides coprocola sp. nov., isolated from human faeces. Int. J. Syst. Evol. Microbiol. 2005, 55, 2143–2147. [Google Scholar] [CrossRef]
- Edwards, R.; Schmieder, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Bolger, A.M.; Usadel, B.; Lohse, M. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Contreras-Moreira, B.; Vinuesa, P. GET_HOMOLOGUES, a Versatile Software Package for Scalable and Robust Microbial Pangenome Analysis. Appl. Environ. Microbiol. 2013, 79, 7696–7701. [Google Scholar] [CrossRef]
- Li, L.; Stoeckert, C.J.; Roos, D.S. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A.; Hoover, P.; Rougemont, J. A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 2008, 57, 758–771. [Google Scholar] [CrossRef] [PubMed]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef]
- Pasolli, E.; Asnicar, F.; Manara, S.; Zolfo, M.; Karcher, N.; Armanini, F.; Beghini, F.; Manghi, P.; Tett, A.; Ghensi, P.; et al. Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle. Cell 2019, 176, 649–662. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J.J.B.B. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Steinegger, M.; Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 2017, 35, 1026–1028. [Google Scholar] [CrossRef]
- Hayaishi, S.; Kawamoto, Y. Low genetic diversity and biased distribution of mitochondrial DNA haplotypes in the Japanese macaque (Macaca fuscata yakui) on Yakushima Island. Primates 2006, 47, 158–164. [Google Scholar] [CrossRef]
- Herlemann, D.P.R.; Labrenz, M.; Jürgens, K.; Bertilsson, S.; Waniek, J.J.; Andersson, A.F. Transitions in bacterial communities along the 2000 km salinity gradient of the Baltic Sea. ISME J. 2011, 5, 1571–1579. [Google Scholar] [CrossRef]
- Rathnayake, R.M.L.D.; Oshiki, M.; Ishii, S.; Segawa, T.; Satoh, H.; Okabe, S. Effects of dissolved oxygen and pH on nitrous oxide production rates in autotrophic partial nitrification granules. Bioresour. Technol. 2015, 197, 15–22. [Google Scholar] [CrossRef]
- Mori, H.; Maruyama, T.; Yano, M.; Yamada, T.; Kurokawa, K.J.B.S.B. VITCOMIC2: Visualization tool for the phylogenetic composition of microbial communities based on 16S rRNA gene amplicons and metagenomic shotgun sequencing. BMC Syst. Biol. 2018, 12, 30. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Ushida, K.; Uwatoko, Y.; Adachi, Y.; Soumah, A.G.; Matsuzawa, T. Isolation of Bifidobacteria from feces of chimpanzees in the wild. J. Gen. Appl. Microbiol. 2010, 56, 57–60. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, S.; Ushida, K. Characterization of intestinal bacterial communities of western lowland gorillas Gorilla gorilla gorilla, central chimpanzees Pan troglodytes troglodytes, and a forest elephant Loxodonta africana cyclotis living in Moukalaba-Doudou National Park in Gabon. Tropics 2015, 23, 175–183. [Google Scholar] [CrossRef]
- Muegge, B.D.; Kuczynski, J.; Knights, D.; Clemente, J.C.; González, A.; Fontana, L.; Henrissat, B.; Knight, R.; Gordon, J.I. Diet Drives Convergence in Gut Microbiome Functions Across Mammalian Phylogeny and Within Humans. Science 2011, 332, 970–974. [Google Scholar] [CrossRef] [PubMed]
- Short, J. Diet and feeding behaviour of the forest elephant. Mammalia 1981, 45, 177–185. [Google Scholar] [CrossRef]
- Yamagiwa, J.; Tsubokawa, K.; Inoue, E.; Ando, C.J.P. Sharing fruit of Treculia africana among western gorillas in the Moukalaba-Doudou National Park, Gabon: Preliminary report. Primates 2015, 56, 3–10. [Google Scholar] [CrossRef]
- Wilfried, E.E.; Yamagiwa, J. Use of tool sets by chimpanzees for multiple purposes in Moukalaba-Doudou National Park, Gabon. Primates 2014, 55, 467–472. [Google Scholar] [CrossRef]
- Ushida, K. Intestinal bacteria of chimpanzees in the wild and in captivity: An application of molecular ecological methodologies. In Primate Parasite Ecology “The Dynamics and Study of Host-Parasite Relationships”; Cambridge University Press: Cambridge, UK, 2009; pp. 283–295. [Google Scholar]
- Uenishi, G.; Fujita, S.; Ohashi, G.; Kato, A.; Yamauchi, S.; Matsuzawa, T.; Ushida, K. Molecular analyses of the intestinal microbiota of chimpanzees in the wild and in captivity. Am. J. Primatol. 2007, 69, 367–376. [Google Scholar] [CrossRef]
- Tsuchida, S.; Maruyama, F.; Ogura, Y.; Toyoda, A.; Hayashi, T.; Okuma, M.; Ushida, K. Genomic Characteristics of Bifidobacterium thermacidophilum Pig Isolates and Wild Boar Isolates Reveal the Unique Presence of a Putative Mobile Genetic Element with tetW for Pig Farm Isolates. Front. Microbiol. 2017, 8, 1540. [Google Scholar] [CrossRef]
- Ballari, S.A.; Barrios-García, M.N. A review of wild boar Sus scrofa diet and factors affecting food selection in native and introduced ranges. Mamm. Rev. 2014, 44, 124–134. [Google Scholar] [CrossRef]
- Cheeke, P.R.; Dierenfeld, E.S. Comparative Animal Nutrition and Metabolism; Cambridge University Press: Cambridge, UK, 2010. [Google Scholar]
- Mann, N. Meat in the human diet: An anthropological perspective. Nutr. Diet. 2007, 64 (Suppl. 4), S102–S107. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Bacterial Taxon | Strain Name | Source of Isolate | Common Name | Location of Collection | Date of Isolation | Collected by | Acession No. |
|---|---|---|---|---|---|---|---|
| Bifidobacterium moukalabense | GG01T (JCM18751, DSM27321) | Gorilla gorilla gorilla | Western Lowland Gorilla | 2°19′38″S 10°34′02″E | 24/11/2010 | K. Ushida and P.P.Nuguma | PRJDB7909, AZMV00000000 |
| GB01 | Gorilla gorilla gorilla | Western Lowland Gorilla | 2°19′53″S 10°34′06″E | 28/10/2009 | K. Ushida and P.P.Nuguma | PRJDB7909 | |
| GB03 | Gorilla gorilla gorilla | Western Lowland Gorilla | 2°19′51″S 10°34′19″E | 01/11/2009 | K. Ushida and P.P.Nuguma | PRJDB7909 | |
| GB04 | Gorilla gorilla gorilla | Western Lowland Gorilla | 2°19′53″S 10°34′21″E | 24/11/2010 | K. Ushida and P.P.Nuguma | PRJDB7909 | |
| GB62 | Gorilla gorilla gorilla | Western Lowland Gorilla | 2°20′02″S 10°34′05″E | 25/11/2010 | K. Ushida and P.P.Nuguma | PRJDB7909 | |
| GB63 | Gorilla gorilla gorilla | Western Lowland Gorilla | 2°19′49″S 10°34′10″E | 26/11/2010 | K. Ushida and P.P.Nuguma | PRJDB7909 | |
| GB65 | Gorilla gorilla gorilla | Western Lowland Gorilla | 2°19′50″S 10°34′05″E | 27/11/2010 | K. Ushida and P.P.Nuguma | PRJDB7909 | |
| CD14 | Pan troglodytes troglodytes | Central chimpanzee | 2°23′08″S 10°33′14″E | 29/11/2011 | S. Tsuchida and P.P Nguema | PRJDB7909 | |
| CD16 | Pan troglodytes troglodytes | Central chimpanzee | 2°23′08″S 10°33′14″E | 29/11/2011 | S. Tsuchida and P.P Nguema | PRJDB7909 | |
| CD33 | Pan troglodytes troglodytes | Central chimpanzee | 2°23′08″S 10°33′14″E | 29/11/2011 | S. Tsuchida and P.P Nguema | PRJDB7909 | |
| EB43 | Loxodonta cyclotis | African forest elephant | 2°20′12″S 10°34′31″E | 01/11/2009 | K. Ushida and P.P.Nuguma | PRJDB7909 | |
| EB44 | Loxodonta cyclotis | African forest elephant | 2°20′12″S 10°34′31″E | 01/11/2009 | K. Ushida and P.P.Nuguma | PRJDB7909 | |
| Bifidobacterium catenulatum | DSM 16992 | Homo sapiens | Human | ID_BIFCAT_00411 | |||
| Bifidobacterium dentium | ATCC 27678 | Homo sapiens | Human | ID_BIFDEN_01570 | |||
| Bifidobacterium dentium | ATCC 27679 | Homo sapiens | Human | ID_HMPREF0168_0306 | |||
| Bifidobacterium dentium | Bd1 | Homo sapiens | Human | ID_BDP_1602 | |||
| Bifidobacterium dentium | JCVIHMP022 | Homo sapiens | Human | ID_HMPREF9003_0584 | |||
| Bifidobacterium longum | DJO10A | Homo sapiens | Human | ID_BLD_0124 | |||
| Bifidobacterium pseudocatenulatum | DSM 20438 | Homo sapiens | Human | ID_BIFPSEUDO_03077 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Segawa, T.; Fukuchi, S.; Bodington, D.; Tsuchida, S.; Mbehang Nguema, P.P.; Mori, H.; Ushida, K. Genomic Analyses of Bifidobacterium moukalabense Reveal Adaptations to Frugivore/Folivore Feeding Behavior. Microorganisms 2019, 7, 99. https://doi.org/10.3390/microorganisms7040099
Segawa T, Fukuchi S, Bodington D, Tsuchida S, Mbehang Nguema PP, Mori H, Ushida K. Genomic Analyses of Bifidobacterium moukalabense Reveal Adaptations to Frugivore/Folivore Feeding Behavior. Microorganisms. 2019; 7(4):99. https://doi.org/10.3390/microorganisms7040099
Chicago/Turabian StyleSegawa, Takahiro, Satoshi Fukuchi, Dylan Bodington, Sayaka Tsuchida, Pierre Philippe Mbehang Nguema, Hiroshi Mori, and Kazunari Ushida. 2019. "Genomic Analyses of Bifidobacterium moukalabense Reveal Adaptations to Frugivore/Folivore Feeding Behavior" Microorganisms 7, no. 4: 99. https://doi.org/10.3390/microorganisms7040099
APA StyleSegawa, T., Fukuchi, S., Bodington, D., Tsuchida, S., Mbehang Nguema, P. P., Mori, H., & Ushida, K. (2019). Genomic Analyses of Bifidobacterium moukalabense Reveal Adaptations to Frugivore/Folivore Feeding Behavior. Microorganisms, 7(4), 99. https://doi.org/10.3390/microorganisms7040099