Probiotics and Prebiotics for the Amelioration of Type 1 Diabetes: Present and Future Perspectives

 ,
,
Abstract
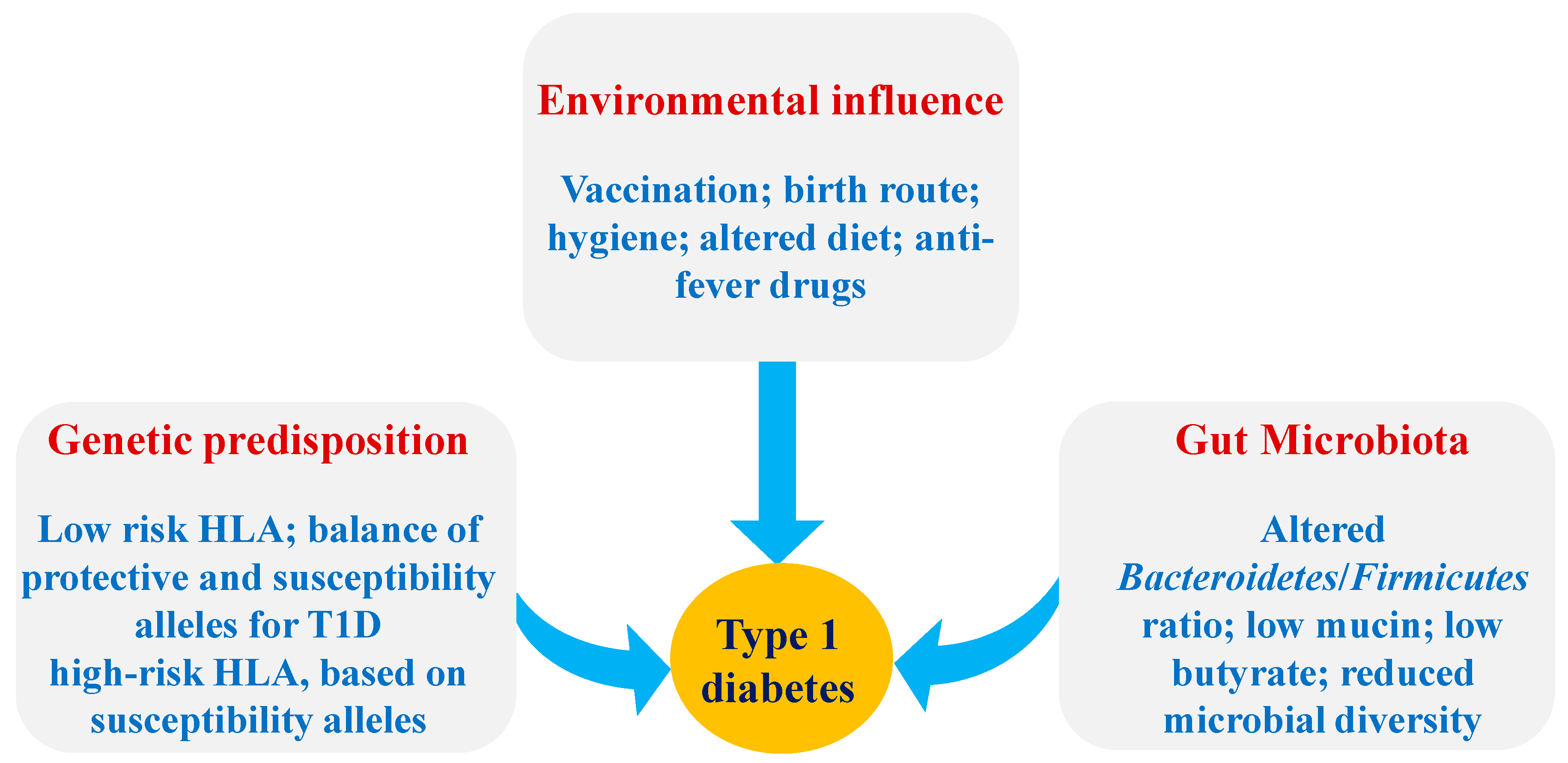
:1. Introduction
2. Role of the Gut Microbiota-Immune Axis in T1D
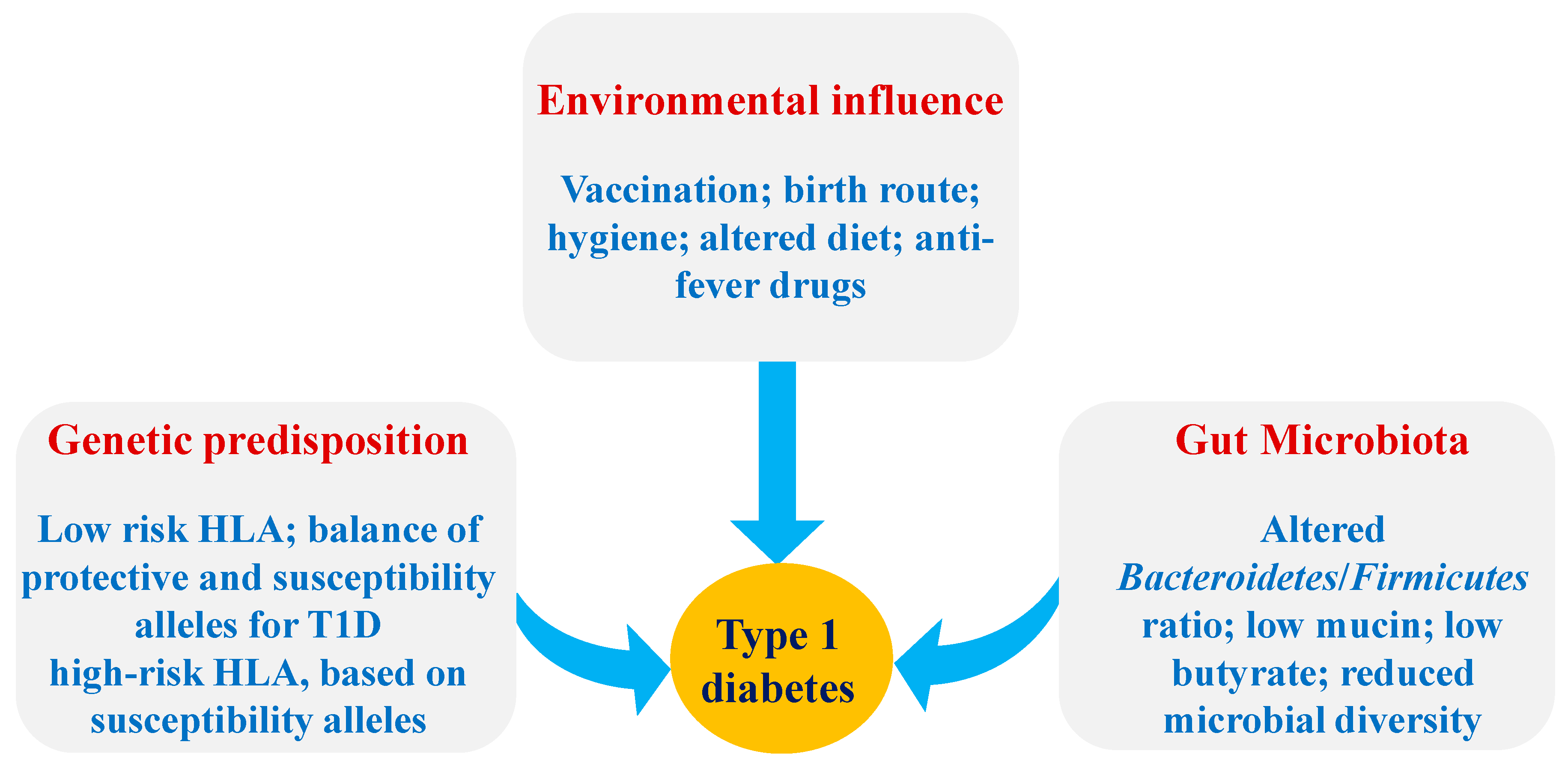
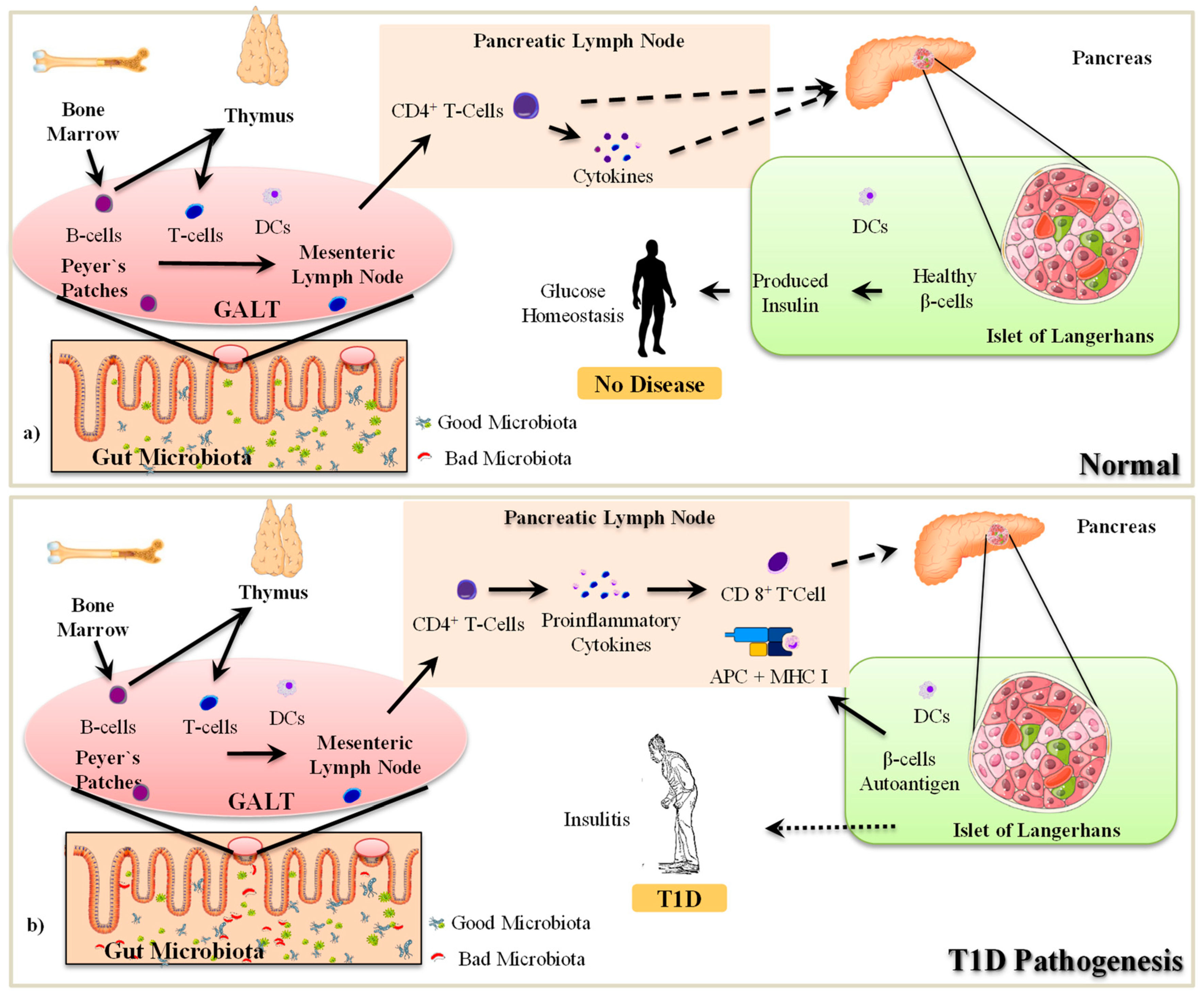
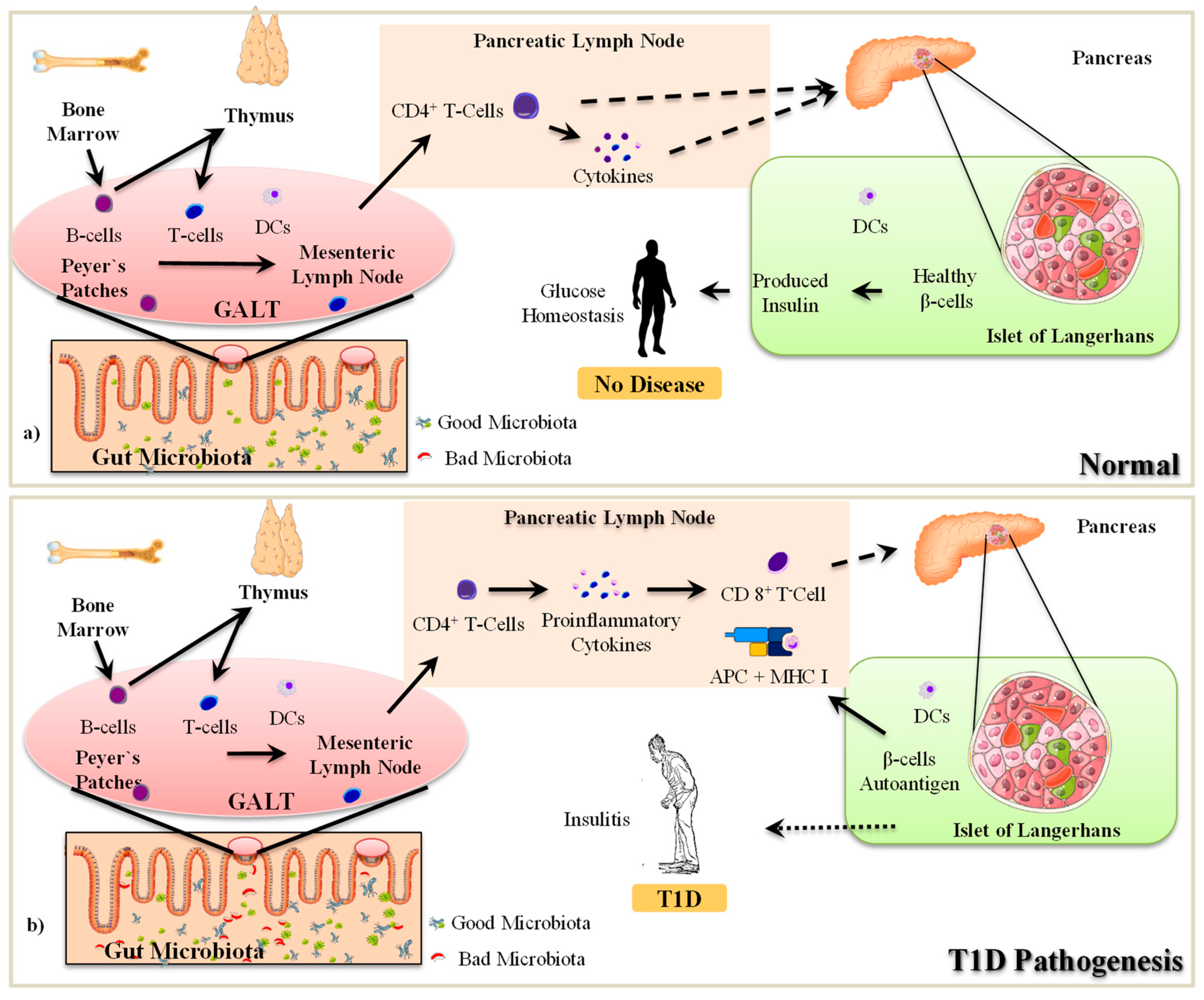
2.1. The Pathogenesis of T1D
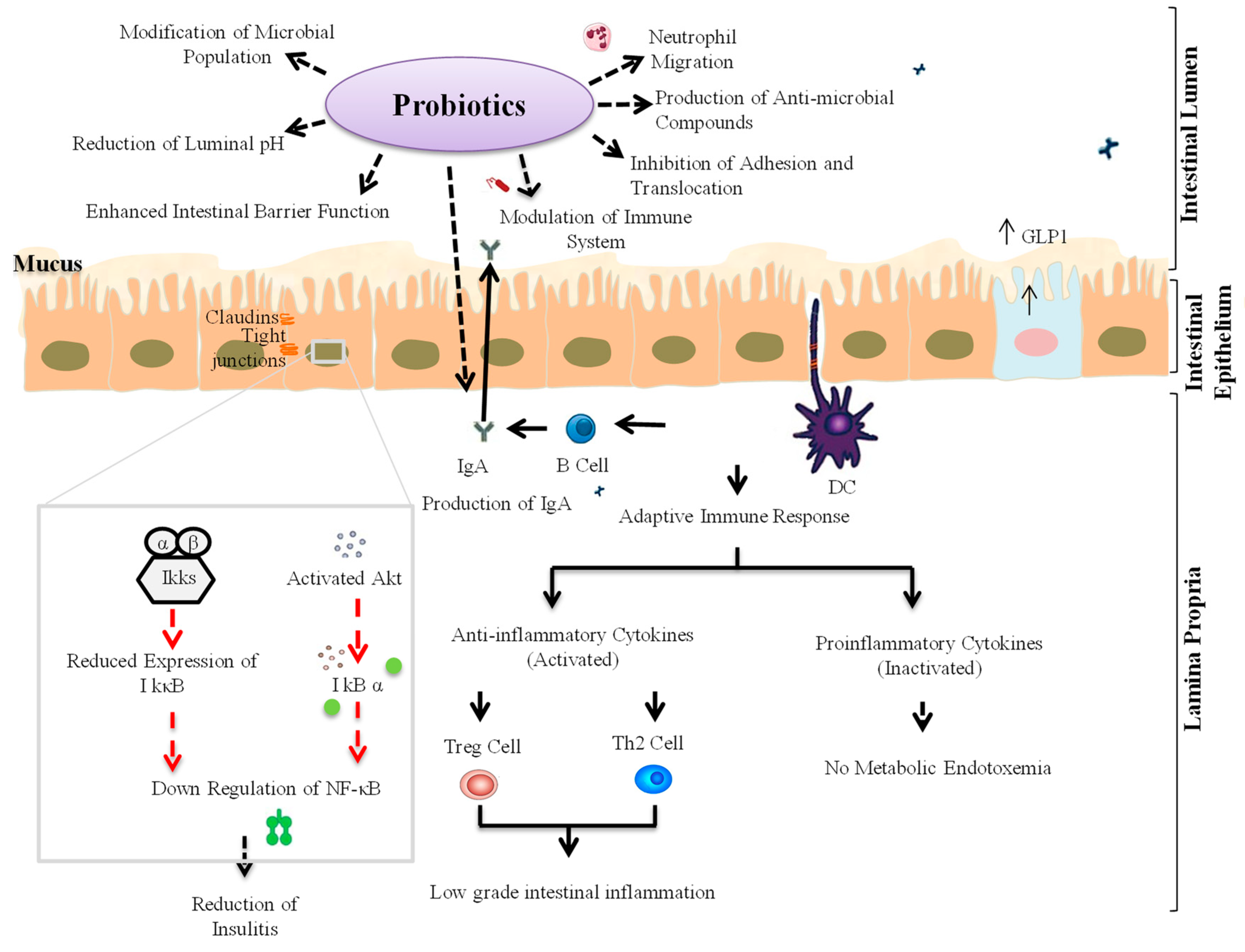
2.2. Gut Microbiota-Immune Interactions in T1D
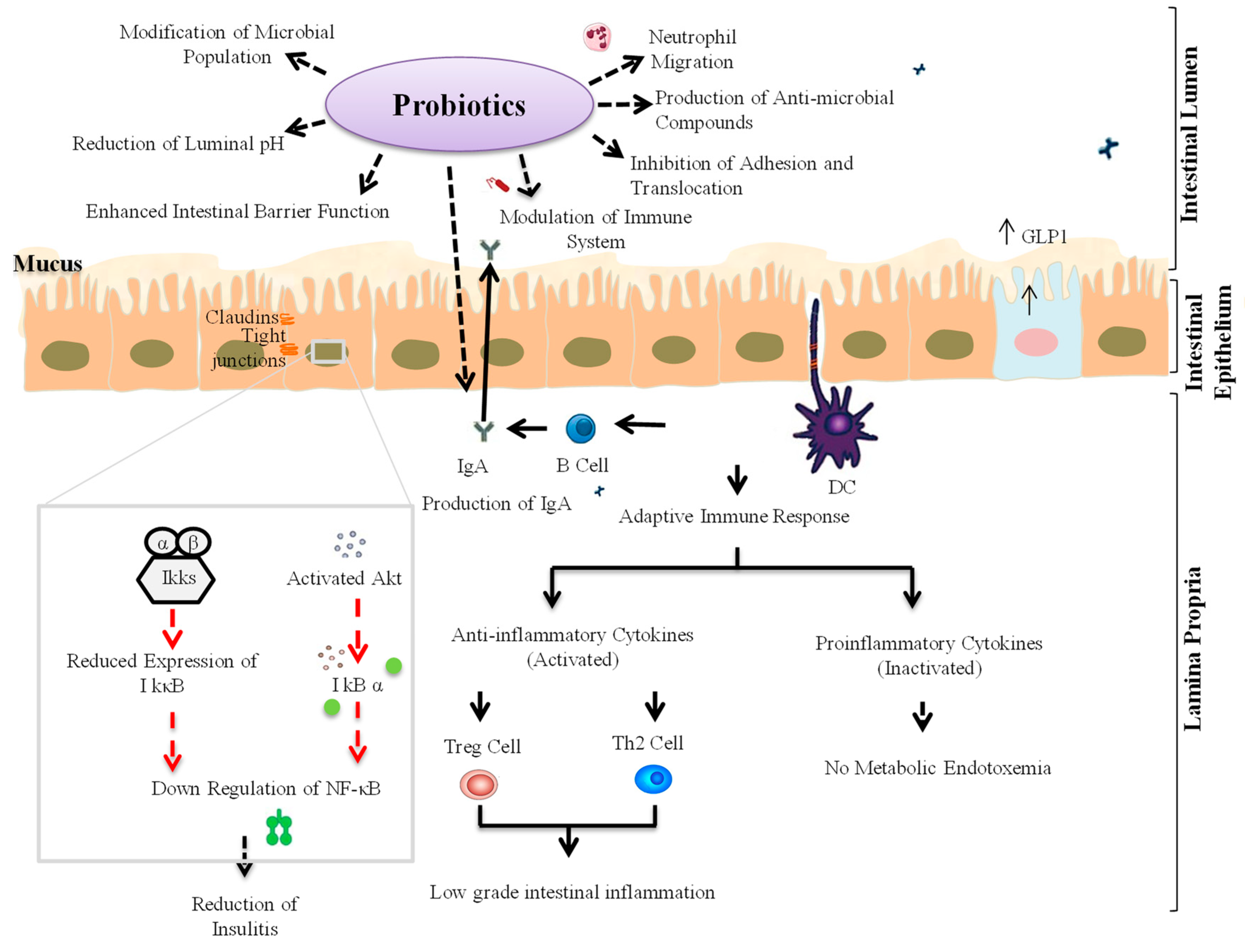
3. Probiotic Interventions to Ameliorate T1D
3.1. Animal Studies
3.2. Human Studies
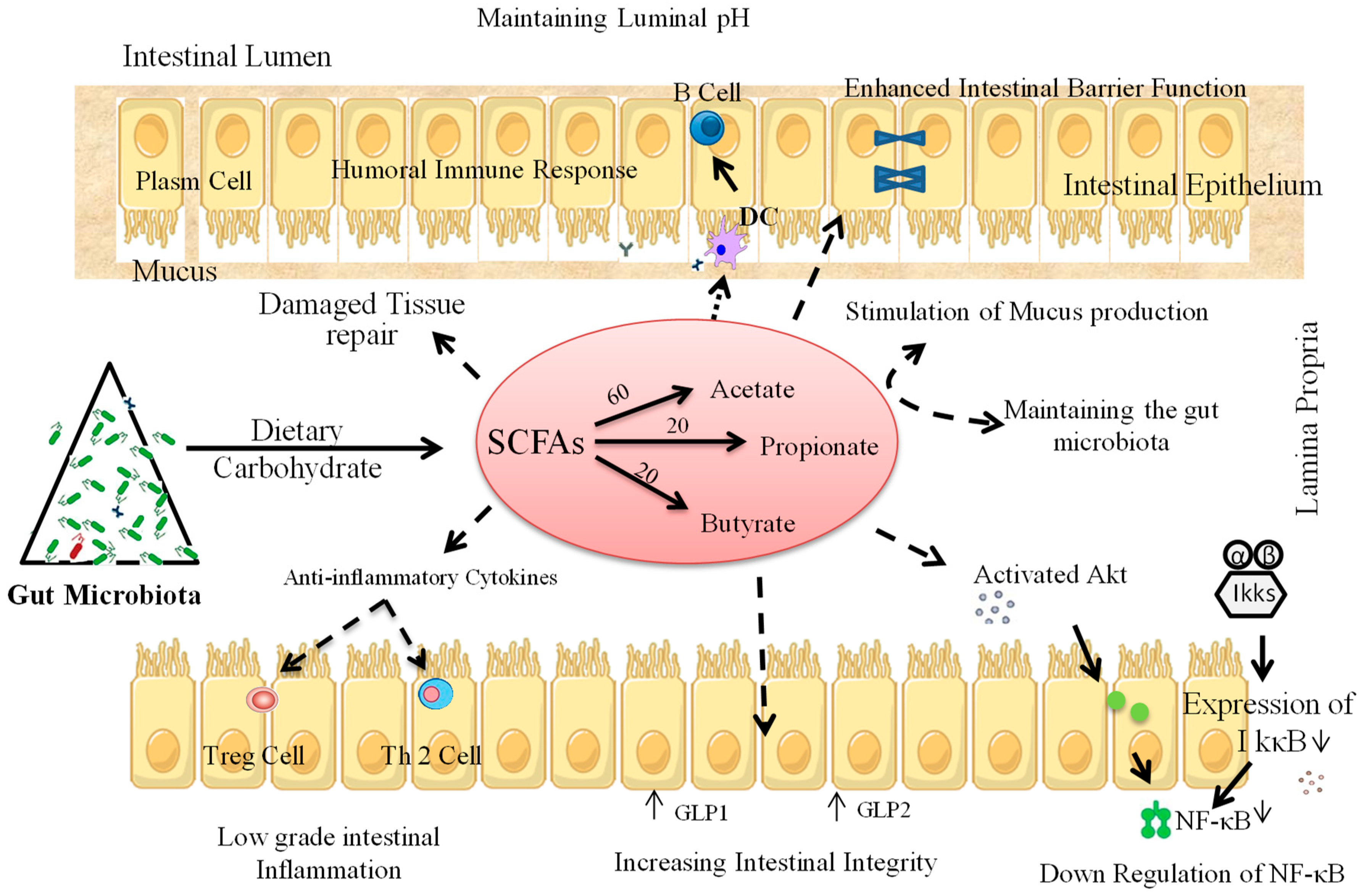
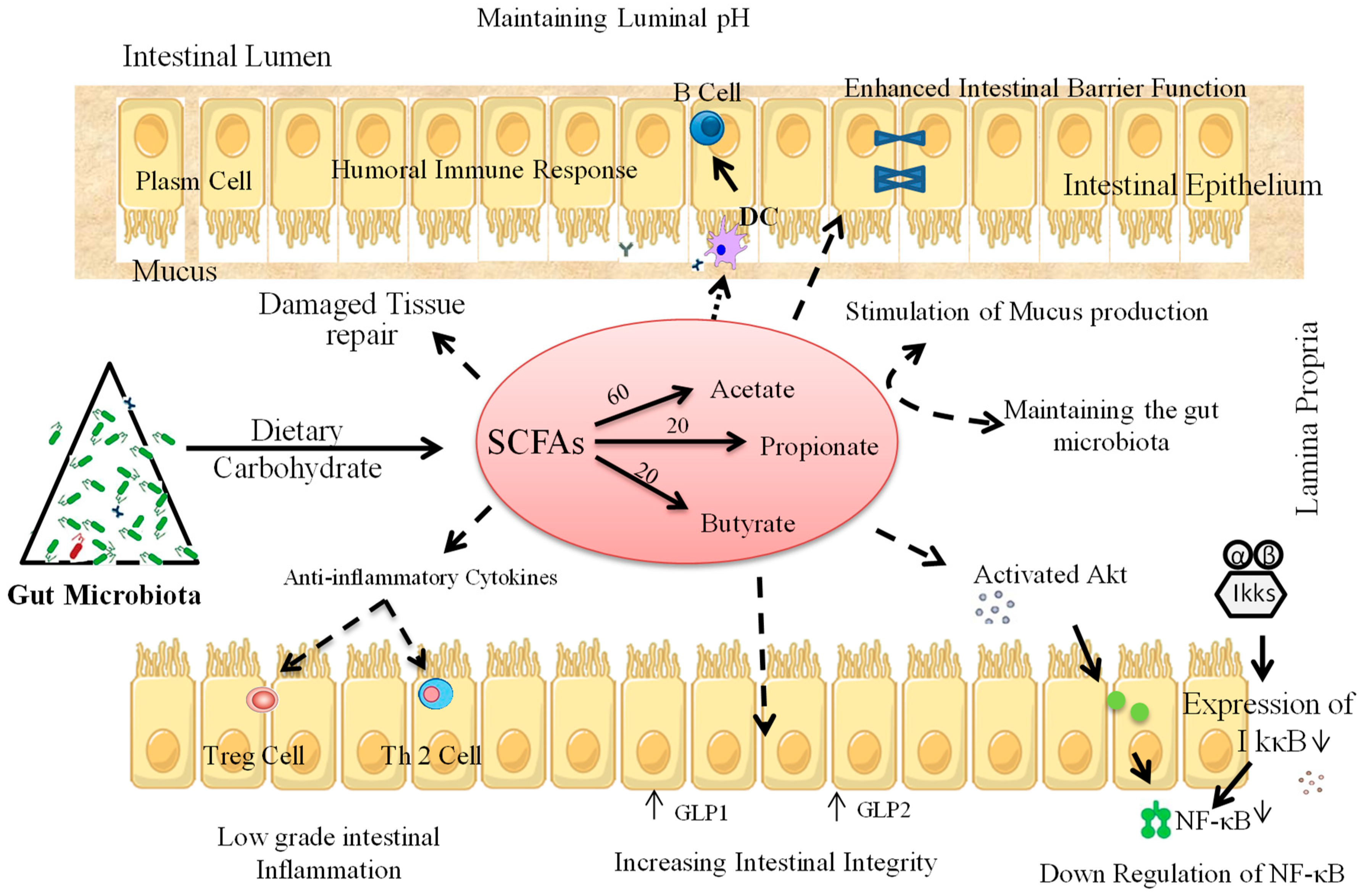
4. Prebiotic Interventions to Ameliorate T1D
4.1. Animal Studies
4.2. Human Studies
5. Other Gut Microbiota Modulators in T1D
6. Conclusions and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Lamichhane, S.; Ahonen, L.; Dyrlund, T.S.; Siljander, H.; Hyoty, H.; Ilonen, J.; Toppari, J.; Veijola, R.; Hyotylainen, T.; Knip, M.; et al. A longitudinal plasma lipidomics dataset from children who developed islet autoimmunity and type 1 diabetes. Sci. Data 2018, 5, 180250. [Google Scholar] [CrossRef] [PubMed]
- Patterson, C.C.; Harjutsalo, V.; Rosenbauer, J.; Neu, A.; Cinek, O.; Skrivarhaug, T.; Rami-Merhar, B.; Soltesz, G.; Svensson, J.; Parslow, R.C.; et al. Trends and cyclical variation in the incidence of childhood type 1 diabetes in 26 European centres in the 25 year period 1989–2013: A multicentre prospective registration study. Diabetologia 2018. [Google Scholar] [CrossRef] [PubMed]
- Rewers, M.; Hyoty, H.; Lernmark, A.; Hagopian, W.; She, J.X.; Schatz, D.; Ziegler, A.G.; Toppari, J.; Akolkar, B.; Krischer, J.; et al. The Environmental Determinants of Diabetes in the Young (TEDDY) Study: 2018 Update. Curr. Diabetes Rep. 2018, 18, 136. [Google Scholar] [CrossRef] [PubMed]
- Battaglia, M.; Atkinson, M.A. The streetlight effect in type 1 diabetes. Diabetes 2015, 64, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Pociot, F.; Lernmark, A. Genetic risk factors for type 1 diabetes. Lancet 2016, 387, 2331–2339. [Google Scholar] [CrossRef]
- Rewers, M.; Ludvigsson, J. Environmental risk factors for type 1 diabetes. Lancet 2016, 387, 2340–2348. [Google Scholar] [CrossRef]
- Knip, M.; Simell, O. Environmental triggers of type 1 diabetes. Cold Spring Harb. Perspect. Med. 2012, 2, a007690. [Google Scholar] [CrossRef] [PubMed]
- Vatanen, T.; Franzosa, E.A.; Schwager, R.; Tripathi, S.; Arthur, T.D.; Vehik, K.; Lernmark, A.; Hagopian, W.A.; Rewers, M.J.; She, J.X.; et al. The human gut microbiome in early-onset type 1 diabetes from the TEDDY study. Nature 2018, 562, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Nageshwar Reddy, D. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787–8803. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Li, Y.; Fang, J.; Liu, G.; Yin, J.; Li, T.; Yin, Y. Gut Microbiota and Type 1 Diabetes. Int. J. Mol. Sci. 2018, 19, 995. [Google Scholar] [CrossRef] [PubMed]
- Schwiertz, A.; Taras, D.; Schafer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in lean and overweight healthy subjects. Obesity (Silver Spring) 2010, 18, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Moya-Perez, A.; Luczynski, P.; Renes, I.B.; Wang, S.; Borre, Y.; Anthony Ryan, C.; Knol, J.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Intervention strategies for cesarean section-induced alterations in the microbiota-gut-brain axis. Nutr. Rev. 2017, 75, 225–240. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, R.; Tsuji, H.; Takahashi, T.; Kawashima, K.; Nagata, S.; Nomoto, K.; Yamashiro, Y. Sensitive Quantitative Analysis of the Meconium Bacterial Microbiota in Healthy Term Infants Born Vaginally or by Cesarean Section. Front. Microbiol. 2016, 7, 1997. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, R.; Kurakawa, T.; Tsuji, H.; Takahashi, T.; Kawashima, K.; Nagata, S.; Nomoto, K.; Yamashiro, Y. Evolution of gut Bifidobacterium population in healthy Japanese infants over the first three years of life: A quantitative assessment. Sci. Rep. 2017, 7, 10097. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, R.; Tsuji, H.; Takahashi, T.; Nomoto, K.; Kawashima, K.; Nagata, S.; Yamashiro, Y. Gut dysbiosis following C-section instigates higher colonisation of toxigenic Clostridium perfringens in infants. Benef. Microbes 2017, 8, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Aw, W.; Fukuda, S. Understanding the role of the gut ecosystem in diabetes mellitus. J. Diabetes Investig. 2018, 9, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Huang, G.; Wang, Z.; Luo, S.; Zheng, P.; Zhou, Z. Epigenetic regulation of Toll-like receptors and its roles in type 1 diabetes. J. Mol. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.K.; Shin, J.S.; Nahm, M.H. NOD-Like Receptors in Infection, Immunity, and Diseases. Yonsei Med. J. 2016, 57, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Priyadarshini, M.; Navarro, G.; Layden, B.T. Gut Microbiota: FFAR Reaching Effects on Islets. Endocrinology 2018, 159, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Ang, Z.; Ding, J.L. GPR41 and GPR43 in Obesity and Inflammation—Protective or Causative? Front. Immunol. 2016, 7, 28. [Google Scholar] [CrossRef] [PubMed]
- Knip, M.; Honkanen, J. Modulation of Type 1 Diabetes Risk by the Intestinal Microbiome. Curr. Diabetes Rep. 2017, 17, 105. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.; Guarner, F.; Reid, G.; Gibson, G.R.; Merenstein, D.J.; Pot, B.; Morelli, L.; Canani, R.B.; Flint, H.J.; Salminen, S.; et al. Expert consensus document. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.R.; Hutkins, R.; Sanders, M.E.; Prescott, S.L.; Reimer, R.A.; Salminen, S.J.; Scott, K.; Stanton, C.; Swanson, K.S.; Cani, P.D.; et al. Expert consensus document: The International Scientific Association for Probiotics and Prebiotics (ISAPP) consensus statement on the definition and scope of prebiotics. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 491–502. [Google Scholar] [CrossRef] [PubMed]
- Zheng, P.; Li, Z.; Zhou, Z. Gut microbiome in type 1 diabetes: A comprehensive review. Diabetes Metab. Res. Rev. 2018, 34, e3043. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.Y.; Kim, S.S. Probiotics and Prebiotics: Present Status and Future Perspectives on Metabolic Disorders. Nutrients 2016, 8, 173. [Google Scholar] [CrossRef] [PubMed]
- Drexhage, H.A.; Dik, W.A.; Leenen, P.J.; Versnel, M.A. The Immune Pathogenesis of Type 1 Diabetes: Not Only Thinking Outside the Cell but Also Outside the Islet and Out of the Box. Diabetes 2016, 65, 2130–2133. [Google Scholar] [CrossRef] [PubMed]
- Pushalkar, S.; Hundeyin, M.; Daley, D.; Zambirinis, C.P.; Kurz, E.; Mishra, A.; Mohan, N.; Aykut, B.; Usyk, M.; Torres, L.E.; et al. The Pancreatic Cancer Microbiome Promotes Oncogenesis by Induction of Innate and Adaptive Immune Suppression. Cancer Discov. 2018, 8, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Paschou, S.A.; Papadopoulou-Marketou, N.; Chrousos, G.P.; Kanaka-Gantenbein, C. On type 1 diabetes mellitus pathogenesis. Endocr. Connect. 2018, 7, R38–R46. [Google Scholar] [CrossRef] [PubMed]
- Campbell-Thompson, M.; Fu, A.; Kaddis, J.S.; Wasserfall, C.; Schatz, D.A.; Pugliese, A.; Atkinson, M.A. Insulitis and beta-Cell Mass in the Natural History of Type 1 Diabetes. Diabetes 2016, 65, 719–731. [Google Scholar] [CrossRef] [PubMed]
- Clark, M.; Kroger, C.J.; Tisch, R.M. Type 1 Diabetes: A Chronic Anti-Self-Inflammatory Response. Front. Immunol. 2017, 8, 1898. [Google Scholar] [CrossRef] [PubMed]
- Burrack, A.L.; Martinov, T.; Fife, B.T. T Cell-Mediated Beta Cell Destruction: Autoimmunity and Alloimmunity in the Context of Type 1 Diabetes. Front. Endocrinol. (Lausanne) 2017, 8, 343. [Google Scholar] [CrossRef] [PubMed]
- Cheung, S.S.; Ou, D.; Metzger, D.L.; Meloche, M.; Ao, Z.; Ng, S.S.; Owen, D.; Warnock, G.L. B7-H4 expression in normal and diseased human islet beta cells. Pancreas 2014, 43, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Van’t Land, B.; van de Worp, W.; Stahl, B.; Folkerts, G.; Garssen, J. Early-Life Nutritional Factors and Mucosal Immunity in the Development of Autoimmune Diabetes. Front. Immunol. 2017, 8, 1219. [Google Scholar] [CrossRef] [PubMed]
- Winer, D.A.; Winer, S.; Dranse, H.J.; Lam, T.K. Immunologic impact of the intestine in metabolic disease. J. Clin. Investig. 2017, 127, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Selmi, C.; Tang, R.; Gershwin, M.E.; Ma, X. The microbiome and autoimmunity: A paradigm from the gut-liver axis. Cell. Mol. Immunol. 2018, 15, 595–609. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.; Hugot, J.P.; Barreau, F. Peyer’s Patches: The Immune Sensors of the Intestine. Int. J. Inflam. 2010, 2010, 823710. [Google Scholar] [CrossRef] [PubMed]
- Ermund, A.; Gustafsson, J.K.; Hansson, G.C.; Keita, A.V. Mucus properties and goblet cell quantification in mouse, rat and human ileal Peyer’s patches. PLoS ONE 2013, 8, e83688. [Google Scholar] [CrossRef] [PubMed]
- Markov, A.G.; Falchuk, E.L.; Kruglova, N.M.; Radloff, J.; Amasheh, S. Claudin expression in follicle-associated epithelium of rat Peyer’s patches defines a major restriction of the paracellular pathway. Acta Physiol. 2016, 216, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Costa, F.R.; Francozo, M.C.; de Oliveira, G.G.; Ignacio, A.; Castoldi, A.; Zamboni, D.S.; Ramos, S.G.; Camara, N.O.; de Zoete, M.R.; Palm, N.W.; et al. Gut microbiota translocation to the pancreatic lymph nodes triggers NOD2 activation and contributes to T1D onset. J. Exp. Med. 2016, 213, 1223–1239. [Google Scholar] [CrossRef] [PubMed]
- Pabst, O.; Mowat, A.M. Oral tolerance to food protein. Mucosal Immunol. 2012, 5, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Winer, D.A.; Luck, H.; Tsai, S.; Winer, S. The Intestinal Immune System in Obesity and Insulin Resistance. Cell Metab. 2016, 23, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Maffeis, C.; Martina, A.; Corradi, M.; Quarella, S.; Nori, N.; Torriani, S.; Plebani, M.; Contreas, G.; Felis, G.E. Association between intestinal permeability and faecal microbiota composition in Italian children with beta cell autoimmunity at risk for type 1 diabetes. Diabetes Metab. Res. Rev. 2016, 32, 700–709. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.; Sun, C.; Gu, W.; Yang, M.; Zhang, X.; Zhai, N.; Lu, Y.; Zhang, Z.; Shou, P.; Zhang, Z.; et al. Free fatty acid receptor 2, a candidate target for type 1 diabetes, induces cell apoptosis through ERK signaling. J. Mol. Endocrinol. 2014, 53, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Ley, R.E.; Volchkov, P.Y.; Stranges, P.B.; Avanesyan, L.; Stonebraker, A.C.; Hu, C.; Wong, F.S.; Szot, G.L.; Bluestone, J.A.; et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature 2008, 455, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Scott, N.A.; Andrusaite, A.; Andersen, P.; Lawson, M.; Alcon-Giner, C.; Leclaire, C.; Caim, S.; Le Gall, G.; Shaw, T.; Connolly, J.P.R.; et al. Antibiotics induce sustained dysregulation of intestinal T cell immunity by perturbing macrophage homeostasis. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Tanca, A.; Palomba, A.; Fraumene, C.; Manghina, V.; Silverman, M.; Uzzau, S. Clostridial Butyrate Biosynthesis Enzymes Are Significantly Depleted in the Gut Microbiota of Nonobese Diabetic Mice. mSphere 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Karumuthil-Melethil, S.; Sofi, M.H.; Gudi, R.; Johnson, B.M.; Perez, N.; Vasu, C. TLR2- and Dectin 1-associated innate immune response modulates T-cell response to pancreatic beta-cell antigen and prevents type 1 diabetes. Diabetes 2015, 64, 1341–1357. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, S.; Dasu, M.R.; Rockwood, J.; Winter, W.; Griffen, S.C.; Jialal, I. Increased toll-like receptor (TLR) 2 and TLR4 expression in monocytes from patients with type 1 diabetes: Further evidence of a proinflammatory state. J. Clin. Endocrinol. Metab. 2008, 93, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Shibasaki, S.; Imagawa, A.; Tauriainen, S.; Iino, M.; Oikarinen, M.; Abiru, H.; Tamaki, K.; Seino, H.; Nishi, K.; Takase, I.; et al. Expression of toll-like receptors in the pancreas of recent-onset fulminant type 1 diabetes. Endocr. J. 2010, 57, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Zipris, D.; Lien, E.; Xie, J.X.; Greiner, D.L.; Mordes, J.P.; Rossini, A.A. TLR Activation Synergizes with Kilham Rat Virus Infection to Induce Diabetes in BBDR Rats. J. Immunol. 2004, 174, 131–142. [Google Scholar] [CrossRef]
- Assmann, T.S.; Brondani Lde, A.; Boucas, A.P.; Canani, L.H.; Crispim, D. Toll-like receptor 3 (TLR3) and the development of type 1 diabetes mellitus. Arch. Endocrinol. Metab. 2015, 59, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.K.; McKenzie, C.; Marino, E.; Macia, L.; Mackay, C.R. Metabolite-Sensing G Protein-Coupled Receptors-Facilitators of Diet-Related Immune Regulation. Annu. Rev. Immunol. 2017, 35, 371–402. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.S.; Li, J.; Krautkramer, K.A.; Badri, M.; Battaglia, T.; Borbet, T.C.; Koh, H.; Ng, S.; Sibley, R.A.; Li, Y.; et al. Antibiotic-induced acceleration of type 1 diabetes alters maturation of innate intestinal immunity. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.J.; Goldsworthy, S.M.; Barnes, A.A.; Eilert, M.M.; Tcheang, L.; Daniels, D.; Muir, A.I.; Wigglesworth, M.J.; Kinghorn, I.; Fraser, N.J.; et al. The Orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. 2003, 278, 11312–11319. [Google Scholar] [CrossRef] [PubMed]
- Bhutia, Y.D.; Ganapathy, V. Short, but Smart: SCFAs Train T Cells in the Gut to Fight Autoimmunity in the Brain. Immunity 2015, 43, 629–631. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Furio, L.; Mecheri, R.; van der Does, A.M.; Lundeberg, E.; Saveanu, L.; Chen, Y.; van Endert, P.; Agerberth, B.; Diana, J. Pancreatic beta-Cells Limit Autoimmune Diabetes via an Immunoregulatory Antimicrobial Peptide Expressed under the Influence of the Gut Microbiota. Immunity 2015, 43, 304–317. [Google Scholar] [CrossRef] [PubMed]
- Ohira, H.; Tsutsui, W.; Fujioka, Y. Are Short Chain Fatty Acids in Gut Microbiota Defensive Players for Inflammation and Atherosclerosis? J. Atheroscler. Thromb. 2017, 24, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Galderisi, A.; Pirillo, P.; Moret, V.; Stocchero, M.; Gucciardi, A.; Perilongo, G.; Moretti, C.; Monciotti, C.; Giordano, G.; Baraldi, E. Metabolomics reveals new metabolic perturbations in children with type 1 diabetes. Pediatr. Diabetes 2018, 19, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Paun, A.; Yau, C.; Danska, J.S. The Influence of the Microbiome on Type 1 Diabetes. J. Immunol. 2017, 198, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Zhang, J. Role of intestinal microbiota and metabolites on gut homeostasis and human diseases. BMC Immunol. 2017, 18, 2. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.G.; Mathews, C.E.; Driver, J.P. The Role of NOD Mice in Type 1 Diabetes Research: Lessons from the Past and Recommendations for the Future. Front. Endocrinol. (Lausanne) 2018, 9, 51. [Google Scholar] [CrossRef] [PubMed]
- Abdelazez, A.; Abdelmotaal, H.; Evivie, S.E.; Melak, S.; Jia, F.F.; Khoso, M.H.; Zhu, Z.T.; Zhang, L.J.; Sami, R.; Meng, X.C. Screening Potential Probiotic Characteristics of Lactobacillus brevis Strains In Vitro and Intervention Effect on Type I Diabetes In Vivo. Biomed. Res. Int. 2018, 2018, 7356173. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.; Khan, S.H.; Mada, S.B.; Meena, S.; Kapila, R.; Kapila, S. Consumption of Probiotic Lactobacillus fermentum MTCC: 5898-Fermented Milk Attenuates Dyslipidemia, Oxidative Stress, and Inflammation in Male Rats Fed on Cholesterol-Enriched Diet. Probiotics Antimicrob. Proteins 2018. [Google Scholar] [CrossRef] [PubMed]
- Raafat, K.; Wurglics, M.; Schubert-Zsilavecz, M. Prunella vulgaris L. active components and their hypoglycemic and antinociceptive effects in alloxan-induced diabetic mice. Biomed. Pharmacother. 2016, 84, 1008–1018. [Google Scholar] [CrossRef] [PubMed]
- Lau, K.; Benitez, P.; Ardissone, A.; Wilson, T.D.; Collins, E.L.; Lorca, G.; Li, N.; Sankar, D.; Wasserfall, C.; Neu, J.; et al. Inhibition of type 1 diabetes correlated to a Lactobacillus johnsonii N6.2-mediated Th17 bias. J. Immunol. 2011, 186, 3538–3546. [Google Scholar] [CrossRef] [PubMed]
- Sarmiento, J.; Wallis, R.H.; Ning, T.; Marandi, L.; Chao, G.; Veillette, A.; Lernmark, A.; Paterson, A.D.; Poussier, P. A functional polymorphism of Ptpn22 is associated with type 1 diabetes in the BioBreeding rat. J. Immunol. 2015, 194, 615–629. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, G.L.V.; Leite, A.Z.; Higuchi, B.S.; Gonzaga, M.I.; Mariano, V.S. Intestinal dysbiosis and probiotic applications in autoimmune diseases. Immunology 2017, 152, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, R.; Wang, S.; Ahmadi, S.; Hayes, J.; Gagliano, J.; Subashchandrabose, S.; Kitzman, D.W.; Becton, T.; Read, R.; Yadav, H. Human-origin probiotic cocktail increases short-chain fatty acid production via modulation of mice and human gut microbiome. Sci. Rep. 2018, 8, 12649. [Google Scholar] [CrossRef] [PubMed]
- Marino, E.; Richards, J.L.; McLeod, K.H.; Stanley, D.; Yap, Y.A.; Knight, J.; McKenzie, C.; Kranich, J.; Oliveira, A.C.; Rossello, F.J.; et al. Gut microbial metabolites limit the frequency of autoimmune T cells and protect against type 1 diabetes. Nat. Immunol. 2017, 18, 552–562. [Google Scholar] [CrossRef] [PubMed]
- Psichas, A.; Sleeth, M.L.; Murphy, K.G.; Brooks, L.; Bewick, G.A.; Hanyaloglu, A.C.; Ghatei, M.A.; Bloom, S.R.; Frost, G. The short chain fatty acid propionate stimulates GLP-1 and PYY secretion via free fatty acid receptor 2 in rodents. Int. J. Obes. (London) 2015, 39, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Christiansen, C.B.; Gabe, M.B.N.; Svendsen, B.; Dragsted, L.O.; Rosenkilde, M.M.; Holst, J.J. The impact of short-chain fatty acids on GLP-1 and PYY secretion from the isolated perfused rat colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2018, 315, G53–G65. [Google Scholar] [CrossRef] [PubMed]
- Le, T.K.; Hosaka, T.; Nguyen, T.T.; Kassu, A.; Dang, T.O.; Tran, H.B.; Pham, T.P.; Tran, Q.B.; Le, T.H.; Pham, X.D. Bifidobacterium species lower serum glucose, increase expressions of insulin signaling proteins, and improve adipokine profile in diabetic mice. Biomed. Res. 2015, 36, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Dolpady, J.; Sorini, C.; Di Pietro, C.; Cosorich, I.; Ferrarese, R.; Saita, D.; Clementi, M.; Canducci, F.; Falcone, M. Oral Probiotic VSL#3 Prevents Autoimmune Diabetes by Modulating Microbiota and Promoting Indoleamine 2,3-Dioxygenase-Enriched Tolerogenic Intestinal Environment. J. Diabetes Res. 2016, 2016, 7569431. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Motyl, K.J.; Irwin, R.; MacDougald, O.A.; Britton, R.A.; McCabe, L.R. Loss of Bone and Wnt10b Expression in Male Type 1 Diabetic Mice Is Blocked by the Probiotic Lactobacillus reuteri. Endocrinology 2015, 156, 3169–3182. [Google Scholar] [CrossRef] [PubMed]
- Mauvais, F.X.; Diana, J.; van Endert, P. Beta cell antigens in type 1 diabetes: Triggers in pathogenesis and therapeutic targets. F1000Res 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- Takiishi, T.; Korf, H.; Van Belle, T.L.; Robert, S.; Grieco, F.A.; Caluwaerts, S.; Galleri, L.; Spagnuolo, I.; Steidler, L.; Van Huynegem, K.; et al. Reversal of autoimmune diabetes by restoration of antigen-specific tolerance using genetically modified Lactococcus lactis in mice. J. Clin. Investig. 2012, 122, 1717–1725. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.A.; Wong, F.S.; Wen, L. The importance of the Non Obese Diabetic (NOD) mouse model in autoimmune diabetes. J. Autoimmun. 2016, 66, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.-H.; Chen, Y.-P.; Chen, M.-J. Selecting probiotics with the abilities of enhancing GLP-1 to mitigate the progression of type 1 diabetes in vitro and in vivo. J. Funct. Foods 2015, 18, 473–486. [Google Scholar] [CrossRef]
- Calcinaro, F.; Dionisi, S.; Marinaro, M.; Candeloro, P.; Bonato, V.; Marzotti, S.; Corneli, R.B.; Ferretti, E.; Gulino, A.; Grasso, F.; et al. Oral probiotic administration induces interleukin-10 production and prevents spontaneous autoimmune diabetes in the non-obese diabetic mouse. Diabetologia 2005, 48, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- Blankenhorn, E.P.; Cort, L.; Greiner, D.L.; Guberski, D.L.; Mordes, J.P. Virus-induced autoimmune diabetes in the LEW.1WR1 rat requires Iddm14 and a genetic locus proximal to the major histocompatibility complex. Diabetes 2009, 58, 2930–2938. [Google Scholar] [CrossRef] [PubMed]
- Alkanani, A.K.; Hara, N.; Gianani, R.; Zipris, D. Kilham Rat Virus-induced type 1 diabetes involves beta cell infection and intra-islet JAK-STAT activation prior to insulitis. Virology 2014, 468-470, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Valladares, R.; Sankar, D.; Li, N.; Williams, E.; Lai, K.K.; Abdelgeliel, A.S.; Gonzalez, C.F.; Wasserfall, C.H.; Larkin, J.; Schatz, D.; et al. Lactobacillus johnsonii N6.2 mitigates the development of type 1 diabetes in BB-DP rats. PLoS ONE 2010, 5, e10507. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.; Dey, D.K.; Vij, R.; Meena, S.; Kapila, R.; Kapila, S. Evaluation of anti-diabetic attributes of Lactobacillus rhamnosus MTCC: 5957, Lactobacillus rhamnosus MTCC: 5897 and Lactobacillus fermentum MTCC: 5898 in streptozotocin induced diabetic rats. Microb. Pathog. 2018, 125, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Bejar, W.; Hamden, K.; Ben Salah, R.; Chouayekh, H. Lactobacillus plantarum TN627 significantly reduces complications of alloxan-induced diabetes in rats. Anaerobe 2013, 24, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Uusitalo, U.; Liu, X.; Yang, J.; Aronsson, C.A.; Hummel, S.; Butterworth, M.; Lernmark, A.; Rewers, M.; Hagopian, W.; She, J.X.; et al. Association of Early Exposure of Probiotics and Islet Autoimmunity in the TEDDY Study. JAMA Pediatr. 2016, 170, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Marcial, G.E.; Ford, A.L.; Haller, M.J.; Gezan, S.A.; Harrison, N.A.; Cai, D.; Meyer, J.L.; Perry, D.J.; Atkinson, M.A.; Wasserfall, C.H.; et al. Lactobacillus johnsonii N6.2 Modulates the Host Immune Responses: A Double-Blind, Randomized Trial in Healthy Adults. Front. Immunol. 2017, 8, 655. [Google Scholar] [CrossRef] [PubMed]
- Weir, G.C.; Bonner-Weir, S. Dreams for Type 1 Diabetes: Shutting Off Autoimmunity and Stimulating β-Cell Regeneration. Endocrinology 2010, 151, 2971–2973. [Google Scholar] [CrossRef] [PubMed]
- Yadav, H.; Lee, J.-H.; Lloyd, J.; Walter, P.; Rane, S.G. Beneficial Metabolic Effects of a Probiotic via Butyrate-induced GLP-1 Hormone Secretion. J. Biol. Chem. 2013, 288, 25088–25097. [Google Scholar] [CrossRef] [PubMed]
- Burrows, M.P.; Volchkov, P.; Kobayashi, K.S.; Chervonsky, A.V. Microbiota regulates type 1 diabetes through Toll-like receptors. Proc. Natl. Acad. Sci. USA 2015, 112, 9973–9977. [Google Scholar] [CrossRef] [PubMed]
- Groele, L.; Szajewska, H.; Szypowska, A. Effects of Lactobacillus rhamnosus GG and Bifidobacterium lactis Bb12 on beta-cell function in children with newly diagnosed type 1 diabetes: Protocol of a randomised controlled trial. BMJ Open 2017, 7, e017178. [Google Scholar] [CrossRef] [PubMed]
- Ahola, A.J.; Harjutsalo, V.; Forsblom, C.; Freese, R.; Makimattila, S.; Groop, P.H. The Self-reported Use of Probiotics is Associated with Better Glycaemic Control and Lower Odds of Metabolic Syndrome and its Components in Type 1 Diabetes. J. Probiotics Health 2017, 05. [Google Scholar] [CrossRef]
- Yang, J.; Tamura, R.N.; Uusitalo, U.M.; Aronsson, C.A.; Silvis, K.; Riikonen, A.; Frank, N.; Joslowski, G.; Winkler, C.; Norris, J.M.; et al. Vitamin D and probiotics supplement use in young children with genetic risk for type 1 diabetes. Eur. J. Clin. Nutr. 2017, 71, 1449–1454. [Google Scholar] [CrossRef] [PubMed]
- Carlson, J.L.; Erickson, J.M.; Lloyd, B.B.; Slavin, J.L. Health Effects and Sources of Prebiotic Dietary Fiber. Curr. Dev. Nutr. 2018, 2, nzy005. [Google Scholar] [CrossRef] [PubMed]
- Wilson, B.; Whelan, K. Prebiotic inulin-type fructans and galacto-oligosaccharides: Definition, specificity, function, and application in gastrointestinal disorders. J. Gastroenterol. Hepatol. 2017, 32 (Suppl. 1), 64–68. [Google Scholar] [CrossRef] [PubMed]
- Shokryazdan, P.; Faseleh Jahromi, M.; Navidshad, B.; Liang, J.B. Effects of prebiotics on immune system and cytokine expression. Med. Microbiol. Immunol. 2017, 206, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kaji, I.; Karaki, S.; Tanaka, R.; Kuwahara, A. Density distribution of free fatty acid receptor 2 (FFA2)-expressing and GLP-1-producing enteroendocrine L cells in human and rat lower intestine, and increased cell numbers after ingestion of fructo-oligosaccharide. J. Mol. Histol. 2011, 42, 27–38. [Google Scholar] [CrossRef] [PubMed]
- den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, S.; Toh, H.; Hase, K.; Oshima, K.; Nakanishi, Y.; Yoshimura, K.; Tobe, T.; Clarke, J.M.; Topping, D.L.; Suzuki, T.; et al. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature 2011, 469, 543–547. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Chen, H.; Faas, M.M.; de Haan, B.J.; Li, J.; Xiao, P.; Zhang, H.; Diana, J.; de Vos, P.; Sun, J. Specific inulin-type fructan fibers protect against autoimmune diabetes by modulating gut immunity, barrier function, and microbiota homeostasis. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Woting, A.; Pfeiffer, N.; Hanske, L.; Loh, G.; Klaus, S.; Blaut, M. Alleviation of high fat diet-induced obesity by oligofructose in gnotobiotic mice is independent of presence of Bifidobacterium longum. Mol. Nutr. Food Res. 2015, 59, 2267–2278. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Neyrinck, A.M.; Fava, F.; Knauf, C.; Burcelin, R.G.; Tuohy, K.M.; Gibson, G.R.; Delzenne, N.M. Selective increases of bifidobacteria in gut microflora improve high-fat-diet-induced diabetes in mice through a mechanism associated with endotoxaemia. Diabetologia 2007, 50, 2374–2383. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Van’t Land, B.; Engen, P.A.; Naqib, A.; Green, S.J.; Nato, A.; Leusink-Muis, T.; Garssen, J.; Keshavarzian, A.; Stahl, B.; et al. Human milk oligosaccharides protect against the development of autoimmune diabetes in NOD-mice. Sci. Rep. 2018, 8, 3829. [Google Scholar] [CrossRef] [PubMed]
- Bach Knudsen, K.E.; Laerke, H.N.; Hedemann, M.S.; Nielsen, T.S.; Ingerslev, A.K.; Gundelund Nielsen, D.S.; Theil, P.K.; Purup, S.; Hald, S.; Schioldan, A.G.; et al. Impact of Diet-Modulated Butyrate Production on Intestinal Barrier Function and Inflammation. Nutrients 2018, 10, 1499. [Google Scholar] [CrossRef] [PubMed]
- Koh, G.Y.; Rowling, M.J.; Schalinske, K.L.; Grapentine, K.; Loo, Y.T. Consumption of Dietary Resistant Starch Partially Corrected the Growth Pattern Despite Hyperglycemia and Compromised Kidney Function in Streptozotocin-Induced Diabetic Rats. J. Agric. Food Chem. 2016, 64, 7540–7545. [Google Scholar] [CrossRef] [PubMed]
- Crookshank, J.A.; Patrick, C.; Wang, G.S.; Noel, J.A.; Scott, F.W. Gut immune deficits in LEW.1AR1-iddm rats partially overcome by feeding a diabetes-protective diet. Immunology 2015, 145, 417–428. [Google Scholar] [CrossRef] [PubMed]
- Gorelick, J.; Yarmolinsky, L.; Budovsky, A.; Khalfin, B.; Klein, J.D.; Pinchasov, Y.; Bushuev, M.A.; Rudchenko, T.; Ben-Shabat, S. The Impact of Diet Wheat Source on the Onset of Type 1 Diabetes Mellitus-Lessons Learned from the Non-Obese Diabetic (NOD) Mouse Model. Nutrients 2017, 9, 482. [Google Scholar] [CrossRef] [PubMed]
- Stenman, L.K.; Waget, A.; Garret, C.; Briand, F.; Burcelin, R.; Sulpice, T.; Lahtinen, S. Probiotic B420 and prebiotic polydextrose improve efficacy of antidiabetic drugs in mice. Diabetol. Metab. Syndr. 2015, 7, 75. [Google Scholar] [CrossRef] [PubMed]
- Prud’homme, G.J.; Glinka, Y.; Kurt, M.; Liu, W.; Wang, Q. The anti-aging protein Klotho is induced by GABA therapy and exerts protective and stimulatory effects on pancreatic beta cells. Biochem. Biophys. Res. Commun. 2017, 493, 1542–1547. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.; Reimer, R.A.; Doulla, M.; Huang, C. Effect of prebiotic intake on gut microbiota, intestinal permeability and glycemic control in children with type 1 diabetes: Study protocol for a randomized controlled trial. Trials 2016, 17, 347. [Google Scholar] [CrossRef] [PubMed]
- Beretta, M.V.; Bernaud, F.R.; Nascimento, C.; Steemburgo, T.; Rodrigues, T.C. Higher fiber intake is associated with lower blood pressure levels in patients with type 1 diabetes. Arch. Endocrinol. Metab. 2018, 62, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Bernaud, F.S.; Beretta, M.V.; do Nascimento, C.; Escobar, F.; Gross, J.L.; Azevedo, M.J.; Rodrigues, T.C. Fiber intake and inflammation in type 1 diabetes. Diabetol. Metab. Syndr. 2014, 6, 66. [Google Scholar] [CrossRef] [PubMed]
- Biester, T.; Aschemeier, B.; Fath, M.; Frey, M.; Scheerer, M.F.; Kordonouri, O.; Danne, T. Effects of dapagliflozin on insulin-requirement, glucose excretion and ss-hydroxybutyrate levels are not related to baseline HbA1c in youth with type 1 diabetes. Diabetes Obes. Metab. 2017, 19, 1635–1639. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Chang, H.W.; Yan, D.; Lee, K.M.; Ucmak, D.; Wong, K.; Abrouk, M.; Farahnik, B.; Nakamura, M.; Zhu, T.H.; et al. Influence of diet on the gut microbiome and implications for human health. J. Transl. Med. 2017, 15, 73. [Google Scholar] [CrossRef] [PubMed]
- Uusitalo, U.; Lee, H.S.; Andren Aronsson, C.; Vehik, K.; Yang, J.; Hummel, S.; Silvis, K.; Lernmark, A.; Rewers, M.; Hagopian, W.; et al. Early Infant Diet and Islet Autoimmunity in the TEDDY Study. Diabetes Care 2018, 41, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Falony, G.; Lazidou, K.; Verschaeren, A.; Weckx, S.; Maes, D.; De Vuyst, L. In vitro kinetic analysis of fermentation of prebiotic inulin-type fructans by Bifidobacterium species reveals four different phenotypes. Appl. Environ. Microbiol. 2009, 75, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Brugman, S.; Ikeda-Ohtsubo, W.; Braber, S.; Folkerts, G.; Pieterse, C.M.J.; Bakker, P. A Comparative Review on Microbiota Manipulation: Lessons From Fish, Plants, Livestock, and Human Research. Front. Nutr. 2018, 5, 80. [Google Scholar] [CrossRef] [PubMed]
- Lund-Blix, N.A.; Dydensborg Sander, S.; Stordal, K.; Nybo Andersen, A.M.; Ronningen, K.S.; Joner, G.; Skrivarhaug, T.; Njolstad, P.R.; Husby, S.; Stene, L.C. Infant Feeding and Risk of Type 1 Diabetes in Two Large Scandinavian Birth Cohorts. Diabetes Care 2017, 40, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Niinisto, S.; Takkinen, H.M.; Erlund, I.; Ahonen, S.; Toppari, J.; Ilonen, J.; Veijola, R.; Knip, M.; Vaarala, O.; Virtanen, S.M. Fatty acid status in infancy is associated with the risk of type 1 diabetes-associated autoimmunity. Diabetologia 2017, 60, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Lamb, M.M.; Miller, M.; Seifert, J.A.; Frederiksen, B.; Kroehl, M.; Rewers, M.; Norris, J.M. The effect of childhood cow’s milk intake and HLA-DR genotype on risk of islet autoimmunity and type 1 diabetes: The Diabetes Autoimmunity Study in the Young. Pediatr. Diabetes 2015, 16, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Takeshima, S.; Matsumoto, Y.; Chen, J.; Yoshida, T.; Mukoyama, H.; Aida, Y. Evidence for cattle major histocompatibility complex (BoLA) class II DQA1 gene heterozygote advantage against clinical mastitis caused by Streptococci and Escherichia species. Tissue Antigens 2008, 72, 525–531. [Google Scholar] [CrossRef] [PubMed]
- Hanninen, A.; Toivonen, R.; Poysti, S.; Belzer, C.; Plovier, H.; Ouwerkerk, J.P.; Emani, R.; Cani, P.D.; De Vos, W.M. Akkermansia muciniphila induces gut microbiota remodelling and controls islet autoimmunity in NOD mice. Gut 2018, 67, 1445–1453. [Google Scholar] [CrossRef] [PubMed]
- Weir, G.C.; Bonner-Weir, S. GABA Signaling Stimulates beta Cell Regeneration in Diabetic Mice. Cell 2017, 168, 7–9. [Google Scholar] [CrossRef] [PubMed]
- Kanikarla-Marie, P.; Jain, S.K. Hyperketonemia (acetoacetate) upregulates NADPH oxidase 4 and elevates oxidative stress, ICAM-1, and monocyte adhesivity in endothelial cells. Cell. Physiol. Biochem. 2015, 35, 364–373. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Probiotics/Prebiotics | Model Type | Mechanism of Action | Major Findings | References |
|---|---|---|---|---|
| Oral Probiotics VSL#3 (Bifidobacteriaceae, Lactobacillaceae, Streptococcus thermophilus) | NOD mice | - Generates more pro-tolerogenic components of inflammasome like indoleamine 2,3-dioxygenase (IDO) and IL-33. - Reduces the synthesis of inflammatory cytokines like IL-1β. - Promotes CD103+ differentiation. - Reduces Teff/Treg cell ratios within the gut mucosa, MLNs and PLNs. | - Modification of gut microbial environment. - Modulating T1D pathogenesis. | [73] |
| Bacterial LPS or Zymosan | NOD mice | - Produces synergetic innate immune response through TLR2 and Dectin-1 signaling. - Eliminates inflammatory immune cells and suppresses autoimmunity. - Triggers the secretion of immune regulatory factors like IL-10, TGF-β1, IL-2 and Raldh1A2. - Increases the numbers of Foxp3+CD4+ T cells in the PLN but not in spleen. | - Used as an immune regulatory adjuvant for promoting β-cell antigen-specific immune modulation. - Reverses the early stages of hyperglycemia in T1D. | [47] |
| Lactobacillus brevis KLDS 1.0727 and KLDS 1.0373 | STZ-induced C57BL/6 T1D mice | - High GABA generating capacity due to the gad gene. - Significant effect in lowering the blood glucose level or insulin in plasma. | - Inhibits the development of T1D in diabetic mice model. | [62] |
| PFM with 1% of Lactobacillus species | STZ-induced albino wistar T1D rats | - Significant decrease in the expression of hepatic gluconeogenesis gene like Glucose-6-phosphatase (G6Pase) and Phosphoenol pyruvate carboxykinase (PEPCK) in the liver. - Significant reduction in serum inflammatory cytokines like IL-6 and TNF-α -Decrease in HbA1c, blood glucose level and serum lipid profile. -Significant increase in the serum insulin level. | - Increases insulin level with significant reduction in blood glucose level. - Improvement in glucose metabolism -Decrease in inflammation, oxidative stress and hepatic gluconeogenesis. | [83] |
| HMOS Prebiotic | NOD Mice | - Increases SCFA concentration in the gut. - Limits autoimmune T-cells and increases the Treg cells. - Induces tolerogenic DC phonotype by induction of MHC II and increases the expression of inhibitory molecules such as PD-L1 and OX40-L. -Increased butyrate production promoting mucin synthesis. - Improves intestinal barrier integrity. -Reduced pancreatic islet destruction by regulating the immune system. | - Modulation and maintaining the α- and β-diversity of the fecal microbiota. - Changes the direct shape of the pancreatic environment, resulting in less insulitis. - Helps in protection against T1D. | [102] |
| Dietary Resistant starch | STZ-induced T1D Sprague-Dawley rats | - Influences the secretion of GLP-1 and PYY hormones. - Proliferation of β-cell and insulin synthesis. - Provides nephron-protection. - No effect on blood glucose level and Vitamin D balance. | - Develops normalized growth pattern in T1D. | [104] |
| CARF extracted from PV | Alloxan-induced T1D Swiss Webster mice | -Decreases α-amylase and α-glucosidase activity. - Reduces HbA1c level. - Elevates serum insulin level. - Increases antioxidant enzyme level. | - Poses anti-diabetogenic, anti-nociceptive and hypoanalgesic properties as therapeutic agents against T1D. | [64] |
| Prebiotic oligofructose | High fat diet induced diabetic C57b16/J mice | -Increases Bifidobacteria number by modifying gut microbiota. - Decreases endotoxemia. -Improves glucose tolerance and regulates glucose-induced insulin secretion. -Increases colonic GLP-1 secretion. | -Pathophysiological regulation of endotoxemia. - Sets the tone of inflammation, glucose tolerance and insulin secretion. | [101] |
| Oral transfer prebiotic Lactobacillus johnsonii N6.2-Mediated | KRV virus induced-BBDP rat | - TH17 lymphocyte biasness within the gut-draining MLN. - Cytokines, like IL-6 and IL-23, were responsible for induction and sustenance of TH17 cells was higher. - Retention of TH17 differentiation state that may prevent T-cell conversion to the diabetogenic phenotype. | - Confirms resistance to T1D. | [65] |
| Probiotic Bifidobacterium spp. | STZ-induced C57BL/6J diabetic mice | - Significant reduction in blood glucose level. - Increases the protein expression of insulin receptor β, insulin receptor substrate 1, (Akt/PKB), IKKα, IκBα. - Decreases the macrophage chemoattractant protein-1 and IL-6 expression. | - Responsible for treating diabetes. | [72] |
| Lactobacillus reuteri | STZ-induced C57BL/6 diabetic mice | - Development of anti-inflammatory property by inhibiting osteoblast TNF-α signaling. - TNF-α modulates the Wnt10b expression in T1D. | - Use of probiotic to benefit bones in T1D patients. | [74] |
| Lactococcus lactis | NOD Mice | - Increases the frequency of local Tregs in the pancreatic islet. - Suppresses immune response in an autoantigen-specific way. - Preserves functional β-cell mass and reduces insulitis. - Secretion of human pro-insulin and IL-10 can stably revert autoimmune diabetes. - Induced Ag-specific Foxp3+ Tregs that prevent diabetes transfer. | - Treatment strategy for T1D in humans. | [76] |
| Lactobacillus kefiranofaciens and Lactobacillus kefiri | STZ-induced C57BL/6 diabetic mice | - Level of IL-10 significantly raised in pancreas. - Increased IL-10 inhibits the secretion of pro-inflammatory cytokines, like TNF-α and TH1 (also IL-1β, IL-2, IL-6). | - Potential ability to stimulate the release of GLP-1. | [78] |
| Bifibobacteria, lactobacilli and Streptococcus salivarius subs. | NOD mice | - Decreases the rate of β cell destruction. - Increases the production of IL-10 from PPs, pancreas and spleen. - Modulates GALT. | -Prevention of autoimmune diabetes. -Induces immunomodulation by a reduction in insulitis severity. | [79] |
| Lactobacillus johnsonii N6.2 | T1D BBDP rats | - Changes in the native gut microbiota. - Induced changes in host mucosal protein and oxidative stress response. - Decreases oxidative response protein in the intestinal mucosa. - Decreases pro-inflammatory cytokines, like IFN-γ. - Higher expression of tight junction proteins, like claudin. | - Delays or inhibits the occurrence of T1D. | [82] |
| Lactobacillus plantarum TN627 | Alloxan induced-diabetic rat | - Improved the immunological parameters of the pancreas. - Reduced the pancreatic and plasmatic α-amylase activity as well as blood glucose level. - Decreased the pancreatic and plasmatic lipase activity, serum triglyceride and LDL-cholesterol rate. - Increases the HDL-cholesterol rate. | - Helpful in preventing diabetic complications in the adult rat. | [84] |
| Low antigen, hydrolyzed casein-based diet | LEW.1AR1-iddm Rat model | - Increased immunoregulatory capacity and gut immune deficits. - Decreased expression of CD3+ T-cells, CD163+ M2 macrophages and Foxp3+ cells in jejunum. - Decrease in CD4+ Foxp3+ regulatory T-cells in PLNs. - IFN-γ expression increase in MLNs. | - Protection against T1D. | [105] |
| Bifidobacterium animalis ssp. lactis 420 (B420) and Metformin | Ketogenic diet-induced C57Bl/6J diabetic mice | - Increases ileum GLP-1 concentration. - Increases the amount of insulin released from pancreatic β-cells. - Significantly decreases the glycemic response and plasma glucose concentration. | - Improves glucose metabolism and insulin secretion. - Improves the efficacy of metformin. | [107] |
| Wheat Flour | NOD mice | - Lacks the epitopes linked with T1D. -Reduction in the level of pro-inflammatory cytokines, like IFN-γ. -Increase in the level of anti-inflammatory cytokine IL-10. | - Reducing the incidence of T1D. | [106] |
| Systemic GABA therapy | STZ-induced C57/BL6 T1D mice | - Increases klotho (anti-aging agent) level expression in serum, pancreatic Islet of Langerhans and kidneys. - Klotho stimulates pancreatic β-cells survival and proliferation. - Increases insulin secretion. - Klotho blocks NF-κB activation by interfering with its nuclear translocation. - Suppresses autoimmune responses. | - Important implications for the treatment of T1D. | [108] |
| Dietary fibers | NOD mice | - Increases CD25+Foxp3+CD4+ Treg and decreases IL17A+CD4+Th17 cells. - Changes the cytokine production profile in the pancreas, spleen and colon. - Enhances tight junction proteins (claudin-2, occludin) and SCFAs. - Enhances Firmicutes/Bacteroidetes ratio as well as Ruminococcaceae and Lactobacilli. | - Modulates T-cell response. - Modulates gut-pancreatic immunity. - Delays the development of T1D. | [99] |
| Probiotics/Prebiotics | Model Type | Mechanism of Action | Major Findings | References |
|---|---|---|---|---|
| Lactobacillus johnsonii N6.2 | 42 healthy adult humans | - Increased serum tryptophan level -Resulted in a decreased kynurenine:tryptophan ratio. - After washout period, monocytes and natural killer cell numbers increase significantly regulated through indoleamine 2,3-dioxygenase (IDO) pathway. - Increases circulatory effector Th1 cells and cytotoxic CD8+ T-cells. - Delay or reduces the apoptosis of memory CD8+ T-cells. | - Responsible for reducing the risk of T1D occurrence. | [86] |
| Lactobacillus rhamnosus GG and Bifidobacterium lactis Bb12 | 96 children aged between 8–17 | - Improved the gut mucosal barrier. - Modulated local and systemic immune responses. - Reduced the risk of autoimmunity. | -Inhibits the growth of pathogens -Preserves the β-cell function. | [90] |
| Probiotics | 1039 adult individuals | - Decrease in obesity, body mass index, waist-to-hip ratio. - Regulated blood pressure, HDL-cholesterol, triglyceride components. - Significantly associated with better glycemic control. | - Beneficial effect on various factor related to the diabetic complications. | [91] |
| Prebiotic (Oligofructose-enriched inulin) | Young children aged between (8–17 years) | -Develops into severe hypoglycemia. -Decreases endotoxemia and reduced insulin resistance. -Improves glycemic control. - Changes gut microbiota, permeability and inflammation. | - A potential and novel agent for treating T1D. | [109] |
| Dietary fiber intake | T1D adult human patients | - Exhibited lower systolic and diastolic blood pressure. - No significant association was found in lipid profile. - Shows lower Body Mass Index (BMI), superior metabolic control of diabetes. - Reduction in the use of medicine to treat diabetes (insulin) and hypertension (ACE inhibitor). | - Association with lower blood pressure in T1D patients. | [110] |
| Dietary fiber | 106 outpatients with T1D | - Develops anti-inflammatory properties. -Decreases the C-reactive protein levels independent of HbA1c value. | - Plays a significant role in reduction of inflammation. - Associated with lowering the risk of coronary heart disease. | [111] |
| Adjunct therapy with DAPA | 33 Youth T1D patients aged between 12–21 years | - Reduced the mean insulin requirement dose for medication. - Increase in urinary glucose excretion. -Leads to a significant reduction in insulin requirements to achieve the target glucose level, irrespective of HbA1c level. | - Offers a future therapeutic agent to the T1D challenged pediatric age group. | [112] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mishra, S.; Wang, S.; Nagpal, R.; Miller, B.; Singh, R.; Taraphder, S.; Yadav, H. Probiotics and Prebiotics for the Amelioration of Type 1 Diabetes: Present and Future Perspectives. Microorganisms 2019, 7, 67. https://doi.org/10.3390/microorganisms7030067
Mishra S, Wang S, Nagpal R, Miller B, Singh R, Taraphder S, Yadav H. Probiotics and Prebiotics for the Amelioration of Type 1 Diabetes: Present and Future Perspectives. Microorganisms. 2019; 7(3):67. https://doi.org/10.3390/microorganisms7030067
Chicago/Turabian StyleMishra, Sidharth, Shaohua Wang, Ravinder Nagpal, Brandi Miller, Ria Singh, Subhash Taraphder, and Hariom Yadav. 2019. "Probiotics and Prebiotics for the Amelioration of Type 1 Diabetes: Present and Future Perspectives" Microorganisms 7, no. 3: 67. https://doi.org/10.3390/microorganisms7030067
APA StyleMishra, S., Wang, S., Nagpal, R., Miller, B., Singh, R., Taraphder, S., & Yadav, H. (2019). Probiotics and Prebiotics for the Amelioration of Type 1 Diabetes: Present and Future Perspectives. Microorganisms, 7(3), 67. https://doi.org/10.3390/microorganisms7030067