A Pyrrhic Victory: The PMN Response to Ocular Bacterial Infections


Abstract
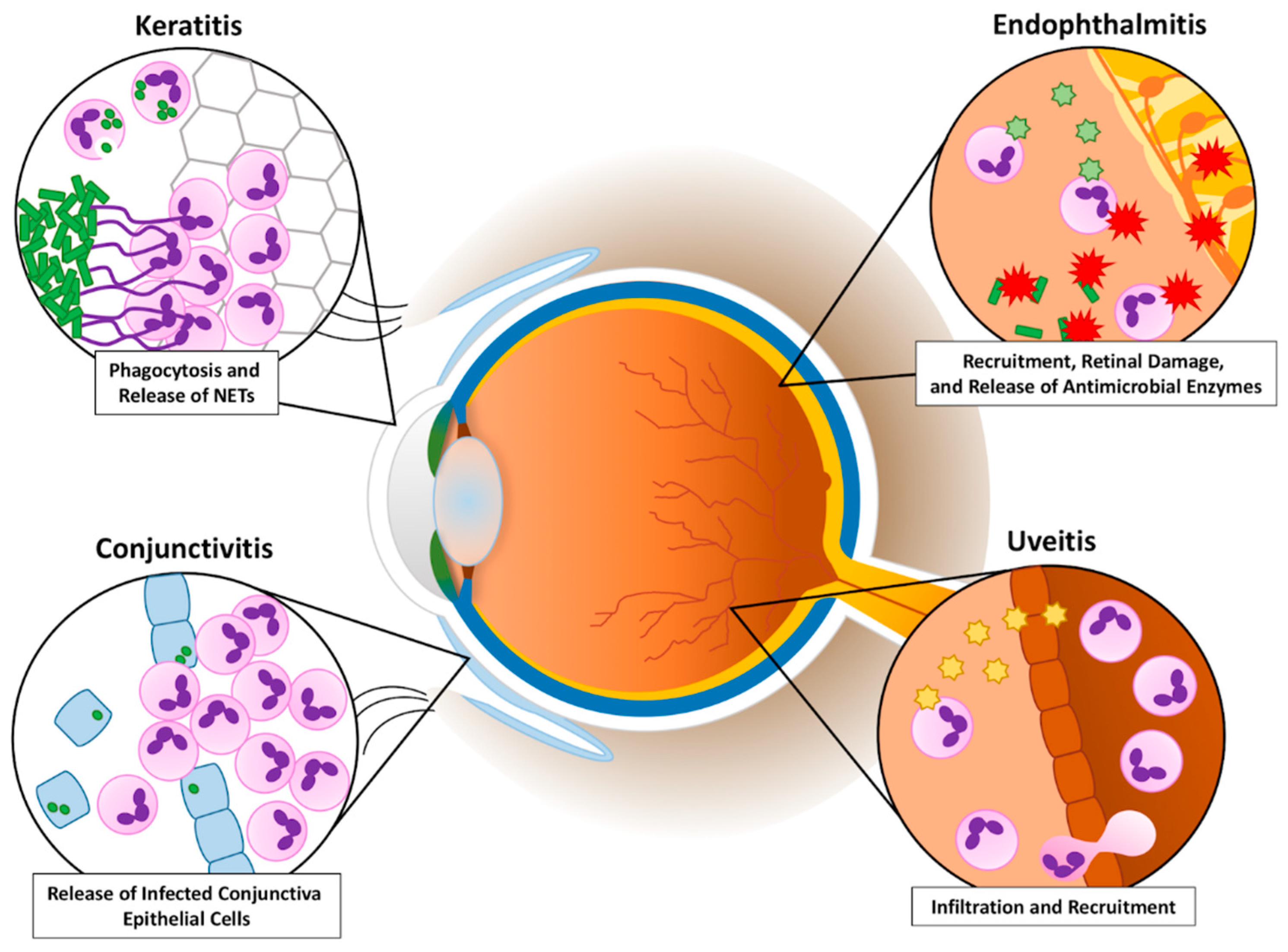
1. Introduction
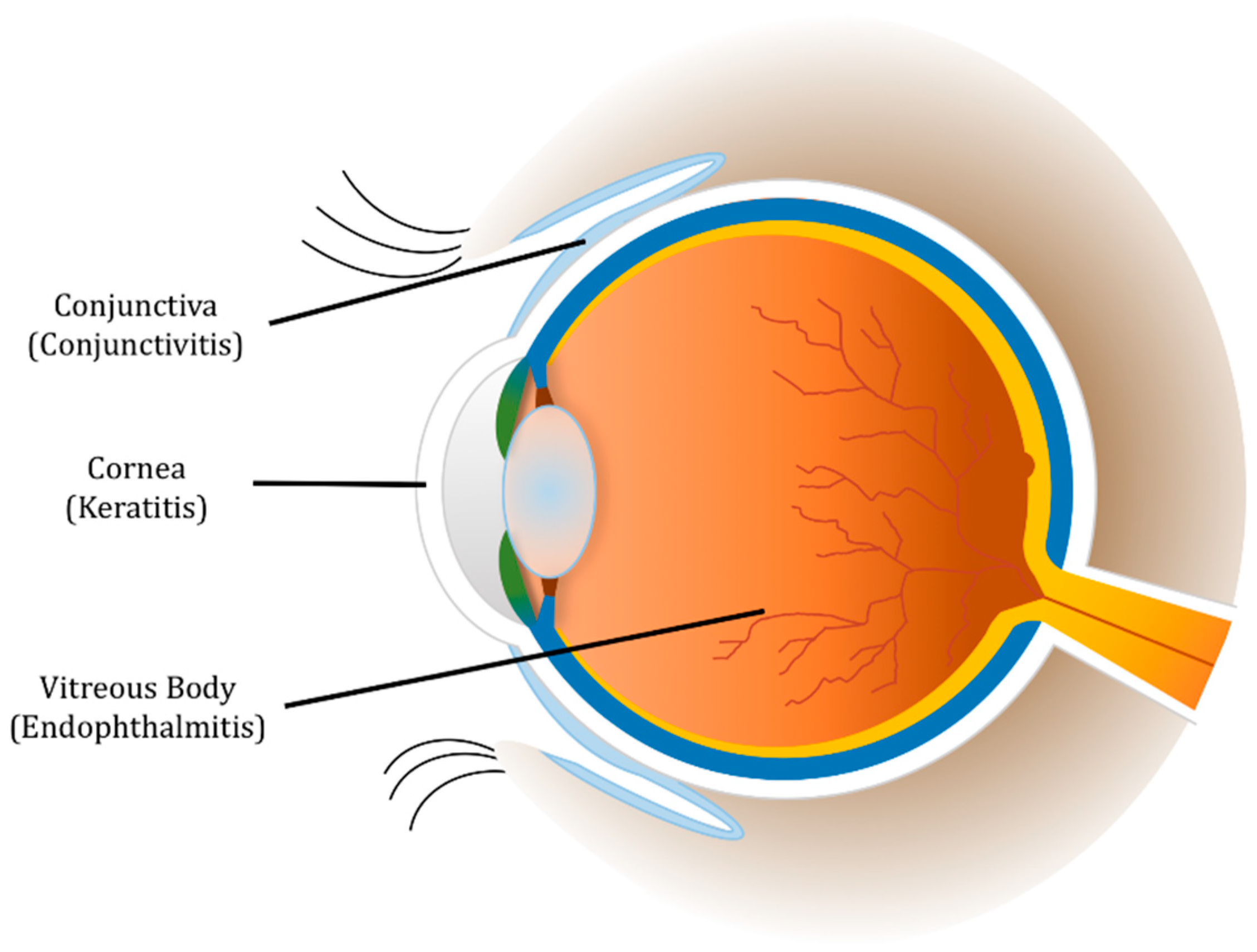
2. Conjunctivitis
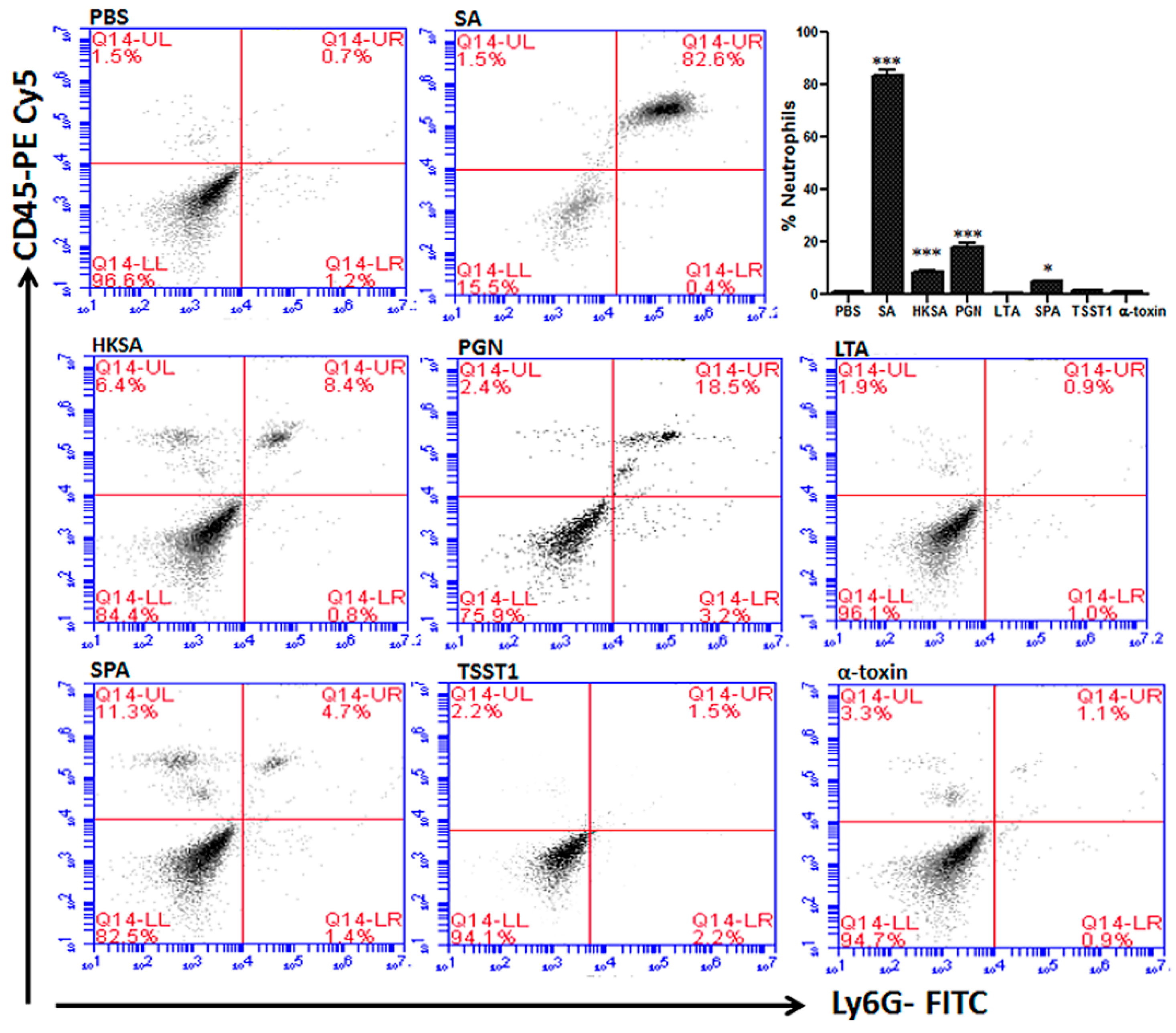
2.1. Staphylococcus aureus Conjunctivitis
2.2. Streptococcus pneumoniae Conjunctivitis
2.3. Chlamydia trachomatis Conjunctivitis
2.4. Conjunctivitis Conclusions
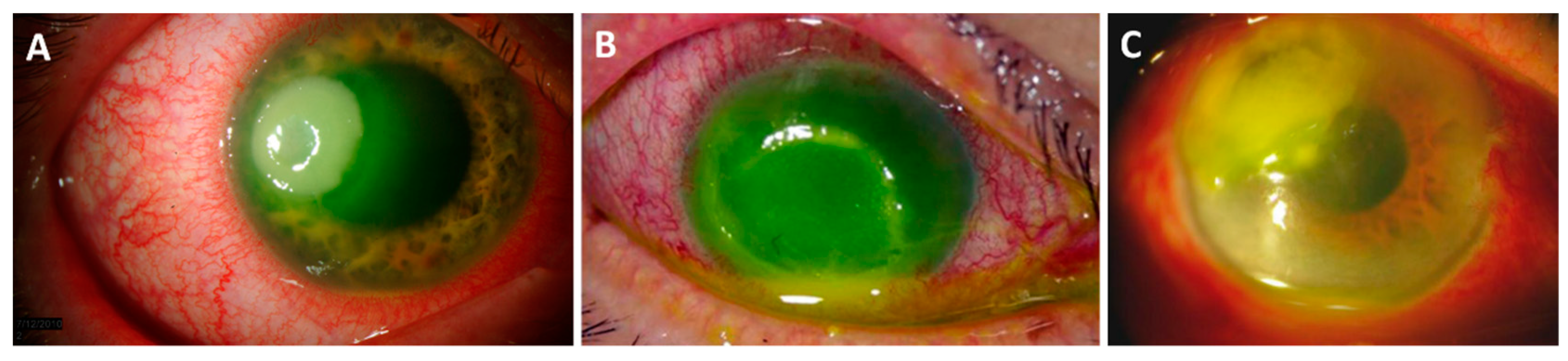
3. Keratitis
3.1. Pseudomonas aeruginosa Keratitis
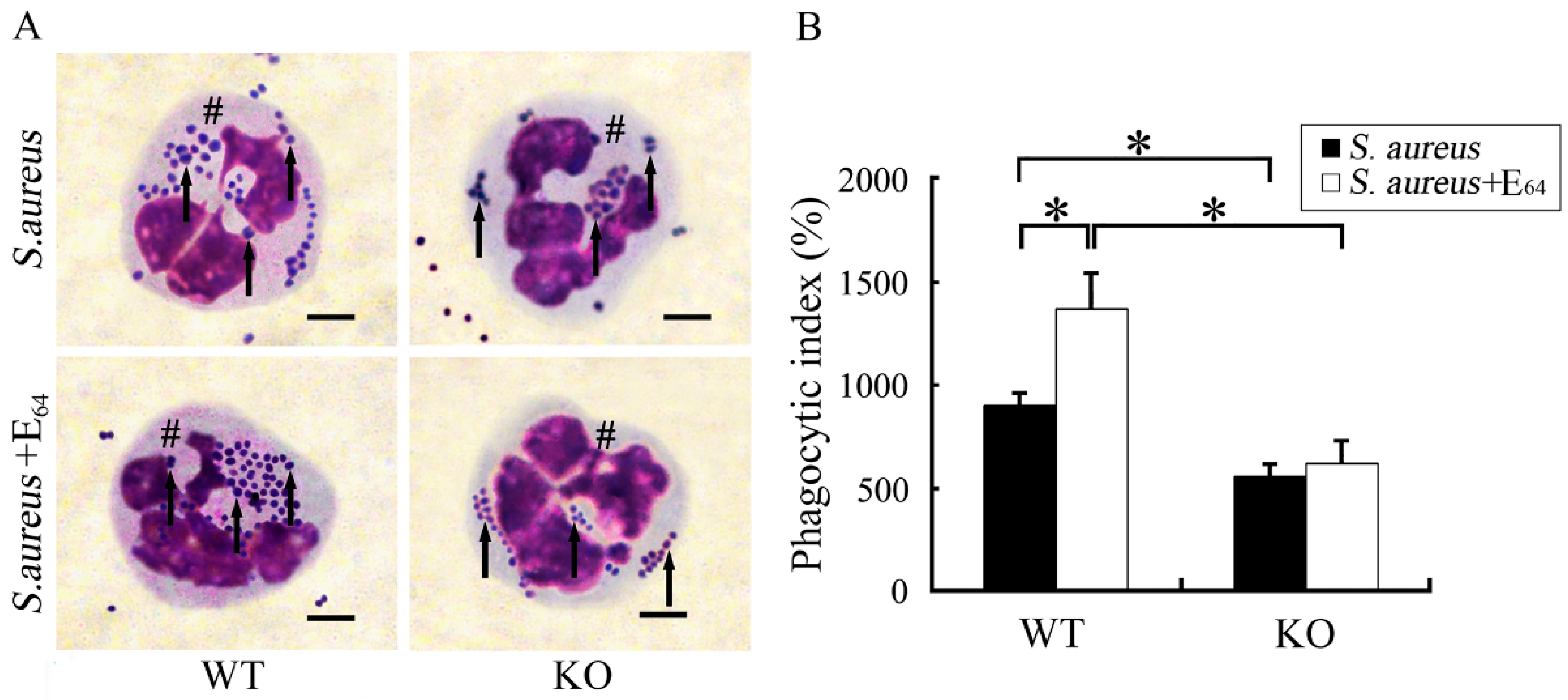
3.2. Staphylococcus aureus Keratitis
3.3. Streptococcus pneumoniae Keratitis
3.4. Keratitis Conclusions
4. Infectious Uveitis Models
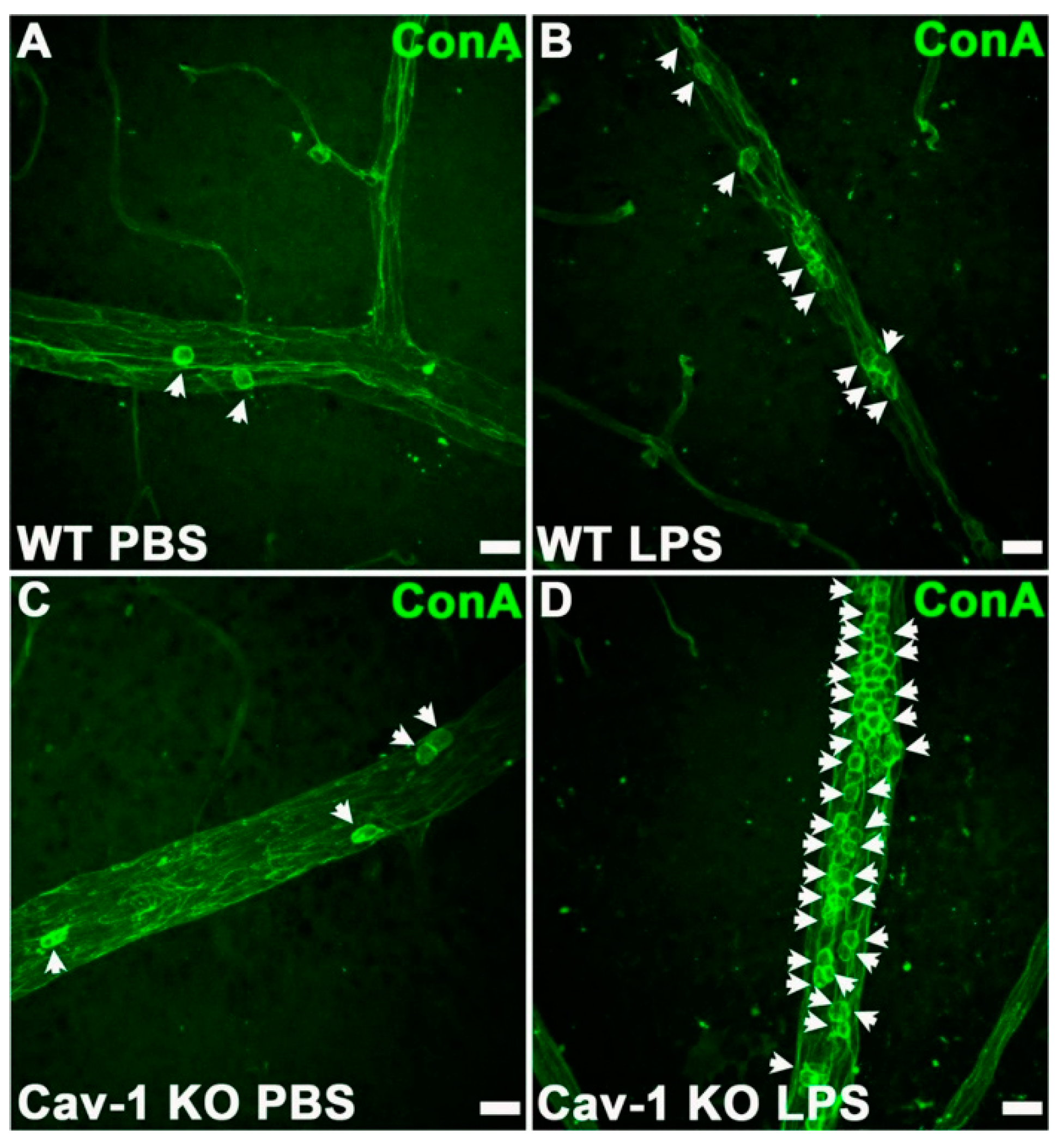
4.1. Endotoxin-Induced Uveitis
4.2. Uveitis Conclusions
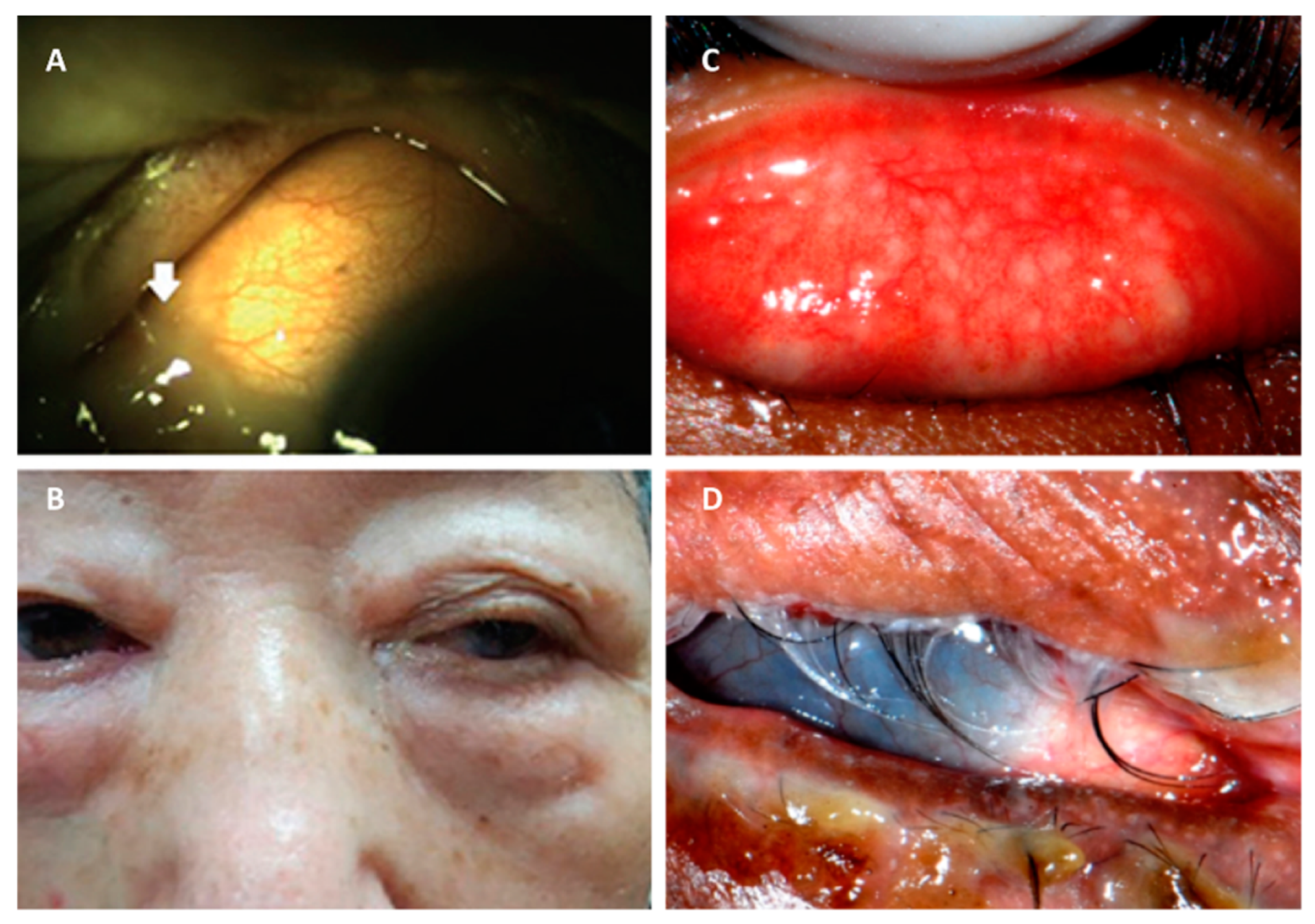
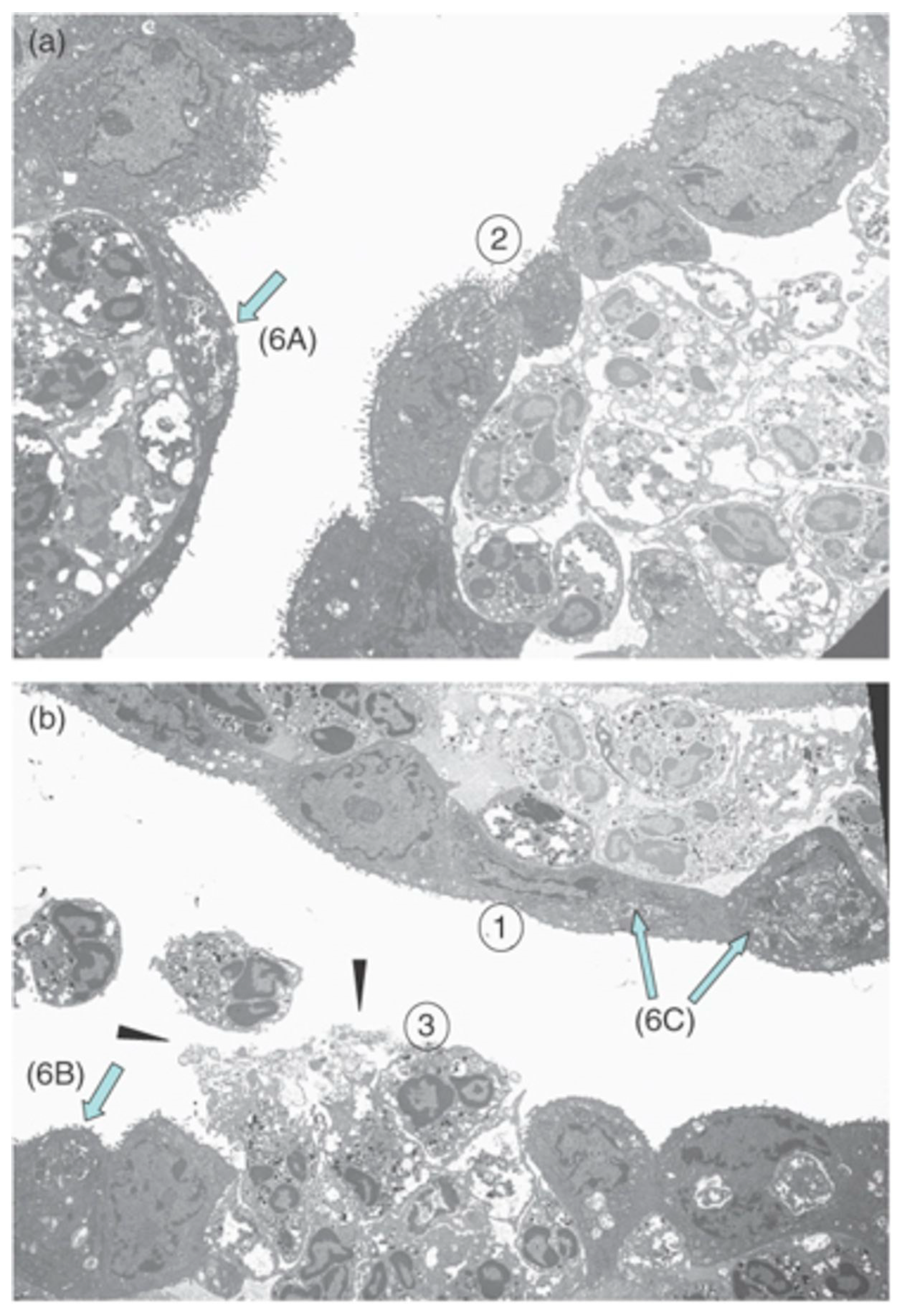
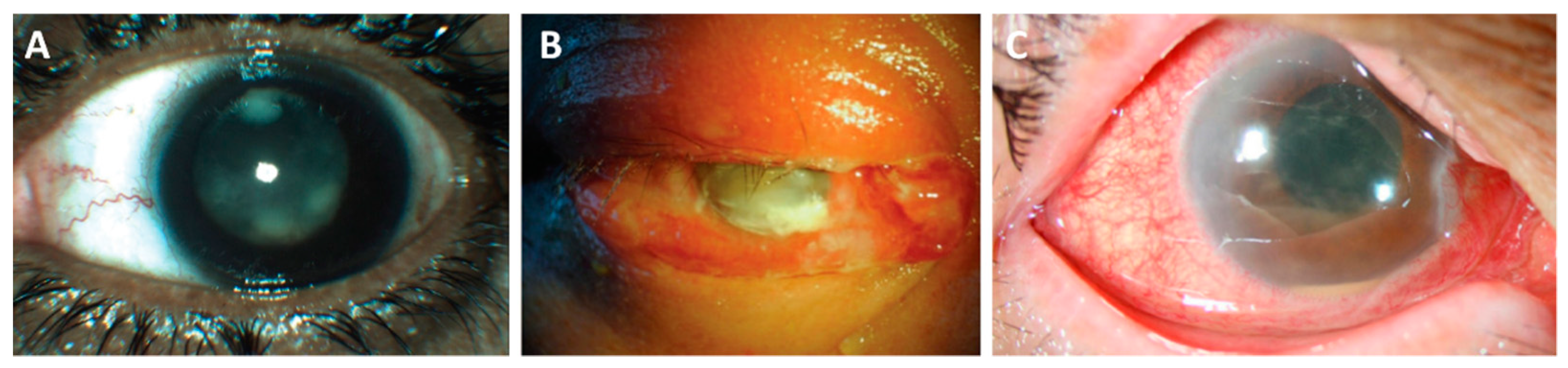
5. Endophthalmitis
5.1. Staphylococcus aureus Endophthalmitis
5.2. Streptococcus pneumoniae Endophthalmitis
5.3. Bacillus Endophthalmitis
5.4. Enterococcus faecalis Endophthalmitis
5.5. Gram-negative Bacterial Endophthalmitis
5.6. Endophthalmitis Conclusions
6. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Medawar, P.B. Immunity to Homologous Grafted Skin. III. The Fate of Skin Homografts Transplanted to the Brain, to Subcutaneous Tissue, and to the Anterior Chamber of the Eye. Br. J. Exp. Pathol. 1948, 29, 58–69. [Google Scholar] [PubMed]
- Stein-Streilein, J. Immune Regulation and the Eye. Cell Press 2008, 29, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.W. Ocular Immunosuppressive Microenvironment. Chem. Immunol. Allergy 2007, 92, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, F.; Di Zazzo, A.; De Simone, L.; Bonini, S. The Intriguing Role of Neuropeptides at the Ocular Surface. Ocul. Surf. 2017, 15, 2–14. [Google Scholar] [CrossRef]
- Foulsham, W.; Coco, G.; Amouzegar, A.; Chauhan, S.K.; Dana, R. When Clarity Is Crucial: Regulating Ocular Surface Immunity. Trends Immunol. 2018, 39, 288–301. [Google Scholar] [CrossRef]
- Garreis, F.; Gottschalt, M.; Paulsen, F.P. Antimicrobial Peptides as a Major Part of the Innate Immune Defense at the Ocular Surface. Dev. Ophthalmol. 2010, 45, 16–22. [Google Scholar] [CrossRef]
- McDermott, A.M. Antimicrobial Compounds in Tears. Exp. Eye Res. 2013, 117, 53–61. [Google Scholar] [CrossRef]
- Aizuss, D.H.; Mondino, B.J.; Sumner, H.L.; Derhlefs, B.A. The Complement System and Host Defense against Pseudomonas Endophthalmitis. Investig. Ophthalmol. Vis. Sci. 1985, 26, 1262–1266. [Google Scholar]
- Engelbert, M.; Gilmore, M.S. Fas Ligand but Not Complement Is Critical for Control of Experimental Staphylococcus aureus Endophthalmitis. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2479–2486. [Google Scholar] [CrossRef]
- Ramadan, R.T.; Ramirez, R.; Novosad, B.D.; Callegan, M.C. Acute Inflammation and Loss of Retinal Architecture and Function during Experimental Bacillus Endophthalmitis. Curr. Eye Res. 2006, 31, 955–965. [Google Scholar] [CrossRef]
- Karthikeyan, R.S.G.; Priya, J.L.; Leal, S.M.; Toska, J.; Rietsch, A.; Prajna, V.; Pearlman, E.; Lalitha, P. Host Response and Bacterial Virulence Factor Expression in Pseudomonas aeruginosa and Streptococcus pneumoniae Corneal Ulcers. PLoS ONE 2013, 8, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Dancey, J.T.; Deubelbeiss, K.A.; Harker, L.A.; Finch, C.A. Neutrophil Kinetics in Man. J. Clin. Investig. 1976, 58, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the Site of Inflammation: The Leukocyte Adhesion Cascade Updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Rosales, C. Neutrophil: A Cell with Many Roles in Inflammation or Several Cell Types? Front. Physiol. 2018, 9, 1–17. [Google Scholar] [CrossRef]
- Steadman, R.; Topley, N.; Jenner, D.E.; Davies, M.; Williams, J.D. Type 1 Fimbriate Escherichia coli Stimulates a Unique Pattern of Degranulation by Human Polymorphonuclear Leukocytes. Infect. Immun. 1988, 56, 815–822. [Google Scholar]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel Cell Death Program Leads to Neutrophil Extracellular Traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar] [CrossRef]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-β: ″N1″ versus ″N2″ TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef]
- Mannis, M.; Plotnik, R. Bacterial Conjunctivitis; Tasman, W., Jaeger, E., Eds.; Lippincott Williams and Wilkins: Philadelphia, PA, USA, 2006. [Google Scholar]
- Sheikh, A.; Hurwitz, B.; van Schayck, C.P.; McLean, S.; Nurmatov, U. Antibiotics versus Placebo for Acute Bacterial Conjunctivitis. Cochrane Database Syst. Rev. 2012, 9, CD001211. [Google Scholar] [CrossRef]
- Alfonso, S.A.; Fawley, J.D.; Alexa Lu, X. Conjunctivitis. Prim. Care 2015, 42, 325–345. [Google Scholar] [CrossRef]
- Smith, A.F.; Waycaster, C. Estimate of the Direct and Indirect Annual Cost of Bacterial Conjunctivitis in the United States. BMC Ophthalmol. 2009, 9, 13. [Google Scholar] [CrossRef]
- Azari, A.A.; Barney, N.P. Conjunctivitis: A Systematic Review of Diagnosis and Treatment. JAMA 2013, 311, 1721–1729. [Google Scholar] [CrossRef] [PubMed]
- Høvding, G. Acute Bacterial Conjunctivitis. Acta Ophthalmol. 2008, 86, 5–17. [Google Scholar] [CrossRef] [PubMed]
- Varu, D.M.; Rhee, M.K.; Akpek, E.K.; Amescua, G.; Farid, M.; Garcia-Ferrer, F.J.; Lin, A.; Musch, D.C.; Mah, F.S.; Dunn, S.P. Conjunctivitis Preferred Practice Pattern®. Ophthalmology 2019, 126, P94–P169. [Google Scholar] [CrossRef] [PubMed]
- Cvenkel, B.; Globocnik, M. Conjunctival Scrapings and Impression Cytology in Chronic Conjunctivitis. Correlation with Microbiology. Eur. J. Ophthalmol. 1997, 7, 19–23. [Google Scholar] [CrossRef]
- Lacy, H.M.; Bowlin, A.K.; Hennings, L.; Scurlock, A.M.; Nagarajan, U.M.; Rank, R.G. Essential Role for Neutrophils in Pathogenesis and Adaptive Immunity in Chlamydia caviae Ocular Infections. Infect. Immun. 2011, 79, 1889–1897. [Google Scholar] [CrossRef]
- Lee, K.W.; Jung, J.W. Systemic Minocycline Treatment of Methicillin-Resistant Staphylococcus aureus in Giant Fornix Syndrome. Korean J. Ophthalmol. 2016, 30, 394–395. [Google Scholar] [CrossRef]
- Hu, V.H.; Holland, M.J.; Burton, M.J. Trachoma: Protective and Pathogenic Ocular Immune Responses to Chlamydia trachomatis. PLoS Negl. Trop. Dis. 2013, 7, e2020. [Google Scholar] [CrossRef]
- Shine, W.E.; Silvany, R.; McCulley, J.P. Relation of Cholesterol-Stimulated Staphylococcus Aureus Growth to Chronic Blepharitis. Investig. Ophthalmol. Vis. Sci. 1993, 34, 2291–2296. [Google Scholar]
- Wong, V.W.Y.; Lai, T.Y.Y.; Chi, S.C.C.; Lam, D.S.C. Pediatric Ocular Surface Infections: A 5-Year Review of Demographics, Clinical Features, Risk Factors, Microbiological Results, and Treatment. Cornea 2011, 30, 995–1002. [Google Scholar] [CrossRef]
- Palmer, M.L.; Hyndiuk, R.A. Contact Lens-Related Infectious Keratitis. Int. Ophthalmol. Clin. 1993, 33, 23–49. [Google Scholar] [CrossRef]
- Kerr, N.; Stern, G.A. Bacterial Keratitis Associated with Vernal Keratoconjunctivitis. Cornea 1992, 11, 355–359. [Google Scholar] [CrossRef] [PubMed]
- Parkunan, S.M.; Callegan, M.C. The Pathogenesis of Bacterial Endophthalmitis. In Endophthalmitis; Durand, M.L., Ed.; Springer International Publishing: Cham, Switzerland, 2016; pp. 17–47. [Google Scholar] [CrossRef]
- Hautala, N.; Koskela, M.; Hautala, T. Major Age Group-Specific Differences in Conjunctival Bacteria and Evolution of Antimicrobial Resistance Revealed by Laboratory Data Surveillance. Curr. Eye Res. 2008, 33, 907–911. [Google Scholar] [CrossRef] [PubMed]
- McCormick, C.C.; Caballero, A.R.; Balzli, C.L.; Tang, A.; Weeks, A.; O′Callaghan, R.J. Diverse Virulence of Staphylococcus aureus Strains for the Conjunctiva. Curr. Eye Res. 2010, 36, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Tang, A.; Balzli, C.L.; Caballero, A.R.; McCormick, C.C.; Taylor, S.D.; O′Callaghan, R.J. Staphylococcus aureus Infection of the Rabbit Cornea Following Topical Administration. Curr. Eye Res. 2012, 37, 1075–1083. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, T.S.; Zaidi, T.; Pier, G.B. Antibodies to Conserved Surface Polysaccharides Protect Mice Against Bacterial Conjunctivitis. Investig. Opthalmol. Vis. Sci. 2018, 59, 2512–2519. [Google Scholar] [CrossRef]
- Martin, M.; Turco, J.H.; Zegans, M.E.; Facklam, R.R.; Sodha, S.; Elliott, J.A.; Pryor, J.H.; Beall, B.; Erdman, D.D.; Baumgartner, Y.Y.; et al. An Outbreak of Conjunctivitis Due to Atypical Streptococcus pneumoniae. N. Engl. J. Med. 2003, 348, 1112–1121. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, M.K.; Oh, J.Y.; Jang, K.C.; Wee, W.R.; Lee, J.H. Outbreak of Gram-Positive Bacterial Keratitis Associated with Epidemic Keratoconjunctivitis in Neonates and Infants. Eye 2009, 23, 1059–1065. [Google Scholar] [CrossRef]
- Crum, N.F.; Barrozo, C.P.; Chapman, F.A.; Ryan, M.A.K.; Russell, K.L. An Outbreak of Conjunctivitis Due to a Novel Unencapsulated Streptococcus pneumoniae among Military Trainees. Clin. Infect. Dis. 2004, 39, 1148–1154. [Google Scholar] [CrossRef]
- Buck, J.M.; Lexau, C.; Shapiro, M.; Glennen, A.; Boxrud, D.J.; Koziol, B.; Whitney, C.G.; Beall, B.; Danila, R.; Lynfield, R. A Community Outbreak of Conjunctivitis Caused by Nontypeable Streptococcus pneumoniae in Minnesota. Pediatr. Infect. Dis. J. 2006, 25, 906–911. [Google Scholar] [CrossRef]
- Patel, P.B.; Diaz, M.C.G.; Bennett, J.E.; Attia, M.W. Clinical Features of Bacterial Conjunctivitis in Children. Acad. Emerg. Med. 2007, 14, 1–5. [Google Scholar] [CrossRef]
- Shayegani, M.; Parsons, L.M.; Gibbons, W.E.; Campbell, D. Characterization of Nontypable Streptococcus Pneumoniae-like Organisms Isolated from Outbreaks of Conjunctivitis. J. Clin. Microbiol. 1982, 16, 8–14. [Google Scholar] [PubMed]
- Hyams, C.; Camberlein, E.; Cohen, J.M.; Bax, K.; Brown, J.S. The Streptococcus pneumoniae Capsule Inhibits Complement Activity and Neutrophil Phagocytosis by Multiple Mechanisms. Infect. Immun. 2010, 78, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.E.; Rubin, L.; Romero-Steiner, S.; Dykes, J.K.; Pais, L.B.; Rizvi, A.; Ades, E.; Carlone, G.M. Correlation of Opsonophagocytosis and Passive Protection Assays Using Human Anticapsular Antibodies in an Infant Mouse Model of Bacteremia for Streptococcus pneumoniae. J. Infect. Dis. 1999, 180, 133–140. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kadioglu, A.; Taylor, S.; Iannelli, F.; Pozzi, G.; Mitchell, T.J.; Andrew, P.W. Upper and Lower Respiratory Tract Infection by Streptococcus pneumoniae Is Affected by Pneumolysin Deficiency and Differences in Capsule Type. Infect. Immun. 2002, 70, 2886–2890. [Google Scholar] [CrossRef]
- Norcross, E.W.; Tullos, N.A.; Taylor, S.D.; Sanders, M.E.; Marquart, M.E. Assessment of Streptococcus pneumoniae Capsule in Conjunctivitis and Keratitis in Vivo Neuraminidase Activity Increases in Nonencapsulated Pneumococci Following Conjunctival Infection. Curr. Eye Res. 2010, 35, 787–798. [Google Scholar] [CrossRef]
- Johnson, M.K.; Boese-Marrazzo, D.; Pierce, W.A. Effects of Pneumolysin on Human Polymorphonuclear Leukocytes and Platelets. Infect. Immun. 1981, 34, 171–176. [Google Scholar]
- Cockeran, R.; Theron, A.J.; Steel, H.C.; Matlola, N.M.; Mitchell, T.J.; Feldman, C.; Anderson, R. Proinflammatory Interactions of Pneumolysin with Human Neutrophils. J. Infect. Dis. 2001, 183, 604–611. [Google Scholar] [CrossRef]
- Valentino, M.D.; McGuire, A.M.; Rosch, J.W.; Bispo, P.J.M.; Burnham, C.; Sanfilippo, C.M.; Carter, R.A.; Zegans, M.E.; Beall, B.; Earl, A.M.; et al. Unencapsulated Streptococcus pneumoniae from Conjunctivitis Encode Variant Traits and Belong to a Distinct Phylogenetic Cluster. Nat. Commun. 2014, 5, 1–12. [Google Scholar] [CrossRef]
- Royle, L.; Matthews, E.; Corfield, A.; Berry, M.; Rudd, P.M.; Dwek, R.A.; Carrington, S.D. Glycan Structures of Ocular Surface Mucins in Man, Rabbit and Dog Display Species Differences. Glycoconj. J. 2008, 25, 763–773. [Google Scholar] [CrossRef]
- Isnard, N.; Bourles-Dagonet, F.; Robert, L.; Renard, G. Studies on Corneal Wound Healing. Ophthalmologica 2005, 219, 324–333. [Google Scholar] [CrossRef]
- Stocks, M.E.; Ogden, S.; Haddad, D.; Addiss, D.G.; Mcguire, C.; Freeman, M.C. Effect of Water, Sanitation, and Hygiene on the Prevention of Trachoma: A Systematic Review and Meta-Analysis. PLoS ONE 2014, 11, 1–29. [Google Scholar] [CrossRef]
- Fitch, C.P.; Rapoza, P.A.; Owens, S.; Murillo-Lopez, F.; Johnson, R.A.; Quinn, T.C.; Pepose, J.S.; Taylor, H.R. Epidemiology and Diagnosis of Acute Conjunctivitis at an Inner-City Hospital. Ophthalmology 1989, 96, 1215–1220. [Google Scholar] [CrossRef]
- Malaty, R.; Dawson, C.R.; Wong, I.; Lyon, C.; Schachter, J. Serum and Tear Antibodies to Chlamydia after Reinfection with Guinea Pig Inclusion Conjunctivitis Agent. Investig. Ophthalmol. Vis. Sci. 1981, 21, 833–841. [Google Scholar]
- Rank, R.G.; Whittimore, J.; Bowlin, A.K.; Dessus-Babus, S.; Wyrick, P.B. Chlamydiae and Polymorphonuclear Leukocytes: Unlikely Allies in the Spread of Chlamydial Infection. FEMS Immunol. Med. Microbiol. 2008, 54, 104–113. [Google Scholar] [CrossRef]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.-K.L.; Flavell, R.A. Transforming Growth Factor-β Regulation of Immune Responses. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef]
- Veena, C. Bacteriological Study of Conjunctivitis. Int. J. Contemp. Med. Res. ISSN 2015, 3, 2393–2915. [Google Scholar]
- Tarabishy, A.B.; Jeng, B.H. Bacterial Conjunctivitis: A Review for Internists. Clevel. Clin. J. Med. 2008, 75, 507–512. [Google Scholar] [CrossRef]
- Wong, R.L.M.; Gangwani, R.A.; Yu, L.W.H.; Lai, J.S.M. New Treatments for Bacterial Keratitis. J. Ophthalmol. 2012, 2012, 1–7. [Google Scholar] [CrossRef]
- Ansari, Z.; Miller, D.; Galor, A. Current Thoughts in Fungal Keratitis: Diagnosis and Treatment. Curr. Fungal Infect. Rep. 2013, 7, 209–218. [Google Scholar] [CrossRef]
- Marquart, M.E.; O′Callaghan, R.J. Infectious Keratitis: Secreted Bacterial Proteins That Mediate Corneal Damage. J. Ophthalmol. 2013, 2013, 1–9. [Google Scholar] [CrossRef]
- Hazlett, L.D.; Rosen, D.; Berk, R.S. Experimental Eye Infections Caused by Pseudomonas aeruginosa. Ophthalmic Res. 1976, 8, 311–318. [Google Scholar] [CrossRef]
- Stapleton, F.; Carnt, N. Contact Lens-Related Microbial Keratitis: How Have Epidemiology and Genetics Helped Us with Pathogenesis and Prophylaxis. Eye 2011, 26, 185–193. [Google Scholar] [CrossRef]
- Musa, F.; Tailor, R.; Gao, A.; Hutley, E.; Rauz, S.; Scott, R.A.H.; Fayyaz, D.; Musa, U. Contact Lens-Related Microbial Keratitis in Deployed British Military Personnel Clinical Science. Br. J. Ophthalmol. 2010, 94, 988–993. [Google Scholar] [CrossRef]
- Lin, A.; Rhee, M.K.; Akpek, E.K.; Amescua, G.; Farid, M.; Garcia-Ferrer, F.J.; Varu, D.M.; Musch, D.C.; Dunn, S.P.; Mah, F.S. Bacterial Keratitis Preferred Practice Pattern®. Ophthalmology 2019, 126, P1–P55. [Google Scholar] [CrossRef]
- Fleiszig, S.M.; Evans, D.J. Pathogenesis of contact lens-associated microbial keratitis. Optom. Vis. Sci. 2010, 87, 225–232. [Google Scholar] [CrossRef]
- Evans, D.J.; McNamara, N.A.; Fleiszig, S.M. Life at the Front: Dissecting Bacterial-Host Interactions at the Ocular Surface. Ocul. Surf. 2007, 5, 213–227. [Google Scholar] [CrossRef]
- Williamson, Y.M.; Gowrisankar, R.; Longo, D.L.; Facklam, R.; Gipson, I.K.; Ades, E.P.; Carlone, G.M.; Sampson, J.S. Adherence of Nontypeable Streptococcus pneumoniae to Human Conjunctival Epithelial Cells. Microb. Pathog. 2008, 44, 175–185. [Google Scholar] [CrossRef]
- Rhem, M.N.; Lech, E.M.; Patti, J.M.; Mcdevitt, D.; Höök, M.; Höök, H.; Höök, H.; Jones, D.B.; Wilhelmus, K.R.; Richardson, S.W. The Collagen-Binding Adhesin Is a Virulence Factor in Staphylococcus aureus Keratitis. Infect. Immun. 2000, 68, 3776–3779. [Google Scholar] [CrossRef]
- Astley, R.; Miller, F.C.; Mursalin, M.H.; Coburn, P.S.; Callegan, M.C. An Eye on Staphylococcus aureus toxins: Roles in Ocular Damage and Inflammation. Toxins 2019, 11, 356. [Google Scholar] [CrossRef]
- Vareechon, C.; Zmina, S.E.; Karmakar, M.; Pearlman, E.; Rietsch, A. Pseudomonas Aeruginosa Effector ExoS Inhibits ROS Production in Human Neutrophils. Cell Host Microbe 2017, 21, 611–618.e5. [Google Scholar] [CrossRef]
- Webeye.ophth.uiowa.edu. Available online: https://webeye.ophth.uiowa.edu/eyeforum/atlas/pages/pseudomonas-keratitis-34.html (accessed on 28 October 2019).
- Lee, K.; Lee, H.; Kim, M. Two Cases of Corneal Ulcer due to Methicillin-Resistant Staphylococcus aureus in High Risk Groups. Korean J. Ophthalmol. 2010, 24, 240–244. [Google Scholar] [CrossRef]
- Alvarruiz-Picazo, J.; Blanco-Marchite, N.; Donate-Tercero, A.; Blanco-Marchite, C.I.; Bautista-Ruescas, V. Streptococcus pneumoniae keratitis, a case report. Arch. Med. 2009, 1, 1–4. [Google Scholar]
- Gerke, J.R.; Magliocco, M.V. Experimental Pseudomonas aeruginosa Infection of the Mouse Cornea. Infect. Immun. 1971, 3, 209–216. [Google Scholar]
- Chusid, M.J.; Davis, S.D. Experimental Bacterial Keratitis in Neutropenic Guinea Pigs: Polymorphonuclear Leukocytes in Corneal Host Defense. Infect. Immun. 1979, 24, 948–952. [Google Scholar]
- Hazlett, L.D.; Rosen, D.; Berk, R.S. Pseudomonas Eye Infections in Cyclophosphamide-Treated Mice. Investig. Opthalmol. Vis. Sci. 1977, 16, 649–652. [Google Scholar]
- Zaidi, T.S.; Zaidi, T.; Pier, G.B. Role of Neutrophils, MyD88-Mediated Neutrophil Recruitment, and Complement in Antibody-Mediated Defense against Pseudomonas aeruginosa Keratitis. Investig. Ophthalmol. Vis. Sci. 2010, 51, 2085–2093. [Google Scholar] [CrossRef]
- Thanabalasuriar, A.; Scott, B.N.V.; Peiseler, M.; Willson, M.E.; Zeng, Z.; Warrener, P.; Keller, A.E.; Surewaard, B.G.J.; Dozier, E.A.; Korhonen, J.T.; et al. Neutrophil Extracellular Traps Confine Pseudomonas aeruginosa Ocular Biofilms and Restrict Brain Invasion. Cell Host Microbe 2019, 25, 526–536. [Google Scholar] [CrossRef]
- Sun, Y.; Karmakar, M.; Taylor, P.R.; Rietsch, A.; Pearlman, E. ExoS and ExoT ADP Ribosyltransferase Activities Mediate Pseudomonas aeruginosa Keratitis by Promoting Neutrophil Apoptosis and Bacterial Survival. J. Immunol. 2012, 188, 1884–1895. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, K.; Ambati, B.; Yu, F.S.X. Toll-like Receptor 5-Mediated Corneal Epithelial Inflammatory Responses to Pseudomonas aeruginosa Flagellin. Investig. Ophthalmol. Vis. Sci. 2003, 44, 4247–4254. [Google Scholar] [CrossRef]
- Karmakar, M.; Sun, Y.; Hise, A.G.; Rietsch, A.; Pearlman, E. IL-1β Processing during Pseudomonas aeruginosa Infection Is Mediated by Neutrophil Serine Proteases and Is Independent of NLRC4 and Caspase-1. J. Immunol. 2012, 189, 4231–4235. [Google Scholar] [CrossRef]
- Xue, M.-L.; Thakur, A.; Cole, N.; Lloyd, A.; Stapleton, F.; Wakefield, D.; Willcox, M.D.P. A Critical Role for CCL2 and CCL3 Chemokines in the Regulation of Polymorphonuclear Neutrophils Recruitment during Corneal Infection in Mice. Immunol. Cell Biol. 2007, 85, 525–531. [Google Scholar] [CrossRef]
- Jeng, B.H.; Gritz, D.C.; Kumar, A.B.; Holsclaw, D.S.; Porco, T.C.; Smith, S.D.; Whitcher, J.P.; Margolis, T.P.; Wong, I.G. Epidemiology of Ulcerative Keratitis in Northern California. Arch. Ophthalmol. 2010, 128, 1022–1028. [Google Scholar] [CrossRef]
- Shanmuganathan, V.A.; Armstrong, M.; Buller, A.; Tullo, A.B. External Ocular Infections Due to Methicillin-Resistant Staphylococcus aureus (MRSA). Eye 2005, 19, 284–291. [Google Scholar] [CrossRef]
- Solomon, R.; Donnenfeld, E.D.; Perry, H.D.; Biser, S. Bilateral Methicillin-Resistant Staphylococcus aureus Keratitis in a Medical Resident Following an Uneventful Bilateral Photorefractive Keratectomy. Eye Contact Lens 2003, 29, 187–189. [Google Scholar] [CrossRef]
- Callegan, M.C.; Engel, L.S.; Hill, J.M.; O′Callaghan, R.J. Corneal Virulence of Staphylococcus aureus: Roles of Alpha-Toxin and Protein A in Pathogenesis. Infect. Immun. 1994, 62, 2478–2482. [Google Scholar]
- Sloop, G.D.; Moreau, J.M.; Conerly, L.L.; Dajcs, J.J.; O′Callaghan, R.J. Acute Inflammation of the Eyelid and Cornea in Staphylococcus Keratitis in the Rabbit. Investig. Ophthalmol. Vis. Sci. 1999, 40, 385–391. [Google Scholar]
- Chusid, M.J.; Nelson, D.B.; Meyer, L.A. The Role of the Polymorphonuclear Leukocyte in the Induction of Corneal Edema. Investig. Ophthalmol. Vis. Sci. 1986, 27, 1466–1469. [Google Scholar]
- O′Callaghan, R.J.; Callegan, M.C.; Moreau, J.M.; Green, L.C.; Foster, T.J.; Hartford, O.M.; Engel, L.S.; Hill, J.M. Specific Roles of Alpha-Toxin and Beta-Toxin during Staphylococcus Aureus Corneal Infection. Infect. Immun. 1997, 65, 1571–1578. [Google Scholar]
- O′Callaghan, R.J.; Girgis, D.O.; Dajcs, J.J.; Sloop, G.D. Host Defense against Bacterial Keratitis. Ocul. Immunol. Inflamm. 2003, 11, 171–181. [Google Scholar] [CrossRef]
- Kumar, A.; Zhang, J.; Yu, F.-S.X. Innate Immune Response of Corneal Epithelial Cells to Staphylococcus aureus Infection: Role of Peptidoglycan in Stimulating Proinflammatory Cytokine Secretion. Investig. Ophthalmol. Vis. Sci. 2004, 45, 3513–3522. [Google Scholar] [CrossRef]
- Sun, Y.; Hise, A.G.; Kalsow, C.M.; Pearlman, E. Staphylococcus aureus-Induced Corneal Inflammation Is Dependent on Toll-Like Receptor 2 and Myeloid Differentiation Factor 88. Infect. Immun. 2006, 74, 5325–5332. [Google Scholar] [CrossRef] [PubMed]
- Cole, N.; Hume, E.B.H.; Khan, S.; Garthwaite, L.; Conibear, T.C.R.; Willcox, M.D.P. The Role of CXC Chemokine Receptor 2 in Staphylococcus aureus Keratitis. Exp. Eye Res. 2014, 12 7, 184–189. [Google Scholar] [CrossRef]
- Khan, S.; Cole, N.; Hume, E.B.; Garthwaite, L.; Conibear, T.C.R.; Miles, D.H.; Aliwaga, Y.; Krockenberger, M.B.; Willcox, M.D.P. The Role of CXC Chemokine Receptor 2 in Pseudomonas aeruginosa Corneal Infection. J. Leukoc. Biol. 2007, 81, 315–318. [Google Scholar] [CrossRef] [PubMed]
- Heimer, S.R.; Evans, D.J.; Mun, J.J.; Stern, M.E.; Fleiszig, S.M. Surfactant protein D contributes to ocular defense against Pseudomonas aeruginosa in a murine model of dry eye disease. PLoS ONE 2013, 8, e65797. [Google Scholar] [CrossRef] [PubMed]
- Mun, J.J.; Tam, C.; Kowbel, D.; Hawgood, S.; Barnett, M.J.; Evans, D.J.; Fleiszig, S.M. Clearance of Pseudomonas aeruginosa from a healthy ocular surface involves surfactant protein D and is compromised by bacterial elastase in a murine null-infection model. Infect. Immun. 2009, 77, 2392–2398. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Abdel-Razek, O.; Hawgood, S.; Wang, G. Protective Role of Surfactant Protein D in Ocular Staphylococcus aureus Infection. PLoS ONE 2015, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Booth, M.C.; Pence, L.M.; Mahasreshti, P.; Callegan, M.C.; Gilmore, M.S. Clonal associations among Staphylococcus aureus isolates from various sites of infection. Infect. Immun. 2001, 69, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Coia, J.E.; Browning, L.; Haines, L.; Birkbeck, T.H.; Platt, D.J. Comparison of Enterotoxins and Haemolysins Produced by Methicillin-Resistant (MRSA) and Sensitive (MSSA) Staphylococcus aureus. J. Med. Microbiol. 1992, 36, 164–171. [Google Scholar] [CrossRef]
- Kwak, Y.K.; Vikstrom, E.; Magnusson, K.E.; Vecsey-Semjen, B.; Colque-Navarro, P.; Mollby, R. The Staphylococcus aureus alpha-toxin perturbs the barrier function in Caco-2 epithelial cell monolayers by altering junctional integrity. Infect. Immun. 2012, 80, 1670–1680. [Google Scholar] [CrossRef]
- Hocke, A.C.; Temmesfeld-Wollbrueck, B.; Schmeck, B.; Berger, K.; Frisch, E.M.; Witzenrath, M.; Brell, B.; Suttorp, N.; Hippenstiel, S. Perturbation of endothelial junction proteins by Staphylococcus aureus alpha-toxin: Inhibition of endothelial gap formation by adrenomullin. Histochem. Cell Biol. 2006, 126, 305–316. [Google Scholar] [CrossRef]
- Dajcs, J.J.; Thibodeaux, B.A.; Girgis, D.O.; O′Callaghan, R.J. Corneal Virulence of Staphylococcus aureus in an Experimental Model of Keratits. DNA Cell Biol. 2004, 21, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Tassopoulos, A.M.; Li, Q.; Yu, F.-S.X. Staphylococcus aureus Protein A Induced Inflammatory Response in Human Corneal Epithelial Cells. Biochem. Biophys. Res. Commun. 2007, 354, 955–961. [Google Scholar] [CrossRef]
- Bharathi, M.J.; Ramakrishnan, R.; Shivakumar, C.; Meenakshi, R.; Lionalraj, D. Etiology and Antibacterial Susceptibility Pattern of Community-Acquired Bacterial Ocular Infections in a Tertiary Eye Care Hospital in South India. Indian J. Ophthalmol. 2010, 58, 497–507. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, S.; Ozturk, I.; Maden, A. Microbial Keratitis in West Anatolia, Turkey: A Retrospective Review. Int. Ophthalmol. 2007, 27, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Parmar, P.; Salman, A.; Kalavathy, C.M.; Jesudasan, C.A.N.; Thomas, P.A. Pneumococcal Keratitis: A Clinical Profile. Clin. Exp. Ophthalmol. 2003, 31, 44–47. [Google Scholar] [CrossRef] [PubMed]
- Gosbell, I.B.; Neville, S.A. Antimicrobial Resistance in Streptococcus pneumoniae: A Decade of Results from South-Western Sydney. Commun. Dis. Intell. 2000, 24, 340–343. [Google Scholar]
- Lu, C.Y.; Lee, P.I.; Hsueh, P.R.; Chang, S.C.; Chiu, T.F.; Lin, H.C.; Lee, C.Y.; Huang, L.M. Penicillin-Nonsusceptible Streptococcus pneumoniae Infections in Children. J. Microbiol. Immunol. Infect. 1999, 32, 179–186. [Google Scholar]
- Cosar, C.B.; Cohen, E.J.; Rapuano, C.J.; Laibson, P.R. Clear Corneal Wound Infection After Phacoemulsification. Arch. Ophthalmol. 2001, 119, 1755–1759. [Google Scholar] [CrossRef]
- Mulet, M.E.; Pérez-Santonja, J.J.; Ferrer, C.; Alió, J.L. Microbial Keratitis After Intrastromal Corneal Ring Segment Implantation. J. Refract. Surg. 2010, 26, 364–369. [Google Scholar] [CrossRef]
- Rehany, U.; Balut, G.; Lefler, E.; Rumelt, S. The Prevalence and Risk Factors for Donor Corneal Button Contamination and Its Association with Ocular Infection after Transplantation. Cornea 2004, 23, 649–654. [Google Scholar] [CrossRef]
- Okonkwo, A.C.O.; Siah, W.F.; Hogg, H.D.J.; Anwar, H.; Figueiredo, F.C. Microbial Keratitis in Corneal Grafts: Predisposing Factors and Outcomes. Eye 2018, 32, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.K.; Hobden, J.A.; Hagenah, M.; O’Callaghan, R.J.; Hill, J.M.; Chen, S. The Role of Pneumolysin in Ocular Infections with Streptococcus pneumoniae. Curr. Eye Res. 1990, 9, 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.C.; Karcioglu, Z.A.; Johnson, M.K. Response of Leukopenic Rabbits to Pneumococcal Toxin. Curr. Eye Res. 1982, 2, 705–710. [Google Scholar] [CrossRef] [PubMed]
- Norcross, E.W.; Sanders, M.E.; Moore, Q.C.; Marquart, M.E.; Marquart, M.E. Pathogenesis of A Clinical Ocular Strain of Streptococcus pneumoniae and the Interaction of Pneumolysin with Corneal Cells. J. Bacteriol. Parasitol. 2011, 2, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.K.; Allen, J.H. The Role of Cytolysin in Pneumococcal Ocular Infection. Am. J. Ophthalmol. 1975, 80, 518–521. [Google Scholar] [CrossRef]
- Johnson, M.K.; Callegan, M.C.; Engel, L.S.; O′Callaghan, R.J.; Hill, J.M.; Hobden, J.A.; Boulnois, G.J.; Andrew, P.W.; Mitchell, T.J. Growth andv virulence of a complement-activation-negative mutant of Streptococcus pneumonia in the rabbit cornea. Curr. Eye Res. 1995, 14, 281–284. [Google Scholar] [CrossRef]
- Karmakar, M.; Katsnelson, M.; Malak, H.A.; Greene, N.G.; Howell, S.J.; Hise, A.G.; Camilli, A.; Kadioglu, A.; Dubyak, G.R.; Pearlman, E. Neutrophil IL-1β Processing Induced by Pneumolysin Is Mediated by the NLRP3/ASC Inflammasome and Caspase-1 Activation, and Is Dependent on K + Efflux. J. Immunol. 2015, 194, 1763–1775. [Google Scholar] [CrossRef]
- Jedrzejas, M.J. Pneumococcal Virulence Factors: Structure and Function. Microbiol. Mol. Biol. Rev. 2001, 65, 187–207. [Google Scholar] [CrossRef]
- Srinivasan, M.; Gonzales, C.A.; George, C.; Cevallos, V.; Mascarenhas, J.M.; Asokan, B.; Wilkins, J.; Smolin, G.; Whitcher, J.P. Epidemiology and Aetiological Diagnosis of Corneal Ulceration in Madurai, South India. Br. J. Ophthalmol. 1997, 81, 965–971. [Google Scholar] [CrossRef]
- Norina, T.J.; Raihan, S.; Bakiah, S.; Ezanee, M.; Liza-Sharmini, A.T.; Wan Hazzabah, W.H. Microbial Keratitis: Aetiological Diagnosis and Clinical Features in Patients Admitted to Hospital Universiti Sains Malaysia. Singap. Med. J. 2008, 49, 67–71. [Google Scholar]
- Bharathi, M.J.; Ramakrishnan, R.; Vasu, S.; Meenakshi, R.; Shivkumar, C.; Palaniappan, R. Epidemiology of Bacterial Keratitis in a Referral Centre in South India. Indian J. Med. Microbiol. 2003, 21, 239–245. [Google Scholar] [PubMed]
- Reed, J.M.; O′Callaghan, R.J.; Girgis, D.O.; McCormick, C.C.; Caballero, A.R.; Marquart, M.E. Ocular Virulence of Capsule-Deficient Streptococcus pneumoniae in a Rabbit Keratitis Model. Investig. Ophthalmol. Vis. Sci. 2005, 46, 604–608. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.B. Decision-Making in the Management of Microbial Keratitis. Ophthalmology 1981, 88, 814–820. [Google Scholar] [CrossRef]
- Solanki, S.; Rathi, M.; Khanduja, S.; Dhull, C.S.; Sachdeva, S.; Phogat, J. Recent Trends: Medical Management of Infectious Keratitis. Oman J. Ophthalmol. 2015, 8, 83–85. [Google Scholar] [CrossRef]
- Rathinam, S.; Namperumalsamy, P. Global Variation and Pattern Changes in Epidemiology of Uveitis. Indian J. Ophthalmol. 2007, 55, 173–183. [Google Scholar] [CrossRef]
- Suhler, E.B.; Lloyd, M.J.; Choi, D.; Rosenbaum, J.T.; Austin, D.F. Incidence and Prevalence of Uveitis in Veterans Affairs Medical Centers of the Pacific Northwest. Am. J. Ophthalmol. 2008, 146, 890–896.e8. [Google Scholar] [CrossRef]
- Lin, P. Infectious Uveitis. Curr. Ophthalmol. Rep. 2015, 3, 170–183. [Google Scholar] [CrossRef]
- Aldave, A.J.; King, J.A.; Cunningham, E.T. Ocular Syphilis. Curr. Opin. Ophthalmol. 2001, 12, 433–441. [Google Scholar] [CrossRef]
- Bernard, A.; Kodjikian, L.; Abukhashabh, A.; Roure-Sobas, C.; Boibieux, A.; Denis, P.; Broussolle, C.; Seve, P. Diagnosis of Lyme-associated Uveitis: Value of Serological Testing in a Tertiary Centre. Br. J. Ophthalmol. 2018, 102, 369–372. [Google Scholar] [CrossRef]
- Bernard, A.; Seve, P.; Abukhashabh, A.; Roure-Sobas, C.; Boibieux, A.; Denis, P.; Broussolle, C.; Mathis, T.; Kodjikian, L. Lyme-Associated Uveitis: Clinical Spectrum and Review of Literature. Eur. J. Ophthalmol. 2019. [Google Scholar] [CrossRef]
- Singh, R.; Gupta, V.; Gupta, A. Pattern of Uveitis in a Referral Eye Clinic in North India. Indian J. Ophthalmol. 2004, 52, 121–125. [Google Scholar] [PubMed]
- Kaimbo Wa Kimbo, D.; Bifuko, A.; Dernouchamps, J.P.; Missotten, L. Chronic Uveitis in Kinshasa (D R Congo). Bull. Soc. Belge Ophtalmol. 1998, 270, 95–100. [Google Scholar] [PubMed]
- Ayo, C. A Toxic Ocular Reaction I. New Property of Shwartzman Toxins. J. Immunol. 1943, 46, 113–132. [Google Scholar]
- Whitcup, S.M.; Wakefield, D.; Li, Q.; Nussenblatt, R.B.; Chan, C.C. Endothelial Leukocyte Adhesion Molecule-1 in Endotoxin-Induced Uveitis. Investig. Ophthalmol. Vis. Sci. 1992, 33, 2626–2630. [Google Scholar]
- Whitcup, S.M.; DeBarge, L.R.; Rosen, H.; Nussenblatt, R.B.; Chan, C.C. Monoclonal Antibody against CD11b/CD18 Inhibits Endotoxin-Induced Uveitis. Investig. Ophthalmol. Vis. Sci. 1993, 34, 673–681. [Google Scholar]
- Li, X.; Gu, X.; Boyce, T.M.; Zheng, M.; Reagan, A.M.; Qi, H.; Mandal, N.; Cohen, A.W.; Callegan, M.C.; Carr, D.J.J.; et al. Caveolin-1 Increases Proinflammatory Chemoattractants and Blood–Retinal Barrier Breakdown but Decreases Leukocyte Recruitment in Inflammation. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6224–6234. [Google Scholar] [CrossRef]
- Hoekzema, R.; Murray, P.I.; van Haren, M.A.; Helle, M.; Kijlstra, A. Analysis of Interleukin-6 in Endotoxin-Induced Uveitis. Investig. Ophthalmol. Vis. Sci. 1991, 32, 88–95. [Google Scholar]
- Granowitz, E.V.; Poutsiaka, D.D.; Cannon, J.G.; Wolff, S.M.; Dinarello, C.A.; Santos, A.A.; Wilmore, D.W. Production of Interleukin-1-Receptor Antagonist during Experimental Endotoxaemia. Lancet 1991, 338, 1423–1424. [Google Scholar] [CrossRef]
- Okumura, A.; Mochizuki, M.; Nishi, M.; Herbort, C.P. Endotoxin-Induced Uveitis (EIU) in the Rat: A Study of Inflammatory and Immunological Mechanisms. Int. Ophthalmol. 1989, 14, 31–36. [Google Scholar] [CrossRef]
- Rosenbaum, J.T.; McDevitt, H.O.; Guss, R.B.; Egbert, P.R. Endotoxin-Induced Uveitis in Rats as a Model for Human Disease. Nature 1980, 286, 611–613. [Google Scholar] [CrossRef]
- Howes, E.L.; Goldyne, M.E.; Perez, H.D.; Goldstein, I.M.; Rosenbaum, J.T. Lipopolysaccharide Tolerance Inhibits Eye Inflammation. Arch. Ophthalmol. 1985, 103, 261–265. [Google Scholar] [CrossRef] [PubMed]
- Bhattacherjee, P.; Parke, A. The Reduction of Inflammatory Responses in Lipopolysaccharide-Tolerant Eyes. Am. J. Pathol. 1986, 122, 268–276. [Google Scholar] [PubMed]
- Chang, J.H.; Hampartzoumian, T.; Everett, B.; Lloyd, A.; McCluskey, P.J.; Wakefield, D. Changes in Toll-like Receptor (TLR)-2 and TLR4 Expression and Function but Not Polymorphisms Are Associated with Acute Anterior Uveitis. Investig. Opthalmol. Vis. Sci. 2007, 48, 1711–1717. [Google Scholar] [CrossRef] [PubMed]
- Mashimo, H.; Ohguro, N.; Nomura, S.; Hashida, N.; Nakai, K.; Tano, Y. Neutrophil Chemotaxis and Local Expression of Interleukin-10 in the Tolerance of Endotoxin-Induced Uveitis. Investig. Opthalmol. Vis. Sci. 2008, 49, 5450–5457. [Google Scholar] [CrossRef]
- Callegan, M.C.; Engelbert, M.; Ii, D.W.P.; Jett, B.D.; Gilmore, M.S. Bacterial Endophthalmitis: Epidemiology, Therapeutics, and Bacterium-Host Interactions. Clin. Microbiol. Rev. 2002, 15, 111–124. [Google Scholar] [CrossRef]
- Callegan, M.C.; Gilmore, M.S.; Gregory, M.; Ramadan, R.T.; Wiskur, B.J.; Moyer, A.L.; Hunt, J.J.; Novosad, B.D. Bacterial Endophthalmitis: Therapeutic Challenges and Host-Pathogen Interactions. Prog. Retin. Eye Res. 2007, 26, 189–203. [Google Scholar] [CrossRef]
- Bhagat, N.; Nagori, S.; Zarbin, M. Post-Traumatic Infectious Endophthalmitis. Surv. Ophthalmol. 2011, 56, 214–251. [Google Scholar] [CrossRef]
- Kattan, H.M.; Flynn, H.W.; Pflugfelder, S.C.; Robertson, C.; Forster, R.K. Nosocomial Endophthalmitis Survey. Current Incidence of Infection after Intraocular Surgery. Ophthalmology 1991, 98, 227–238. [Google Scholar] [CrossRef]
- Vision 2020. The Right to Sight. Global Initiative for the Elimination of Avoidable Blindness. Available online: https://www.who.int/blindness/Vision2020_report.pdf (accessed on 25 June 2019).
- Kresloff, M.S.; Castellarin, A.A.; Zarbin, M.A. Endophthalmitis. Surv. Ophthalmol. 1998, 43, 193–224. [Google Scholar] [CrossRef]
- Kernt, M.; Kampik, A.; Kernt, M. Clinical Ophthalmology Endophthalmitis: Pathogenesis, Clinical Presentation, Management, and Perspectives. Clin. Ophthalmol. 2010, 4, 121–135. [Google Scholar] [CrossRef]
- Results of the Endophthalmitis Vitrectomy Study. A Randomized Trial of Immediate Vitrectomy and of Intravenous Antibiotics for the Treatment of Postoperative Bacterial Endophthalmitis. Endophthalmitis Vitrectomy Study Group. Arch. Ophthalmol. 1995, 113, 1479–1496. [Google Scholar] [CrossRef]
- Seal, D.; Reischl, U.; Behr, A.; Ferrer, C.; Alió, J.; Koerner, R.J.; Barry, P.; ESCRS Endophthalmitis Study Group. Laboratory Diagnosis of Endophthalmitis: Comparison of Microbiology and Molecular Methods in the European Society of Cataract; Refractive Surgeons Multicenter Study and Susceptibility Testing. J. Cataract Refract. Surg. 2008, 34, 1439–1450. [Google Scholar] [CrossRef] [PubMed]
- Scott, I.U.; Flynn, H.W.; Feuer, W.; Pflugfelder, S.C.; Alfonso, E.C.; Forster, R.K.; Miller, D. Endophthalmitis Associated with Microbial Keratitis. Ophthalmology 1996, 103, 1864–1870. [Google Scholar] [CrossRef]
- Maitray, A.; Rishi, E.; Rishi, P.; Gopal, L.; Bhende, P.; Ray, R.; Therese, K.L. Endogenous endophthalmitis in children and adolescents: Case series and literature review. Indian J. Ophthalmol. 2019, 67, 795–800. [Google Scholar] [PubMed]
- Pan, Q.; Liu, Y.; Wang, R.; Chen, T.; Yang, Z.; Deng, Y.; Zhao, Z.; Hu, X.; Chen, X.; Wei, W.; et al. Treatment of Bacillus cereus endophthalmitis with endoscopy-assisted vitrectomy. Medicine 2017, 50, 1–6. [Google Scholar] [CrossRef]
- Ahn, M.W.; Shin, M.K.; Park, S.W.; Lee, J.E. Two Cases of Recurrent Enterococcus Faecalis Endophthalmitis after Cataract Surgery. J. Korean Ophthalmol. Soc. 2015, 4, 632–637. [Google Scholar] [CrossRef][Green Version]
- Callegan, M.C.; Booth, M.C.; Jett, B.D.; Gilmore, M.S. Pathogenesis of Gram-Positive Bacterial Endophthalmitis. Infect. Immun. 1999, 67, 3348–3356. [Google Scholar]
- Callegan, M.C.; Cochran, D.C.; Kane, S.T.; Gilmore, M.S.; Gominet, M.; Lereclus, D. Contribution of Membrane-Damaging Toxins to Bacillus Endophthalmitis Pathogenesis. Infect. Immun. 2002, 70, 5381–5389. [Google Scholar] [CrossRef]
- Callegan, M.C.; Kane, S.T.; Cochran, D.C.; Gilmore, M.S. Bacteria Molecular Mechanisms of Bacillus Endophthalmitis Pathogenesis. DNA Cell Biol. 2004, 21, 367–373. [Google Scholar] [CrossRef]
- Giese, M.J.; Berliner, J.A.; Riesner, A.; Wagar, E.A.; Mondino, B.J. A Comparison of the Early Inflammatory Effects of an Agr-/Sar-versus a Wild Type Strain of Staphylococcus aureus in a Rat Model of Endophthalmitis. Curr. Eye Res. 1999, 18, 177–185. [Google Scholar] [CrossRef]
- Weiss, S.J. Tissue Destruction by Neutrophils. N. Engl. J. Med. 1989, 320, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Affeldt, J.C.; Flynn, H.W.; Forster, R.K.; Mandelbaum, S.; Clarkson, J.G.; Jarus, G.D. Microbial Endophthalmitis Resulting from Ocular Trauma. Ophthalmology 1987, 94, 407–413. [Google Scholar] [CrossRef]
- Olson, J.C.; Flynn, H.W.; Forster, R.K.; Culbertson, W.W. Results in the Treatment of Postoperative Endophthalmitis. Ophthalmology 1983, 90, 692–699. [Google Scholar] [CrossRef]
- Rowsey, J.J.; Newsom, D.L.; Sexton, D.J.; Harms, W.K. Endophthalmitis: Current Approaches. Ophthalmology 1982, 89, 1055–1066. [Google Scholar] [CrossRef]
- Major, J.C.; Engelbert, M.; Flynn, H.W.; Miller, D.; Smiddy, W.E.; Davis, J.L. Staphylococcus aureus Endophthalmitis: Antibiotic Susceptibilities, Methicillin Resistance, and Clinical Outcomes. Am. J. Ophthalmol. 2010, 149, 278–283. [Google Scholar] [CrossRef]
- Abbey, M.A.; Darlene, M.; Flynn, H.W. Staphylococcus aureus Endophthalmitis: Antibiotic Susceptibilities, Methicillin Resistance, and Clinical Outcomes. Investig. Ophthalmol. Vis. Sci. 2012, 53, 1689. [Google Scholar]
- Suzuki, T.; Campbell, J.; Swoboda, J.G.; Walker, S.; Gilmore, M.S. Role of wall teichoic acids in Staphylococcus aureus endophthalmitis. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3187–3192. [Google Scholar] [CrossRef]
- Kumar, A.; Kumar, A. Role of Staphylococcus aureus Virulence Factors in Inducing Inflammation and Vascular Permeability in a Mouse Model of Bacterial Endophthalmitis. PLoS ONE 2015, 10, 1–17. [Google Scholar] [CrossRef]
- Siqueira, J.A.; Speeg-Schatz, C.; Freitas, F.I.S.; Sahel, J.; Monteil, H.; Prevost, G. Channel-Forming Leucotoxins from Staphylococcus aureus Cause Severe Inflammatory Reactions in a Rabbit Eye Model. J. Med. Microbiol. 1997, 46, 486–494. [Google Scholar] [CrossRef]
- Jenul, C.; Horswill, A.R. Regulation of Staphylococcus aureus Virulence. Microbiol. Spectr. 2018, 6, 1–34. [Google Scholar] [CrossRef]
- Booth, M.C.; Atkuri, R.V.; Nanda, S.K.; Iandolo, J.J.; Gilmore, M.S. Accessory Gene Regulator Controls Staphylococcus aureus Virulence in Endophthalmitis. Infect. Immun. 1995, 36, 1828–1836. [Google Scholar]
- Booth, M.C.; Cheung, A.L.; Hatter, K.L.; Jett, B.D.; Callegan, M.C.; Gilmore, M.S. Staphylococcal Accessory Regulator (Sar) in Conjunction with Agr Contributes to Staphylococcus aureus Virulence in Endophthalmitis. Infect. Immun. 1997, 65, 1550–1556. [Google Scholar] [PubMed]
- Bhakdi, S.; Tranum-Jensen, J. Alpha-Toxin of Staphylococcus aureus. Microbiol. Rev. 1991, 55, 733–751. [Google Scholar] [PubMed]
- Giese, M.J.; Rayner, S.A.; Fardin, B.; Sumner, H.L.; Rozengurt, N.; Mondino, B.J.; Gordon, L.K. Mitigation of Neutrophil Infiltration in a Rat Model of Early Staphylococcus aureus Endophthalmitis. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3077–3082. [Google Scholar] [CrossRef]
- Rajamani, D.; Singh, P.K.; Rottmann, B.G.; Singh, N.; Bhasin, M.K.; Kumar, A. Temporal Retinal Transcriptome and Systems Biology Analysis Identifies Key Pathways and Hub Genes in Staphylococcus aureus Endophthalmitis. Sci. Rep. 2016, 6, 1–15. [Google Scholar] [CrossRef]
- Kumar, A.; Giri, S.; Kumar, A. 5-Aminoimidazole-4-Carboxamide Ribonucleosidemediated Adenosine Monophosphate-Activated Protein Kinase Activation Induces Protective Innate Responses in Bacterial Endophthalmitis. Cell. Microbiol. 2016, 18, 1815–1830. [Google Scholar] [CrossRef]
- Cho, J.S.; Pietras, E.M.; Garcia, N.C.; Ramos, R.I.; Farzam, D.M.; Monroe, H.R.; Magorien, J.E.; Blauvelt, A.; Kolls, J.K.; Cheung, A.L.; et al. IL-17 Is Essential for Host Defense against Cutaneous Staphylococcus aureus Infection in Mice. J. Clin. Investig. 2010, 120, 1762–1773. [Google Scholar] [CrossRef]
- Minegishi, Y.; Saito, M.; Nagasawa, M.; Takada, H.; Hara, T.; Tsuchiya, S.; Agematsu, K.; Yamada, M.; Kawamura, N.; Ariga, T.; et al. Molecular Explanation for the Contradiction between Systemic Th17 Defect and Localized Bacterial Infection in Hyper-IgE Syndrome. J. Exp. Med. 2009, 206, 1291–1301. [Google Scholar] [CrossRef]
- Karthikeyan, R.S.; Vareechon, C.; Prajna, N.V.; Dharmalingam, K.; Pearlman, E.; Lalitha, P. Interleukin 17 Expression in Peripheral Blood Neutrophils from Fungal Keratitis Patients and Healthy Cohorts in Southern India. J. Infect. Dis. 2015, 211, 130–134. [Google Scholar] [CrossRef]
- Taylor, P.R.; Roy, S.; Leal, S.M.; Sun, Y.; Howell, S.J.; Cobb, B.A.; Li, X.; Pearlman, E. Activation of Neutrophils by Autocrine IL-17A–IL-17RC Interactions during Fungal Infection Is Regulated by IL-6, IL-23, RORγt and Dectin-2. Nat. Immunol. 2014, 15, 143–151. [Google Scholar] [CrossRef]
- Miller, J.J.; Scott, I.U.; Flynn, H.W.; Smiddy, W.E.; Corey, R.P.; Miller, D. Endophthalmitis Caused by Streptococcus pneumoniae. Am. J. Ophthalmol. 2004, 138, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Mao, L.K.; Flynn, H.W.; Miller, D.; Pflugfelder, S.C. Endophthalmitis Caused by Streptococcal Species. Arch. Ophthalmol. 1992, 110, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Nouri, M.; Terada, H.; Alfonso, E.C.; Foster, C.S.; Durand, M.L.; Dohlman, C.H. Endophthalmitis After Keratoprosthesis. Arch. Ophthalmol. 2001, 119, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Soriano, F.; Pérez-Trallero, E.; Pallarés, R.; Meseguer, M.A.; Fleites, A.; Gené, A.; González, A.; Liñares, J.; Esteban, J.; Baquero, F.; et al. Streptococcus pneumoniae Endophthalmitis: A Study of 36 Cases with Special Reference to Antibiotic Resistance and Treatment Options. Clin. Microbiol. Infect. 2006, 12, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Mandelbaum, S.; Forster, R.K.; Gelender, H.; Culbertson, W. Late Onset Endophthalmitis Associated with Filtering Blebs. Ophthalmology 1985, 92, 964–972. [Google Scholar] [CrossRef]
- Katz, L.J.; Cantor, L.B.; Spaeth, G.L. Complications of Surgery in Glaucoma. Early and Late Bacterial Endophthalmitis Following Glaucoma Filtering Surgery. Ophthalmology 1985, 92, 959–963. [Google Scholar] [CrossRef]
- McCannel, C.A. Meta-analysis of Endophthalmitis After Intravitreal Injection of Anti–vascular Endothelial Growth Factor Agents. Retina 2011, 31, 654–661. [Google Scholar] [CrossRef]
- Sanders, M.E.; Norcross, E.W.; Moore, Q.C.; Onwubiko, C.; King, L.B.; Fratkin, J.; Marquart, M.E.; Marquart, M.E. A Comparison of Pneumolysin Activity and Concentration in Vitro and in Vivo in a Rabbit Endophthalmitis Model. Clin. Ophthalmol. 2008, 2, 793–800. [Google Scholar] [CrossRef][Green Version]
- Sanders, M.E.; Norcross, E.W.; Robertson, Z.M.; Moore, Q.C.; Fratkin, J.; Marquart, M.E. The Streptococcus pneumoniae Capsule Is Required for Full Virulence in Pneumococcal Endophthalmitis. Investig. Ophthalmol. Vis. Sci. 2011, 52, 865–872. [Google Scholar] [CrossRef]
- Llull, D.; López, R.; García, E. Genetic Bases and Medical Relevance of Capsular Polysaccharide Biosynthesis in Pathogenic Streptococci. Curr. Mol. Med. 2001, 1, 475–491. [Google Scholar] [CrossRef]
- Sanders, M.E.; Norcross, E.W.; Moore, Q.C.; Fratkin, J.; Thompson, H.; Marquart, M.E. Immunization with Pneumolysin Protects against Both Retinal and Global Damage Caused by Streptococcus pneumoniae Endophthalmitis. J. Ocul. Pharmacol. Ther. 2010, 26, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Ng, E.W.; Samiy, N.; Rubins, J.B.; Cousins, F.V.; Ruoff, K.L.; Baker, A.S.; D′Amico, D.J. Implication of Pneumolysin as a Virulence Factor in Streptococcus pneumoniae Endophthalmitis. Retina 1997, 17, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Ng, E.; Costa, J.R.; Samiy, N.; Ruoff, K.L.; Connolly, E.; Cousins, F.V.; D′Amico, D.J. Contribution of Pneumolysin and Autolysin to the Pathogenesis of Experimental Pneumococcal Endophthalmitis. Retina 2002, 22, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Benton, A.H.; Marquart, M.E. The Role of Pneumococcal Virulence Factors in Ocular Infectious Diseases. Interdiscip. Perspect. Infect. Dis. 2018, 2018, 1–9. [Google Scholar] [CrossRef]
- Sanders, M.E.; Taylor, S.; Tullos, N.; Norcross, E.W.; Moore, Q.C.; Thompson, H.; King, L.B.; Marquart, M.E. Passive Immunization with Pneumovax® 23 and Pneumolysin in Combination with Vancomycin for Pneumococcal Endophthalmitis. BMC Ophthalmol. 2013, 13, 1–8. [Google Scholar] [CrossRef]
- Tuomanen, E.; Liu, H.; Hengstler, B.; Zak, O.; Tomasz, A. The Induction of Meningeal Inflammation by Components of the Pneumococcal Cell Wall. J. Infect. Dis. 1985, 151, 859–868. [Google Scholar] [CrossRef]
- Mitchell, T.J.; Alexander, J.E.; Morgan, P.J.; Andrew, P.W. Molecular Analysis of Virulence Factors of Streptococcus pneumoniae. J. Appl. Microbiol. 1997, 83, 62S–71S. [Google Scholar] [CrossRef]
- Cowan, C.L.; Madden, W.M.; Hatem, G.F.; Merritt, J.C. Endogenous Bacillus cereus Panophthalmitis. Ann. Ophthalmol. 1987, 19, 65–68. [Google Scholar]
- Davey, R.T.; Tauber, W.B. Posttraumatic Endophthalmitis: The Emerging Role of Bacillus cereus Infection. Rev. Infect. Dis. 1987, 9, 110–123. [Google Scholar] [CrossRef]
- David, D.B.; Kirkby, G.R.; Noble, B.A. Bacillus cereus Endophthalmitis. Br. J. Ophthalmol. 1994, 78, 577–580. [Google Scholar] [CrossRef]
- Callegan, M.C.; Cochran, D.C.; Kane, S.T.; Ramadan, R.T.; Chodosh, J.; McLean, C.; Stroman, D.W. Virulence Factor Profiles and Antimicrobial Susceptibilities of Ocular Bacillus Isolates. Curr. Eye Res. 2006, 31, 693–702. [Google Scholar] [CrossRef] [PubMed]
- Bennouna, S.; Denkers, E.Y. Microbial Antigen Triggers Rapid Mobilization of TNF-α to the Surface of Mouse Neutrophils Transforming Them into Inducers of High-Level Dendritic Cell TNF-α Production. J. Immunol. 2005, 174, 4845–4851. [Google Scholar] [CrossRef] [PubMed]
- Ramadan, R.T.; Moyer, A.L.; Callegan, M.C. A Role for Tumor Necrosis Factor-α in Experimental Bacillus cereus Endophthalmitis Pathogenesis. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4482–4489. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Parkunan, S.M.; Randall, C.B.; Astley, R.A.; Furtado, G.C.; Lira, S.A.; Callegan, M.C. CXCL1, but Not IL-6, Significantly Impacts Intraocular Inflammation during Infection. J. Leukoc. Biol. 2016, 100, 1125–1134. [Google Scholar] [CrossRef] [PubMed]
- Novosad, B.D.; Astley, R.A.; Callegan, M.C. Role of Toll-like Receptor (TLR) 2 in Experimental Bacillus cereus Endophthalmitis. PLoS ONE 2011, 6, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Parkunan, S.M.; Randall, C.B.; Coburn, P.S.; Astley, R.A.; Staats, R.L.; Callegana, M.C. Unexpected Roles for Toll-like Receptor 4 and TRIF in Intraocular Infection with Gram-Positive Bacteria. Infect. Immun. 2015, 83, 3926–3936. [Google Scholar] [CrossRef] [PubMed]
- Coburn, P.S.; Miller, F.C.; LaGrow, A.L.; Parkunan, S.M.; Blake Randall, C.; Staats, R.L.; Callegan, M.C. TLR4 Modulates Inflammatory Gene Targets in the Retina during Bacillus cereus Endophthalmitis. BMC Ophthalmol. 2018, 18, 1–11. [Google Scholar] [CrossRef]
- Beecher, D.J.; Pulido, J.S.; Barney, N.P.; Lee Wong, A.C. Extracellular Virulence Factors in Bacillus cereus Endophthalmitis: Methods and Implication of Involvement of Hemolysin BL. Infect. Immun. 1995, 63, 632–639. [Google Scholar] [CrossRef]
- Callegan, M.C.; Jett, B.D.; Hancock, L.E.; Gilmore, M.S. Role of Hemolysin BL in the Pathogenesis of Extraintestinal Bacillus cereus Infection Assessed in an Endophthalmitis Model. Infect. Immun. 1999, 67, 3357–3366. [Google Scholar]
- Beecher, D.J.; Olsen, T.W.; Somers, E.B.; Wong, A.C.L. Evidence for Contribution of Tripartite Hemolysin BL, Phosphatidylcholine-Preferring Phospholipase C, and Collagenase to Virulence of Bacillus cereus Endophthalmitis. Infect. Immun. 2000, 68, 5269–5276. [Google Scholar] [CrossRef]
- Callegan, M.C.; Kane, S.T.; Cochran, D.C.; Gilmore, M.S.; Gominet, M.; Lereclus, D. Relationship of PlcR-Regulated Factors to Bacillus Endophthalmitis Virulence. Infect. Immun. 2003, 71, 3116–3124. [Google Scholar] [CrossRef] [PubMed]
- Callegan, M.C.; Kane, S.T.; Cochran, D.C.; Novosad, B.; Gilmore, M.S.; Gominet, M.; Lereclus, D. Bacillus Endophthalmitis: Roles of Bacterial Toxins and Motility during Infection. Investig. Ophthalmol. Vis. Sci. 2005, 46, 3233–3238. [Google Scholar] [CrossRef] [PubMed]
- Mursalin, M.H.; Coburn, P.S.; Livingston, E.; Miller, F.C.; Astley, R.; Fouet, A.; Callegan, M.C. S-Layer Impacts the Virulence of Bacillus in Endophthalmitis. Investig. Opthalmol. Vis. Sci. 2019, 60, 3727–3739. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, M.; Coburn, P.; Nallapareddy, S.; Murray, B. Enterococcal Virulence. In The Enterococci: Pathogenesis, Molecular Biology, and Antibiotic Resistance; Gilmore, C., Courvalin, D., Murray, R., Eds.; American Society of Microbiology: Washington, DC, USA, 2002; pp. 301–354. [Google Scholar] [CrossRef]
- Antibiotic Resistance Threats in the United States. Available online: https://www.cdc.gov/drugresistance/pdf/ar-threats-2013–508.pdf (accessed on 19 August 2019).
- Stevens, S.X.; Jensen, H.G.; Jett, B.D.; Gilmore, M.S. A Hemolysin-Encoding Plasmid Contributes to Bacterial Virulence in Experimental Enterococcus Faecalis Endophthalmitis. Investig. Ophthalmol. Vis. Sci. 1992, 33, 1650–1656. [Google Scholar]
- Jett, B.D.; Jensen, H.G.; Nordquist, R.E.; Gilmore, M.S. Contribution of the PAD1-Encoded Cytolysin to the Severity of Experimental Enterococcus faecalis Endophthalmitis. Infect. Immun. 1992, 60, 2445–2452. [Google Scholar]
- Engelbert, M.; Mylonakis, E.; Ausubel, F.M.; Calderwood, S.B.; Gilmore, M.S. Contribution of Gelatinase, Serine Protease, and Fsr to the Pathogenesis of Enterococcus faecalis Endophthalmitis. Infect. Immun. 2004, 72, 3628–3633. [Google Scholar] [CrossRef] [PubMed]
- Mylonakis, E.; Engelbert, M.; Qin, X.; Sifri, C.D.; Murray, B.E.; Ausubel, F.M.; Gilmore, M.S.; Calderwood, S.B. The Enterococcus faecalis FsrB Gene, a Key Component of the Fsr Quorum-Sensing System, Is Associated with Virulence in the Rabbit Endophthalmitis Model. Infect. Immun. 2002, 70, 4678–4681. [Google Scholar] [CrossRef]
- Suzuki, T.; Wada, T.; Kozai, S.; Ike, Y.; Gilmore, M.S.; Ohashi, Y. Contribution of Secreted Proteases to the Pathogenesis of Postoperative Enterococcus faecalis Endophthalmitis. J. Cataract Refract. Surg. 2008, 34, 1776–1784. [Google Scholar] [CrossRef]
- LaGrow, A.L.; Coburn, P.S.; Miller, F.C.; Land, C.; Parkunan, S.M.; Luk, B.T.; Gao, W.; Zhang, L.; Callegan, M.C. A Novel Biomimetic Nanosponge Protects the Retina from the Enterococcus faecalis Cytolysin. mSphere 2017, 2, e00335-17. [Google Scholar] [CrossRef]
- Coburn, P.S.; Miller, F.C.; LaGrow, A.L.; Land, C.; Mursalin, M.H.; Livingston, E.; Amayem, O.; Chen, Y.; Gao, W.; Zhang, L.; et al. Disarming Pore-Forming Toxins with Biomimetic Nanosponges in Intraocular Infections. mSphere 2019, 4, e00262-19. [Google Scholar] [CrossRef]
- Jackson, T.L.; Eykyn, S.J.; Graham, E.M.; Stanford, M.R. Endogenous Bacterial Endophthalmitis: A 17-Year Prospective Series and Review of 267 Reported Cases. Surv. Ophthalmol. 2003, 48, 403–423. [Google Scholar] [CrossRef]
- Jackson, T.L.; Paraskevopoulos, T.; Georgalas, I. Systematic Review of 342 Cases of Endogenous Bacterial Endophthalmitis. Surv. Ophthalmol. 2014, 59, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Podschun, R.; Ullmann, U. Klebsiella Spp. as Nosocomial Pathogens: Epidemiology, Taxonomy, Typing Methods, and Pathogenicity Factors. Clin. Microbiol. Rev. 1998, 11, 589–603. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.-L.; Ko, W.-C.; Cheng, K.-C.; Lee, H.-C.; Ke, D.-S.; Lee, C.-C.; Fung, C.-P.; Chuang, Y.-C. Association between RmpA and MagA Genes and Clinical Syndromes Caused by Klebsiella pneumoniae in Taiwan. Clin. Infect. Dis. 2006, 42, 1351–1358. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.-T.; Chuang, Y.-P.; Shun, C.-T.; Chang, S.-C.; Wang, J.-T. A Novel Virulence Gene in Klebsiella pneumoniae Strains Causing Primary Liver Abscess and Septic Metastatic Complications. J. Exp. Med. 2004, 199, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Chiu, C.-T.; Lin, D.-Y.; Liaw, Y.-F. Metastatic Septic Endophthalmitis in Pyogenic Liver Abscess. J. Clin. Gastroenterol. 1988, 10, 524–527. [Google Scholar] [CrossRef]
- Cheng, D.L.; Liu, Y.C.; Yen, M.Y.; Liu, C.Y.; Wang, R.S. Septic Metastatic Lesions of Pyogenic Liver Abscess. Their Association with Klebsiella pneumoniae Bacteremia in Diabetic Patients. Arch. Intern. Med. 1991, 151, 1557–1559. [Google Scholar] [CrossRef]
- Struve, C.; Bojer, M.; Nielsen, E.M.; Hansen, D.S.; Krogfelt, K.A. Investigation of the Putative Virulence Gene MagA in a Worldwide Collection of 495 Klebsiella Isolates: MagA Is Restricted to the Gene Cluster of Klebsiella pneumoniae Capsule Serotype K1. J. Med. Microbiol. 2005, 54, 1111–1113. [Google Scholar] [CrossRef]
- Fang, F.C.; Sandler, N.; Libby, S.J. Liver Abscess Caused by MagA+ Klebsiella pneumoniae in North America. J. Clin. Microbiol. 2005, 43, 991–992. [Google Scholar] [CrossRef]
- Fung, C.-P.; Chang, F.-Y.; Lee, S.-C.; Hu, B.-S.; Kuo, B.I.-T.; Liu, C.-Y.; Ho, M.; Siu, L.K. A Global Emerging Disease of Klebsiella pneumoniae Liver Abscess: Is Serotype K1 an Important Factor for Complicated Endophthalmitis? Gut 2002, 50, 420–424. [Google Scholar] [CrossRef]
- Chee, S.P.; Ang, C.L. Endogenous Klebsiella Endophthalmitis—A Case Series. Ann. Acad. Med. Singap. 1995, 24, 473–478. [Google Scholar] [PubMed]
- Ang, L.P.-K.; Lee, H.-M.; Eong, K.-G.A.; Yap, E.-Y.; Lim, A.T.-H. Endogenous Klebsiella Endophthalmitis. Eye 2000, 14, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Astley, R.A.; Coburn, P.S.; Madhumathi Parkunan, S.; Callegan, M.C. Modeling Intraocular Bacterial Infections. Prog. Retin. Eye Res. 2016, 54, 30–48. [Google Scholar] [CrossRef] [PubMed]
- Wiskur, B.J.; Hunt, J.J.; Callegan, M.C. Hypermucoviscosity as a Virulence Factor in Experimental Klebsiella pneumoniae Endophthalmitis. Investig. Ophthalmol. Vis. Sci. 2008, 49, 4931–4938. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hunt, J.J.; Wang, J.-T.; Callegan, M.C. Contribution of Mucoviscosity-Associated Gene A (MagA) to Virulence in Experimental Klebsiella pneumoniae Endophthalmitis. Investig. Ophthalmol. Vis. Sci. 2011, 6340, 233–243. [Google Scholar]
- Hunt, J.J.; Astley, R.; Wheatley, N.; Wang, J.; Michelle, C. TLR4 Contributes to the Host Response to Klebsiella Intraocular Infection. Curr. Eye Res. 2015, 39, 790–802. [Google Scholar] [CrossRef]
- Eifrig, C.W.; Scott, I.U.; Flynn, H.W.; Miller, D. Endophthalmitis Caused by Pseudomonas aeruginosa. Ophthalmology 2003, 110, 1714–1717. [Google Scholar] [CrossRef]
- Pinna, A.; Usai, D.; Sechi, L.A.; Zanetti, S.; Jesudasan, N.C.A.; Thomas, P.A.; Kaliamurthy, J. An Outbreak of Post-Cataract Surgery Endophthalmitis Caused by Pseudomonas aeruginosa. Ophthalmology 2009, 116, 2321–2326. [Google Scholar] [CrossRef]
- Chen, K.-J.; Sun, M.-H.; Lai, C.-C.; Wu, W.-C.; Chen, T.-L.; Kuo, Y.-H.; Chao, A.-N.; Hwang, Y.-S.; Chen, Y.-P.; Wang, N.-K.; et al. Endophthalmitis Caused by Pseudomonas aeruginosa in Taiwan. Retina 2011, 31, 1193–1198. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bacterial Pathogen | Bacterial Components | PMN Response | Inflammatory Pathways | Cytokines and Chemokines |
|---|---|---|---|---|
| Conjunctivitis | ||||
| S. aureus | PNAG [37] | Infiltration [25,35,36,37] | ||
| S. pneumoniae | Polysaccharide capsule [47,50] | Infiltration [47] | ||
| C. trachomatis | Infiltration [26,55,56] Damage to epithelia [26] | TGF-β and IL-5 [26] | ||
| Keratitis | ||||
| P. aeruginosa | T3SS [80] ExoS and ExoT [72,81] Flagellum [82] | Infiltration [63,76,77,78,79,80,82,84] NETosis [80] Apoptosis [81] | MyD88 [79] NF-κB [82] TLR5 [82] IκB-α [82] SP-D [97,98] | IL-6, IL-8, and 1β [82] CCL2 and CCL3 [84] |
| S. aureus | Peptidoglycan [93] Cysteine proteases [99] α-toxin [88,91,100,101] β-toxin [91] γ-toxin [104] Protein A [88,105] | Infiltration [88,91,94,95,96,104] Phagocytosis [99] | TLR2 [93,94] MyD88 [94] MAPKs [93] NF-κB [93] ICAM-1 [95] SP-D [99] | TNFα [93] IL-6, IL-8, TNF-α [93,96] CXCR2 [95,96] |
| S. pneumoniae | Pneumolysin [11,115,116,117,118,119,120] | Infiltration [116,117] Corneal damage [115] | NLRP3/ASC [120] Caspase-1 [120] | IL-1β [120] |
| Endophthalmitis | ||||
| S. aureus | Lipotechoic acids [171,172] α-toxin [148] β-toxin [148] γ-toxin [148,173] Peptidoglycan [172] Protein A [172] TSST-1 [172] PVL [173] | Infiltration [161,172,173,178] | FasL [9] Complement [9] | IL-6 and 1β [172] TNFα [172] KC [172] MIP2 [172] |
| S. pneumoniae | Polysaccharide capsule [193,199] Pneumolysin [192,196,197,199] Autolysin [197] | Infiltration [192,193] | ||
| Bacillus | Hemolysin BL [212,213] PC-PLC [162,214] PI-PLC [162] S-layer [217] | Infiltration [10,148,207] | TLR2 [209] TLR4 [210] MyD88 [210] TRIF [210] NOD2, NLRP3 [239] | TNFα [10,207] IL-6 and IL-1β [207,208,210] MIP-1α [207] CXCL1 [208,210] |
| E. faecalis | Gelatinase [222,224] Cytolysin [220,221,225] Serine protease [222,224] | Infiltration [221,222,223,224] | ||
| K. pneumoniae | HMV phenotype [230,236,240,241,242] | Infiltration [238,239,241] | TLR4 [242] | CXCL1, TNF, MIP-1 [242] |
| P. aeruginosa | Infiltration [8] | |||
| E. coli | Infiltration [239] | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Livingston, E.T.; Mursalin, M.H.; Callegan, M.C. A Pyrrhic Victory: The PMN Response to Ocular Bacterial Infections. Microorganisms 2019, 7, 537. https://doi.org/10.3390/microorganisms7110537
Livingston ET, Mursalin MH, Callegan MC. A Pyrrhic Victory: The PMN Response to Ocular Bacterial Infections. Microorganisms. 2019; 7(11):537. https://doi.org/10.3390/microorganisms7110537
Chicago/Turabian StyleLivingston, Erin T., Md Huzzatul Mursalin, and Michelle C. Callegan. 2019. "A Pyrrhic Victory: The PMN Response to Ocular Bacterial Infections" Microorganisms 7, no. 11: 537. https://doi.org/10.3390/microorganisms7110537
APA StyleLivingston, E. T., Mursalin, M. H., & Callegan, M. C. (2019). A Pyrrhic Victory: The PMN Response to Ocular Bacterial Infections. Microorganisms, 7(11), 537. https://doi.org/10.3390/microorganisms7110537