Socioeconomic Status and the Gut Microbiome: A TwinsUK Cohort Study

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Data
Ethics Approval and Consent to Participate
2.2. Microbiota Sample Processing
2.3. Measures
2.3.1. Socioeconomic Status (SES)
2.3.2. Covariates
2.4. Microbiome analysis
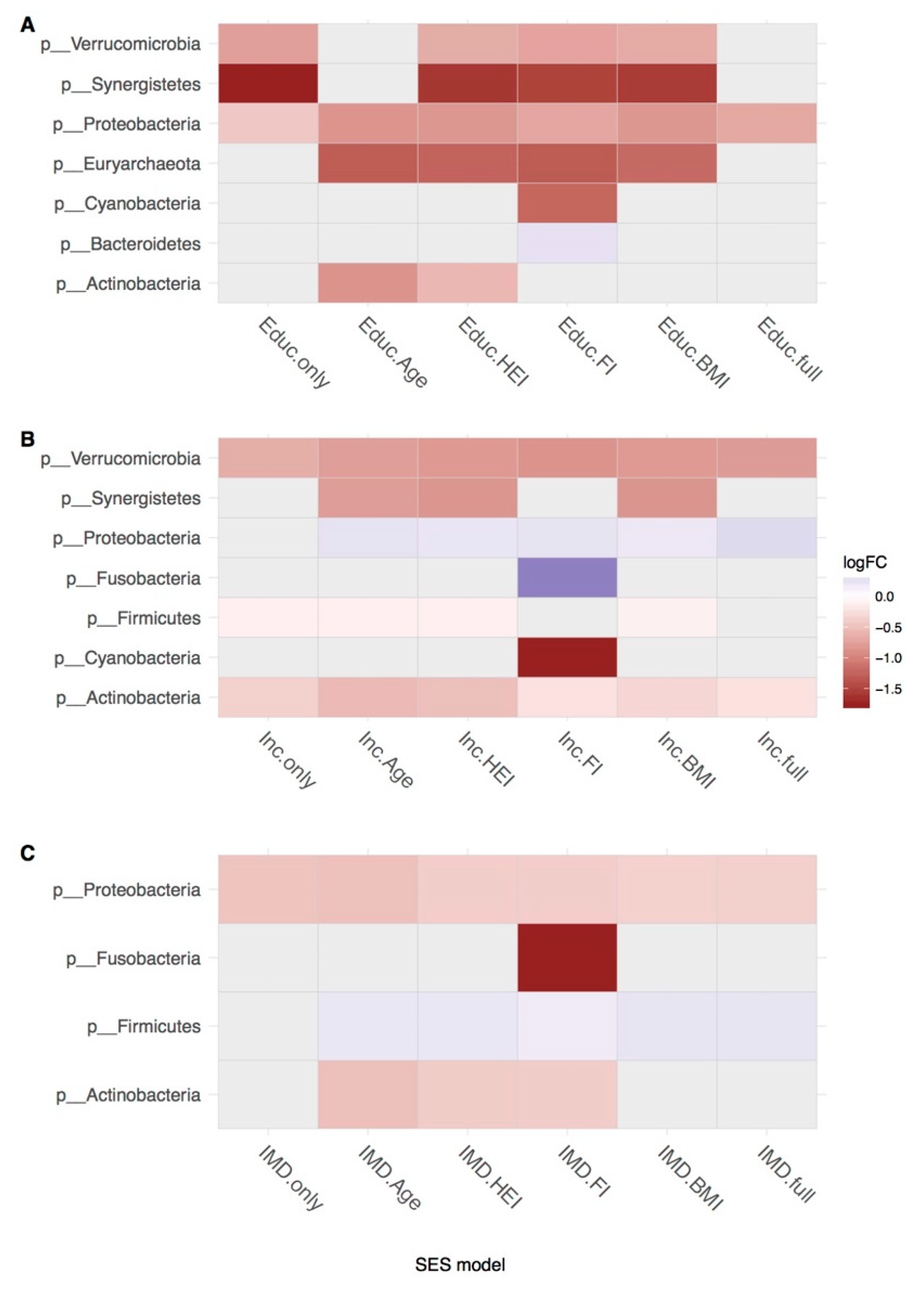
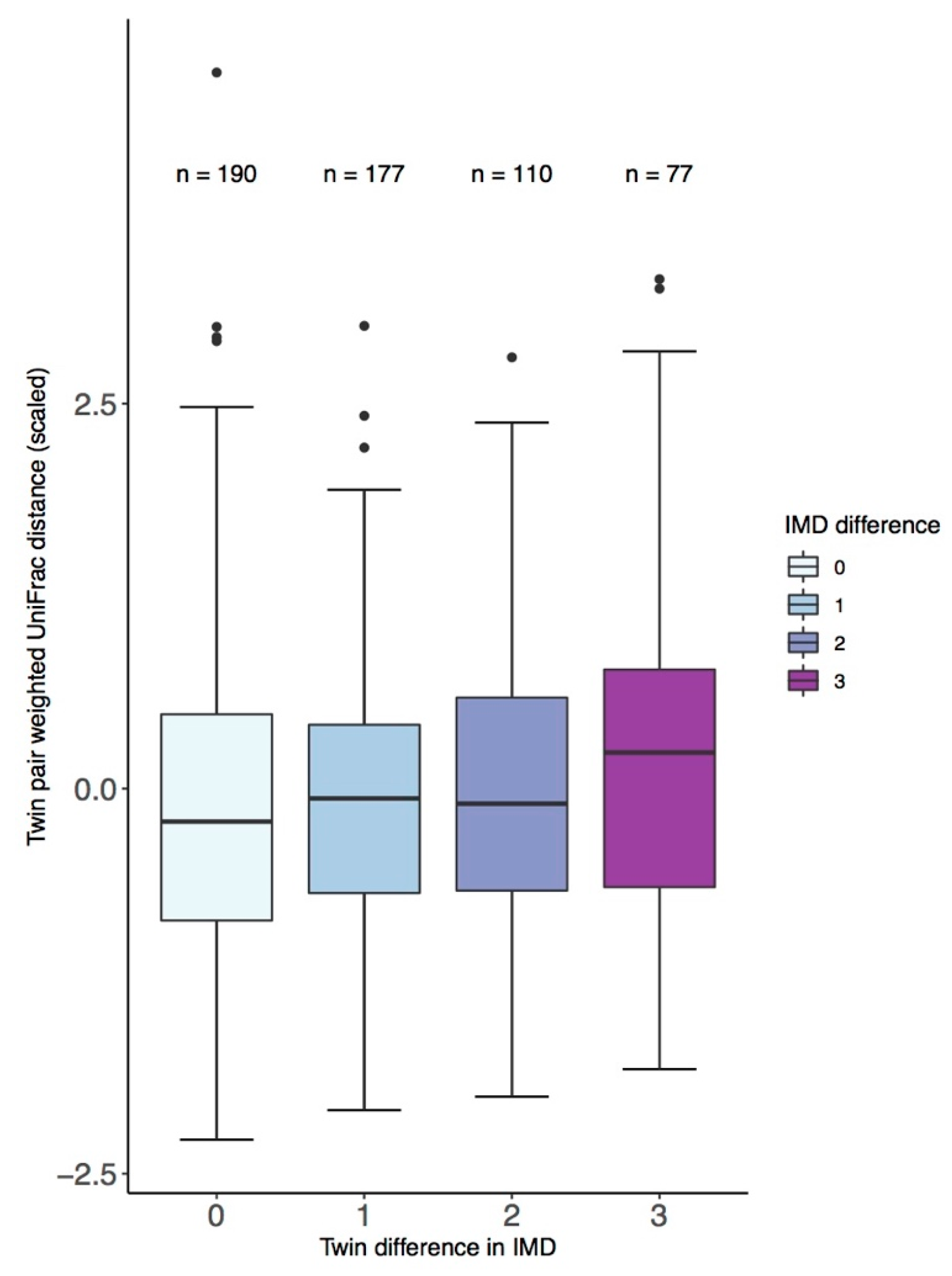
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Foster, J.A.; McVey Neufeld, K.-A. Gut-brain axis: How the microbiome influences anxiety and depression. Trends Neurosci. 2013, 36, 305–312. [Google Scholar] [CrossRef] [PubMed]
- Shreiner, A.B.; Kao, J.Y.; Young, V.B. The gut microbiome in health and in disease. Curr. Opin. Gastroenterol. 2015, 31, 69. [Google Scholar] [CrossRef] [PubMed]
- Jackson, M.A.; Jeffery, I.B.; Beaumont, M.; Bell, J.T.; Clark, A.G.; Ley, R.E.; O’Toole, P.W.; Spector, T.D.; Steves, C.J. Signatures of early frailty in the gut microbiota. Genome Med. 2016, 8, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothschild, D.; Weissbrod, O.; Barkan, E.; Kurilshikov, A.; Korem, T.; Zeevi, D.; Costea, P.I.; Godneva, A.; Kalka, I.N.; Bar, N. Environment dominates over host genetics in shaping human gut microbiota. Nature 2018, 555, 210. [Google Scholar] [CrossRef] [PubMed]
- Llewellyn, S.R.; Britton, G.J.; Contijoch, E.J.; Vennaro, O.H.; Mortha, A.; Colombel, J.F.; Grinspan, A.; Clemente, J.C.; Merad, M.; Faith, J.J. Interactions Between Diet and the Intestinal Microbiota Alter Intestinal Permeability and Colitis Severity in Mice. Gastroenterology 2018, 154, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Leng, J.; Peruluswami, P.; Bari, S.; Gaur, S.; Radparvar, F.; Parvez, F.; Chen, Y.; Flores, C.; Gany, F. South Asian Health: Inflammation, Infection, Exposure, and the Human Microbiome. J. Immigrant Minority Health 2017, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Herd, P.; Palloni, A.; Rey, F.; Dowd, J.B. Social and population health science approaches to understand the human microbiome. Nat. Hum. Behav. 2018, 2, 808–815. [Google Scholar] [CrossRef]
- Dowd, J.; Renson, A. “Under the Skin” and into the Gut. Curr. Epidemiol. Rep. 2018, 5, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Dennis, C.L. Breastfeeding initiation and duration: A 1990–2000 literature review. J. Obstet. Gynecol. Neonatal Nurs. 2002, 31, 12–32. [Google Scholar] [CrossRef]
- Dominguez-Bello, M.G.; Costello, E.K.; Contreras, M.; Magris, M.; Hidalgo, G.; Fierer, N.; Knight, R. Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. USA 2010, 107, 11971–11975. [Google Scholar] [CrossRef] [Green Version]
- Mueller, N.T.; Bakacs, E.; Combellick, J.; Grigoryan, Z.; Dominguez-Bello, M.G. The infant microbiome development: Mom matters. Trends Mol. Med. 2015, 21, 109–117. [Google Scholar] [CrossRef]
- Moeller, A.H.; Foerster, S.; Wilson, M.L.; Pusey, A.E.; Hahn, B.H.; Ochman, H. Social behavior shapes the chimpanzee pan-microbiome. Sci. Adv. 2016, 2, e1500997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amato, K.R.; Van Belle, S.; Di Fiore, A.; Estrada, A.; Stumpf, R.; White, B.; Nelson, K.E.; Knight, R.; Leigh, S.R. Patterns in Gut Microbiota Similarity Associated with Degree of Sociality among Sex Classes of a Neotropical Primate. Microb. Ecol. 2017, 74, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Amaral, W.Z.; Lubach, G.R.; Proctor, A.; Lyte, M.; Phillips, G.J.; Coe, C.L. Social Influences on Prevotella and the Gut Microbiome of Young Monkeys. Psychosom. Med. 2017, 79, 888–897. [Google Scholar] [CrossRef] [PubMed]
- Tung, J.; Barreiro, L.B.; Burns, M.B.; Grenier, J.-C.; Lynch, J.; Grieneisen, L.E.; Altmann, J.; Alberts, S.C.; Blekhman, R.; Archie, E.A. Social networks predict gut microbiome composition in wild baboons. Elife 2015, 4, e05224. [Google Scholar] [CrossRef]
- Lax, S.; Smith, D.P.; Hampton-Marcell, J.; Owens, S.M.; Handley, K.M.; Scott, N.M.; Gibbons, S.M.; Larsen, P.; Shogan, B.D.; Weiss, S.; et al. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science 2014, 345, 1048–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Claesson, M.J.; Jeffery, I.B.; Conde, S.; Power, S.E.; O’Connor, E.M.; Cusack, S.; Harris, H.M.B.; Coakley, M.; Lakshminarayanan, B.; O’Sullivan, O.; et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 2012, 488, 178. [Google Scholar] [CrossRef]
- Bailey, M.T. Psychological stress, immunity, and the effects on indigenous microflora. In Microbial Endocrinology: Interkingdom Signaling in Infectious Disease and Health; Springer: New York, NY, USA, 2016; pp. 225–246. [Google Scholar]
- Grzywacz, J.G.; Almeida, D.M.; Neupert, S.D.; Ettner, S.L. Socioeconomic status and health: A micro-level analysis of exposure and vulnerability to daily stressors. J. Health Soc. Behav. 2004, 45, 1–16. [Google Scholar] [CrossRef]
- Bailey, M. Influence of Stressor-Induced Nervous System Activation on the Intestinal Microbiota and the Importance for Immunomodulation. In Microbial Endocrinology: The Microbiota-Gut-Brain Axis in Health and Disease; Lyte, M., Cryan, J.F., Eds.; Springer: New York, NY, USA, 2014; pp. 255–276. [Google Scholar]
- Kingston, D.; Sword, W.; Krueger, P.; Hanna, S.; Markle-Reid, M. Life Course Pathways to Prenatal Maternal Stress. J. Obst. Gyn. Neonatal 2012, 41, 609–626. [Google Scholar] [CrossRef]
- Bailey, M.T.; Dowd, S.E.; Galley, J.D.; Hufnagle, A.R.; Allen, R.G.; Lyte, M. Exposure to a social stressor alters the structure of the intestinal microbiota: Implications for stressor-induced immunomodulation. Brain Behav. Immun. 2011, 25, 397–407. [Google Scholar] [CrossRef] [Green Version]
- O’Mahony, S.M.; Marchesi, J.R.; Scully, P.; Codling, C.; Ceolho, A.-M.; Quigley, E.M.M.; Cryan, J.F.; Dinan, T.G. Early Life Stress Alters Behavior, Immunity, and Microbiota in Rats: Implications for Irritable Bowel Syndrome and Psychiatric Illnesses. Biol. Psychiatry 2009, 65, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Jašarević, E.; Howerton, C.L.; Howard, C.D.; Bale, T.L. Alterations in the Vaginal Microbiome by Maternal Stress Are Associated With Metabolic Reprogramming of the Offspring Gut and Brain. Endocrinology 2015, 156, 3265–3276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, M.T.; Coe, C.L. Maternal separation disrupts the integrity of the intestinal microflora in infant rhesus monkeys. Dev. Psychobiol. 1999, 35, 146–155. [Google Scholar] [CrossRef]
- Stilling, R.M.; Moloney, G.M.; Ryan, F.J.; Hoban, A.E.; Bastiaanssen, T.F.S.; Shanahan, F.; Clarke, G.; Claesson, M.J.; Dinan, T.G.; Cryan, J.F. Social interaction-induced activation of RNA splicing in the amygdala of microbiome-deficient mice. Elife 2018, 7, e33070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, G.E.; Engen, P.A.; Gillevet, P.M.; Shaikh, M.; Sikaroodi, M.; Forsyth, C.B.; Mutlu, E.; Keshavarzian, A. Lower Neighborhood Socioeconomic Status Associated with Reduced Diversity of the Colonic Microbiota in Healthy Adults. PLoS ONE 2016, 11, e0148952. [Google Scholar] [CrossRef] [PubMed]
- Moayyeri, A.; Hammond, C.J.; Valdes, A.M.; Spector, T.D. Cohort profile: TwinsUK and healthy ageing twin study. Int. J. Epidemiol. 2013, 42, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, J.K.; Davenport, E.R.; Beaumont, M.; Jackson, M.A.; Knight, R.; Ober, C.; Spector, T.D.; Bell, J.T.; Clark, A.G.; Ley, R.E. Genetic Determinants of the Gut Microbiome in UK Twins. Cell Host Microbe 2016, 19, 731–743. [Google Scholar] [CrossRef] [PubMed]
- The English Indices of Deprivation. 2015. Available online: https://www.gov.uk/government/uploads/system/uploads/attachment_data/file/465791/English_Indices_of_Deprivation_2015_-_Statistical_Release.pdf (accessed on 30 January 2018).
- RStudio. RStudio: Integrated Development for R; RStudio Inc.: Boston, MA, USA, 2015. [Google Scholar]
- Team, QGIS Developement. QGIS Geographic Information System. Open Source Geospatial Foundation Project; Team, Open Source Geospatial Foundation: Chigaco, IL, USA, 2016. [Google Scholar]
- Searle, S.D.; Mitnitski, A.; Gahbauer, E.A.; Gill, T.M.; Rockwood, K. A standard procedure for creating a frailty index. BMC Geriatr. 2008, 8, 1–10. [Google Scholar] [CrossRef]
- Bowyer, R.C.E.; Jackson, M.A.; Pallister, T.; Skinner, J.; Spector, T.D.; Welch, A.A.; Steves, C.J. Use of dietary indices to control for diet in human gut microbiota studies. Microbiome 2018, 6, 77. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S.; Kindt, R.; Legendre, P.; O’Hara, R.B. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Bates, D.; Mächler, M.; Bolker, B.; Walker, S. Fitting Linear Mixed-Effects Models Using lme4. J. Stat. Softw. 2015, 67, 1–48. [Google Scholar] [CrossRef]
- Kuznetsova, A.; Brockhoff, P.B.; Christensen, R.H.B. LmerTest Package: Tests in Linear Mixed Effects Models. J. Stat. Softw. 2017, 82, 26. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y. Waste Not, Want Not: Why Rarefying Microbiome Data Is Inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Kindt, R.; Legendre, P.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; Stevens, M.H.H.; Helene, W. Vegan: Community Ecology Package. Available online: http://cran.r/project.org/package=vegan (accessed on 31 October 2018).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [PubMed]
- Pasco, J.A.; Holloway, K.L.; Dobbins, A.G.; Kotowicz, M.A.; Williams, L.J.; Brennan, S.L. Body mass index and measures of body fat for defining obesity and underweight: A cross-sectional, population-based study. BMC Obes. 2014, 1, 9. [Google Scholar] [CrossRef] [PubMed]
- Rockwood, K.; Andrew, M.; Mitnitski, A. A comparison of two approaches to measuring frailty in elderly people. J. Gerontol. Biol. 2007, 62, 738–743. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Vineis, P.; Kelly-Irving, M.; Rappaport, S.; Stringhini, S. The biological embedding of social differences in ageing trajectories. J. Epidemiol. Commun. Health 2016, 70, 111–113. [Google Scholar] [CrossRef]
- Adler, N.; Marmot, M.; McEwen, B.; Stewart, J. Socio-economic status and health in industrialised nations: Social, psychological and biological pathways. Ann. N.Y. Acad. Sci. 1999, 896, 1–500. [Google Scholar]
- Walter, J.; Ley, R. The Human Gut Microbiome: Ecology and Recent Evolutionary Changes. Annu. Rev. Microbiol. 2011, 65, 411–429. [Google Scholar] [CrossRef]
- Azad, M.B.; Konya, T.; Maughan, H.; Guttman, D.S.; Field, C.J.; Sears, M.R.; Becker, A.B.; Scott, J.A.; Kozyrskyj, A.L.; Investigators, C.S. Infant gut microbiota and the hygiene hypothesis of allergic disease: Impact of household pets and siblings on microbiota composition and diversity. Allergy Asthma Clin. Immunol. 2013, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Flies, E.J.; Skelly, C.; Negi, S.S.; Prabhakaran, P.; Liu, Q.Y.; Liu, K.K.; Goldizen, F.C.; Lease, C.; Weinstein, P. Biodiverse green spaces: A prescription for global urban health. Front. Ecol. Environ. 2017, 15, 510–516. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steves, C.J.; Jackson, S.H.D.; Spector, T.D. Cognitive Change in Older Women Using a Computerised Battery: A Longitudinal Quantitative Genetic Twin Study. Behav. Genet. 2013, 43, 468–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schloss, P.D.; Westcott, S.L. Assessing and Improving Methods Used in Operational Taxonomic Unit-Based Approaches for 16S rRNA Gene Sequence Analysis. Appl. Environ. Microbiol. 2011, 77, 3219–3226. [Google Scholar] [CrossRef] [Green Version]
- Quévrain, E.; Maubert, M.A.; Michon, C.; Chain, F.; Marquant, R.; Tailhades, J.; Miquel, S.; Carlier, L.; Bermúdez-Humarán, L.G.; Pigneur, B.; et al. Identification of an anti-inflammatory protein from Faecalibacterium prausnitzii, a commensal bacterium deficient in Crohn’s disease. Gut 2016, 65, 415–425. [Google Scholar] [CrossRef]
- Zhou, K.Q. Strategies to promote abundance of Akkermansia muciniphila, an emerging probiotics in the gut, evidence from dietary intervention studies. J. Funct. Food 2017, 33, 194–201. [Google Scholar] [CrossRef]
- Pasolli, E.; Truong, D.T.; Malik, F.; Waldron, L.; Segata, N. Machine Learning Meta-analysis of Large Metagenomic Datasets: Tools and Biological Insights. PLoS Computat. Biol. 2016, 12, e1004977. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Leon, F.; John, B. The Importance of Cognitive Development in Middle Childhood for Adulthood Socioeconomic Status, Mental Health, and Problem Behavior. Child Dev. 2004, 75, 1329–1339. [Google Scholar]
- Laforest-Lapointe, I.; Arrieta, M.C. Patterns of early-Life Gut Microbial Colonization during Human immune Development: An ecological Perspective. Front. Immunol. 2017, 8, 788. [Google Scholar] [CrossRef] [PubMed]
- Lukovac, S.; Belzer, C.; Pellis, L.; Keijser, B.J.; de Vos, W.M.; Montijn, R.C.; Roeselers, G. Differential Modulation by Akkermansia muciniphila and Faecalibacterium prausnitzii of Host Peripheral Lipid Metabolism and Histone Acetylation in Mouse Gut Organoids. mBio 2014, 5, e01438-14. [Google Scholar] [CrossRef] [PubMed]
- Whisner, C.M.; Martin, B.R.; Nakatsu, C.H.; McCabe, G.P.; McCabe, L.D.; Peacock, M.; Weaver, C.M. Soluble maize fibre affects short-term calcium absorption in adolescent boys and girls: A randomised controlled trial using dual stable isotopic tracers. Br. J. Nutr. 2014, 112, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Arrazuria, R.; Elguezabal, N.; Juste, R.A.; Derakhshani, H.; Khafipour, E. Mycobacterium avium Subspecies paratuberculosis Infection Modifies Gut Microbiota under Different Dietary Conditions in a Rabbit Model. Front. Microbiol. 2016, 7, 446. [Google Scholar] [CrossRef] [PubMed]
- Belzer, C.; Chia, L.W.; Aalvink, S.; Chamlagain, B.; Piironen, V.; Knol, J.; de Vos, W.M. Microbial Metabolic Networks at the Mucus Layer Lead to Diet-Independent Butyrate and Vitamin B12 Production by Intestinal Symbionts. mBio 2017, 8, e00770-17. [Google Scholar] [CrossRef] [PubMed]
- Strati, F.; Cavalieri, D.; Albanese, D.; De Felice, C.; Donati, C.; Hayek, J.; Jousson, O.; Leoncini, S.; Pindo, M.; Renzi, D.; et al. Altered gut microbiota in Rett syndrome. Microbiome 2016, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Schwalm, N.D.; Groisman, E.A. Navigating the Gut Buffet: Control of Polysaccharide Utilization in Bacteroides spp. Trends Microbiol. 2017, 25, 1005–1015. [Google Scholar] [CrossRef]
- Tuncil, Y.E.; Xiao, Y.; Porter, N.T.; Reuhs, B.L.; Martens, E.C.; Hamaker, B.R. Reciprocal Prioritization to Dietary Glycans by Gut Bacteria in a Competitive Environment Promotes Stable Coexistence. mBio 2017, 8, e01068-17. [Google Scholar] [CrossRef]
- Frank, D.N.; St. Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [Green Version]
- Torres, J.; Bao, X.; Goel, A.; Colombel, J.F.; Pekow, J.; Jabri, B.; Williams, K.M.; Castillo, A.; Odin, J.A.; Meckel, K.; et al. The features of mucosa-associated microbiota in primary sclerosing cholangitis. Aliment. Pharmacol. Therapeut. 2016, 43, 790–801. [Google Scholar] [CrossRef] [Green Version]
- Hopfner, F.; Künstner, A.; Müller, S.H.; Künzel, S.; Zeuner, K.E.; Margraf, N.G.; Deuschl, G.; Baines, J.F.; Kuhlenbäumer, G. Gut microbiota in Parkinson disease in a northern German cohort. Brain Res. 2017, 1667, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Montassier, E.; Al-Ghalith, G.A.; Ward, T.; Corvec, S.; Gastinne, T.; Potel, G.; Moreau, P.; Cochetiere, M.F.d.l.; Batard, E.; Knights, D. Pretreatment gut microbiome predicts chemotherapy-related bloodstream infection. Genome Med. 2016, 8, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woting, A.; Pfeiffer, N.; Loh, G.; Klaus, S.; Blaut, M. Clostridium ramosum Promotes High-Fat Diet-Induced Obesity in Gnotobiotic Mouse Models. mBio 2014, 5, e01530-14. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Liu, F.; Ling, Z.; Tong, X.; Xiang, C. Human Intestinal Lumen and Mucosa-Associated Microbiota in Patients with Colorectal Cancer. PLoS ONE 2012, 7, e39743. [Google Scholar] [CrossRef] [PubMed]
- Kasai, C.; Sugimoto, K.; Moritani, I.; Tanaka, J.; Oya, Y.; Inoue, H.; Tameda, M.; Shiraki, K.; Ito, M.; Takei, Y.; et al. Comparison of the gut microbiota composition between obese and non-obese individuals in a Japanese population, as analyzed by terminal restriction fragment length polymorphism and next-generation sequencing. Bmc Gastroenterol. 2015, 15, 100. [Google Scholar] [CrossRef]
- Biagi, E.; Franceschi, C.; Rampelli, S.; Severgnini, M.; Ostan, R.; Turroni, S.; Consolandi, C.; Quercia, S.; Scurti, M.; Monti, D.; et al. Gut Microbiota and Extreme Longevity. Curr. Biol. 2016, 26, 1480–1485. [Google Scholar] [CrossRef]
- Stenman, L.K.; Burcelin, R.; Lahtinen, S. Establishing a causal link between gut microbes, body weight gain and glucose metabolism in humans—Towards treatment with probiotics. Benef. Microbes 2015, 7, 11–22. [Google Scholar] [CrossRef]
- Oki, K.; Toyama, M.; Banno, T.; Chonan, O.; Benno, Y.; Watanabe, K. Comprehensive analysis of the fecal microbiota of healthy Japanese adults reveals a new bacterial lineage associated with a phenotype characterized by a high frequency of bowel movements and a lean body type. BMC Microbiol. 2016, 16, 284. [Google Scholar] [CrossRef]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vázquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef]
- Ahn, J.; Sinha, R.; Pei, Z.; Dominianni, C.; Wu, J.; Shi, J.; Goedert, J.J.; Hayes, R.B.; Yang, L. Human Gut Microbiome and Risk for Colorectal Cancer. J. Natl. Cancer Inst. 2013, 105, 1907–1911. [Google Scholar] [CrossRef] [Green Version]
- Skuja, V.; Derovs, A.; Pekarska, K.; Rudzite, D.; Lavrinovica, E.; Piekuse, L.; Kempa, I.; Straume, Z.; Eglite, J.; Lejnieks, A.; et al. Gut colonization with extended-spectrum β-lactamase-producing: An interim analysis. Eur. J. Gastroenterol. Hepatol. 2018, 30, 92–100. [Google Scholar] [CrossRef]
- Alcántar-Curiel, M.D.; Girón, J.A. Klebsiella pneumoniae and the pyogenic liver abscess: Implications and association of the presence of rpmA genes and expression of hypermucoviscosity. Virulence 2015, 6, 407–409. [Google Scholar] [CrossRef] [PubMed]
- Pallister, T.; Jackson, M.A.; Martin, T.C.; Glastonbury, C.A.; Jennings, A.; Beaumont, M.; Mohney, R.P.; Small, K.S.; MacGregor, A.; Steves, C.J.; et al. Untangling the relationship between diet and visceral fat mass through blood metabolomics and gut microbiome profiling. Int. J. Obes. 2017, 41, 1106–1113. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Bäckhed, F.; Fulton, L.; Gordon, J.I. Diet-Induced Obesity Is Linked to Marked but Reversible Alterations in the Mouse Distal Gut Microbiome. Cell Host Microbe 2008, 3, 213–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilg, H.; Moschen, A.R.; Kaser, A. Obesity and the Microbiota. Gastroenterology 2009, 136, 1476–1483. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.T.; Duncan, S.H.; Stams, A.J.M.; Dijl, J.M.v.; Flint, H.J.; Harmsen, H.J.M. The gut anaerobe. ISME J. 2012, 6, 1578. [Google Scholar] [CrossRef]
- Louis, P.; Flint, H.J. Diversity, metabolism and microbial ecology of butyrate-producing bacteria from the human large intestine. FEMS Microbiol. Lett. 2009, 294, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golombos, D.M.; Ayangbesan, A.; O’Malley, P.; Lewicki, P.; Barlow, L.; Barbieri, C.E.; Chan, C.; DuLong, C.; Abu-Ali, G.; Huttenhower, C.; et al. The Role of Gut Microbiome in the Pathogenesis of Prostate Cancer: A Prospective, Pilot Study. Urology 2018, 111, 122–128. [Google Scholar] [CrossRef]
- Reeves, A.E.; Koenigsknecht, M.J.; Bergin, I.L.; Young, V.B. Suppression of Clostridium difficile in the Gastrointestinal Tracts of Germfree Mice Inoculated with a Murine Isolate from the Family Lachnospiraceae. Infect. Immun. 2012, 80, 3786–3794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kameyama, K.; Itoh, K. Intestinal Colonization by a Lachnospiraceae Bacterium Contributes to the Development of Diabetes in Obese Mice. Microbes Environ. 2014, 29, 427–430. [Google Scholar] [CrossRef] [Green Version]
- Ordiz, M.I.; May, T.D.; Mihindukulasuriya, K.; Martin, J.; Crowley, J.; Tarr, P.I.; Ryan, K.; Mortimer, E.; Gopalsamy, G.; Maleta, K.; et al. The effect of dietary resistant starch type 2 on the microbiota and markers of gut inflammation in rural Malawi children. Microbiome 2015, 3, 37. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.-L.; Inserra, A.; Lewis, M.D.; Mastronardi, C.A.; Leong, L.; Choo, J.; Kentish, S.; Xie, P.; Morrison, M.; Wesselingh, S.L.; et al. Inflammasome signaling affects anxiety- and depressive-like behavior and gut microbiome composition. Mol. Psychiatry 2016, 21, 797. [Google Scholar] [CrossRef] [PubMed]
- Furet, J.-P.; Kong, L.-C.; Tap, J.; Poitou, C.; Basdevant, A.; Bouillot, J.-L.; Mariat, D.; Corthier, G.; Doré, J.; Henegar, C.; et al. Differential adaptation of human gut microbiota to bariatric surgery-induced weight loss: Links with metabolic and low-grade inflammation markers. Diabetes 2010, 59, 3049–3057. [Google Scholar] [CrossRef] [PubMed]
- Scher, J.U.; Sczesnak, A.; Longman, R.S.; Segata, N.; Ubeda, C.; Bielski, C.; Rostron, T.; Cerundolo, V.; Pamer, E.G.; Abramson, S.B.; et al. Expansion of intestinal Prevotella copri correlates with enhanced susceptibility to arthritis. eLife 2013, 2, e01202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasai, C.; Sugimoto, K.; Moritani, I.; Tanaka, J.; Oya, Y.; Inoue, H.; Tameda, M.; Shiraki, K.; Ito, M.; Takei, Y.; et al. Comparison of human gut microbiota in control subjects and patients with colorectal carcinoma in adenoma: Terminal restriction fragment length polymorphism and next-generation sequencing analyses. Oncol. Rep. 2016, 35, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Kuehn, L.A.; Bono, J.L.; Berry, E.D.; Kalchayanand, N.; Freetly, H.C.; Benson, A.K.; Wells, J.E. The impact of the bovine faecal microbiome on Escherichia coli O157:H7 prevalence and enumeration in naturally infected cattle. J. Appl. Microbiol. 2017, 123, 1027–1042. [Google Scholar] [CrossRef] [PubMed]
- Dinh, D.M.; Volpe, G.E.; Duffalo, C.; Bhalchandra, S.; Tai, A.K.; Kane, A.V.; Wanke, C.A.; Ward, H.D. Intestinal Microbiota, Microbial Translocation, and Systemic Inflammation in Chronic HIV Infection. J. Infect. Dis. 2015, 211, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Alard, J.; Lehrter, V.; Rhimi, M.; Mangin, I.; Peucelle, V.; Abraham, A.L.; Mariadassou, M.; Maguin, E.; Waligora-Dupriet, A.-J.; Pot, B.; et al. Beneficial metabolic effects of selected probiotics on diet-induced obesity and insulin resistance in mice are associated with improvement of dysbiotic gut microbiota. Environ. Microbiol. 2018, 18, 1484–1497. [Google Scholar] [CrossRef]
- Tap, J.; Mondot, S.; Levenez, F.; Pelletier, E.; Caron, C.; Furet, J.-P.; Ugarte, E.; Muñoz-Tamayo, R.; Paslier, D.L.E.; Nalin, R.; et al. Towards the human intestinal microbiota phylogenetic core. Environ. Microbiol. 2009, 11, 2574–2584. [Google Scholar] [CrossRef]
- Walker, A.W.; Ince, J.; Duncan, S.H.; Webster, L.M.; Holtrop, G.; Ze, X.; Brown, D.; Stares, M.D.; Scott, P.; Bergerat, A.; et al. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. 2011, 5, 220–230. [Google Scholar] [CrossRef]
- Willing, B.P.; Dicksved, J.; Halfvarson, J.; Andersson, A.F.; Lucio, M.; Zheng, Z.; Järnerot, G.; Tysk, C.; Jansson, J.K.; Engstrand, L. A Pyrosequencing Study in Twins Shows That Gastrointestinal Microbial Profiles Vary With Inflammatory Bowel Disease Phenotypes. Gastroenterology 2010, 139, 1844–1854. [Google Scholar] [CrossRef] [PubMed]
- Crost, E.H.; Tailford, L.E.; Monestier, M.; Swarbreck, D.; Henrissat, B.; Crossman, L.C.; Juge, N. The mucin-degradation strategy of Ruminococcus gnavus: The importance of intramolecular trans-sialidases. Gut Microbes 2016, 7, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zeng, B.; Zhang, J.; Li, W.; Mou, F.; Wang, H.; Zou, Q.; Zhong, B.; Wu, L.; Wei, H.; et al. Role of the Gut Microbiome in Modulating Arthritis Progression in Mice. Sci. Rep. 2016, 6, 30594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogier, R.; Evans-Marin, H.; Manasson, J.; van der Kraan, P.M.; Walgreen, B.; Helsen, M.M.; van den Bersselaar, L.A.; van de Loo, F.A.; van Lent, P.L.; Abramson, S.B.; et al. Alteration of the intestinal microbiome characterizes preclinical inflammatory arthritis in mice and its modulation attenuates established arthritis. Sci. Rep. 2017, 7, 15613. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Cai, G.; Qiu, Y.; Fei, N.; Zhang, M.; Pang, X.; Jia, W.; Cai, S.; Zhao, L. Structural segregation of gut microbiota between colorectal cancer patients and healthy volunteers. ISME J. 2012, 6, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Wang, Q.; Huang, Z.; Song, A.; Peng, Y.; Hou, S.; Guo, S.; Zhu, W.; Yan, S.; Lin, Z.; et al. Gastric Bypass Surgery Reverses Diabetic Phenotypes in Bdnf-Deficient Mice. Am. J. Pathol. 2016, 186, 2117–2128. [Google Scholar] [CrossRef]

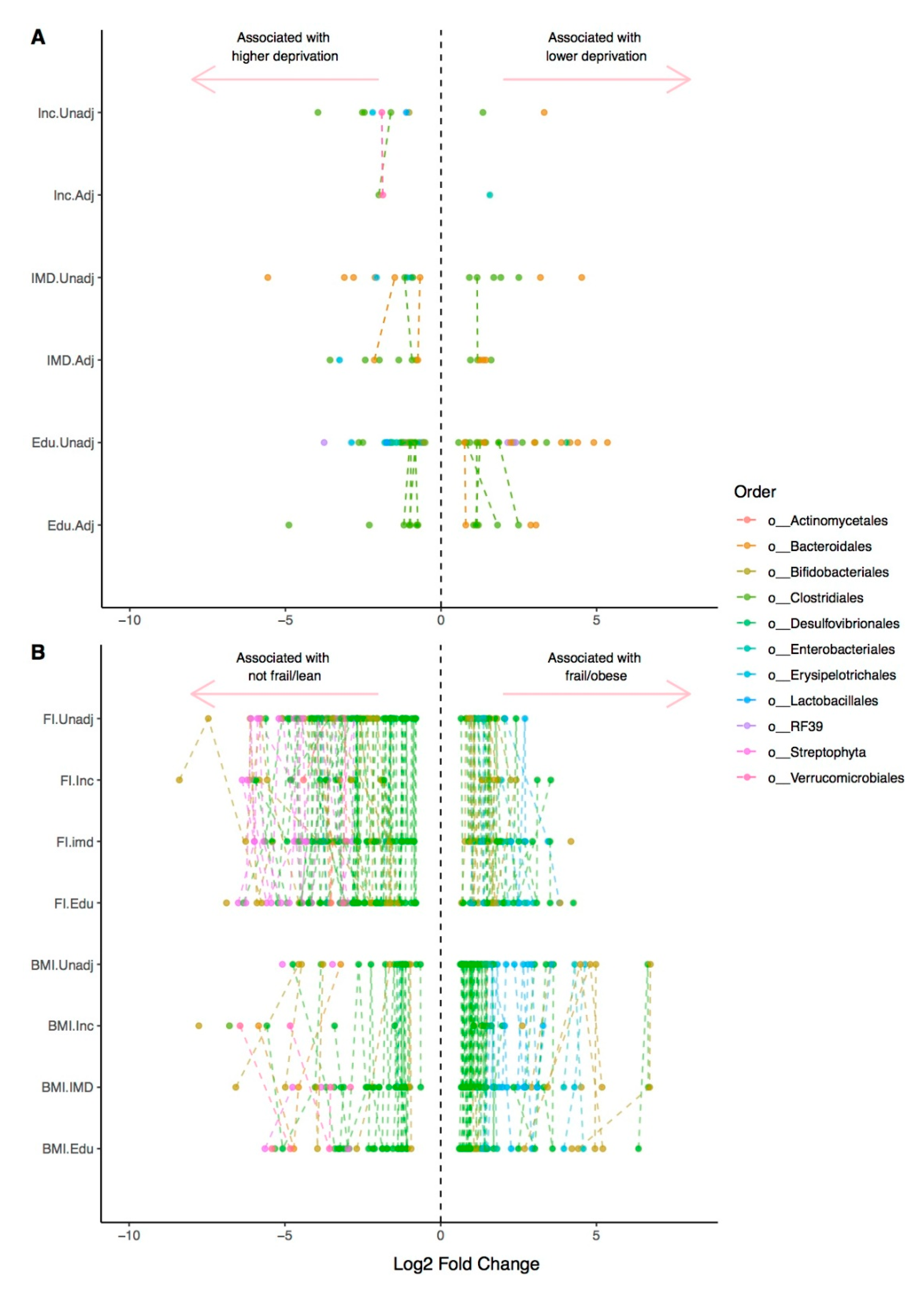


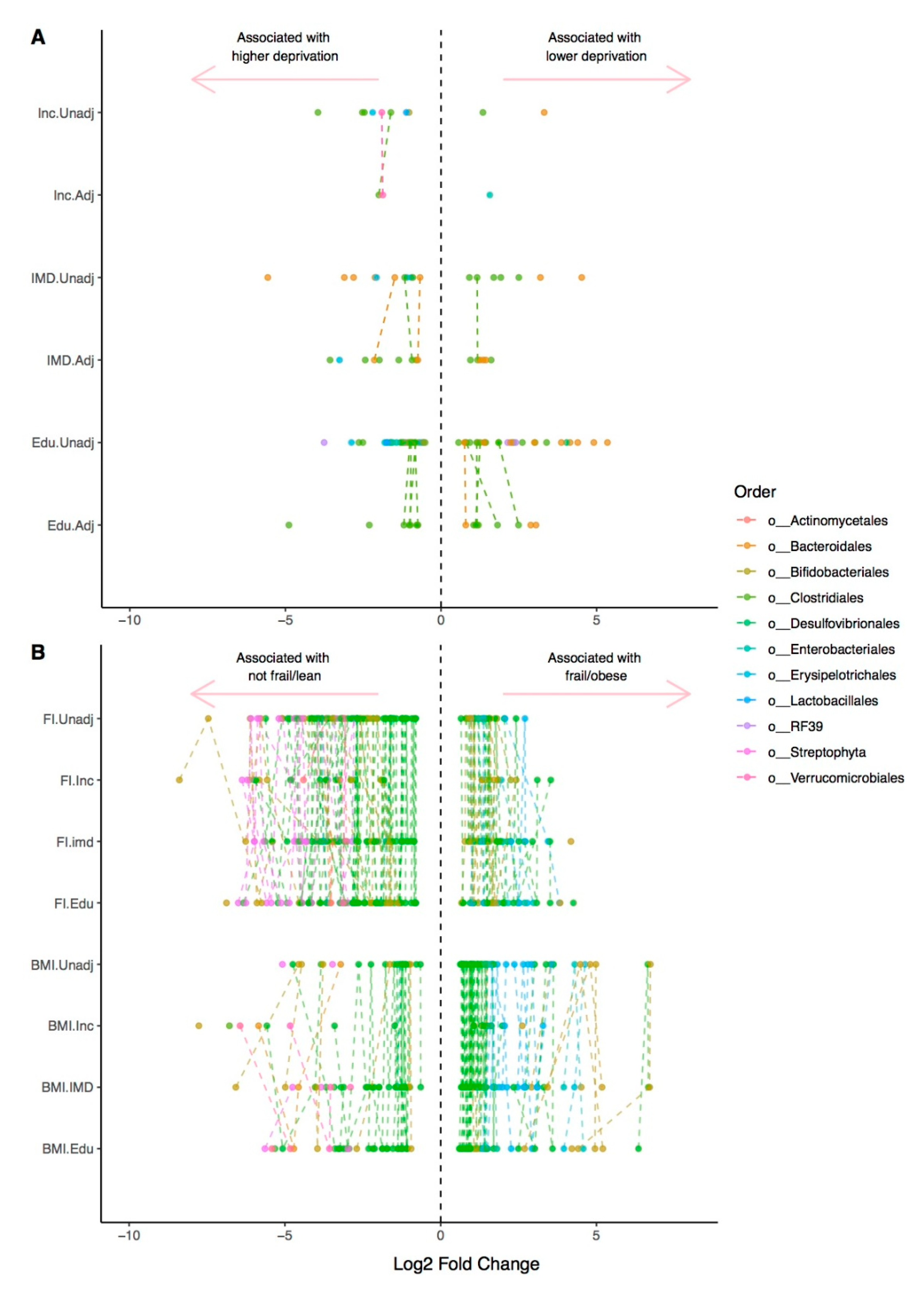
{kind=link}
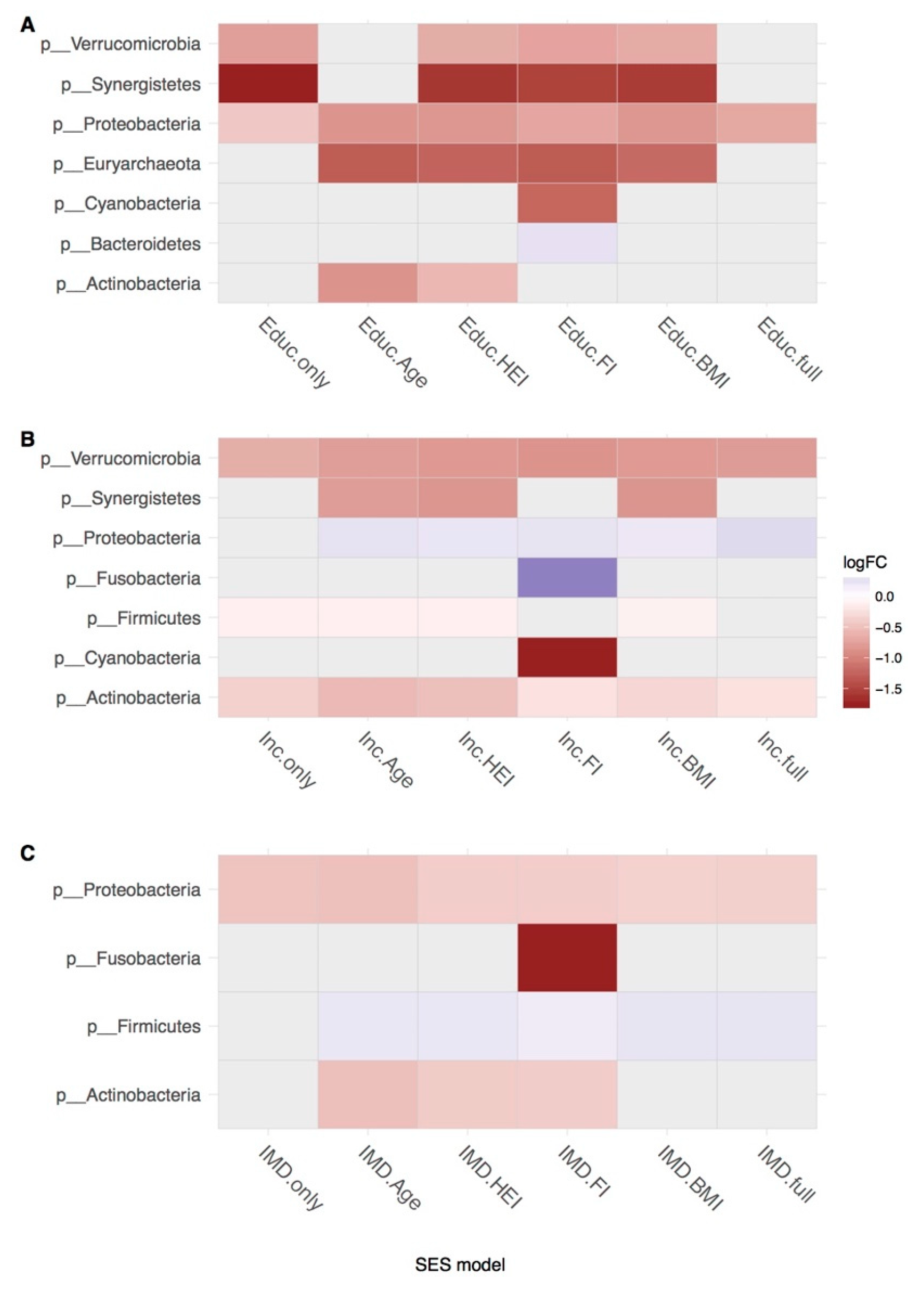
{kind=link}
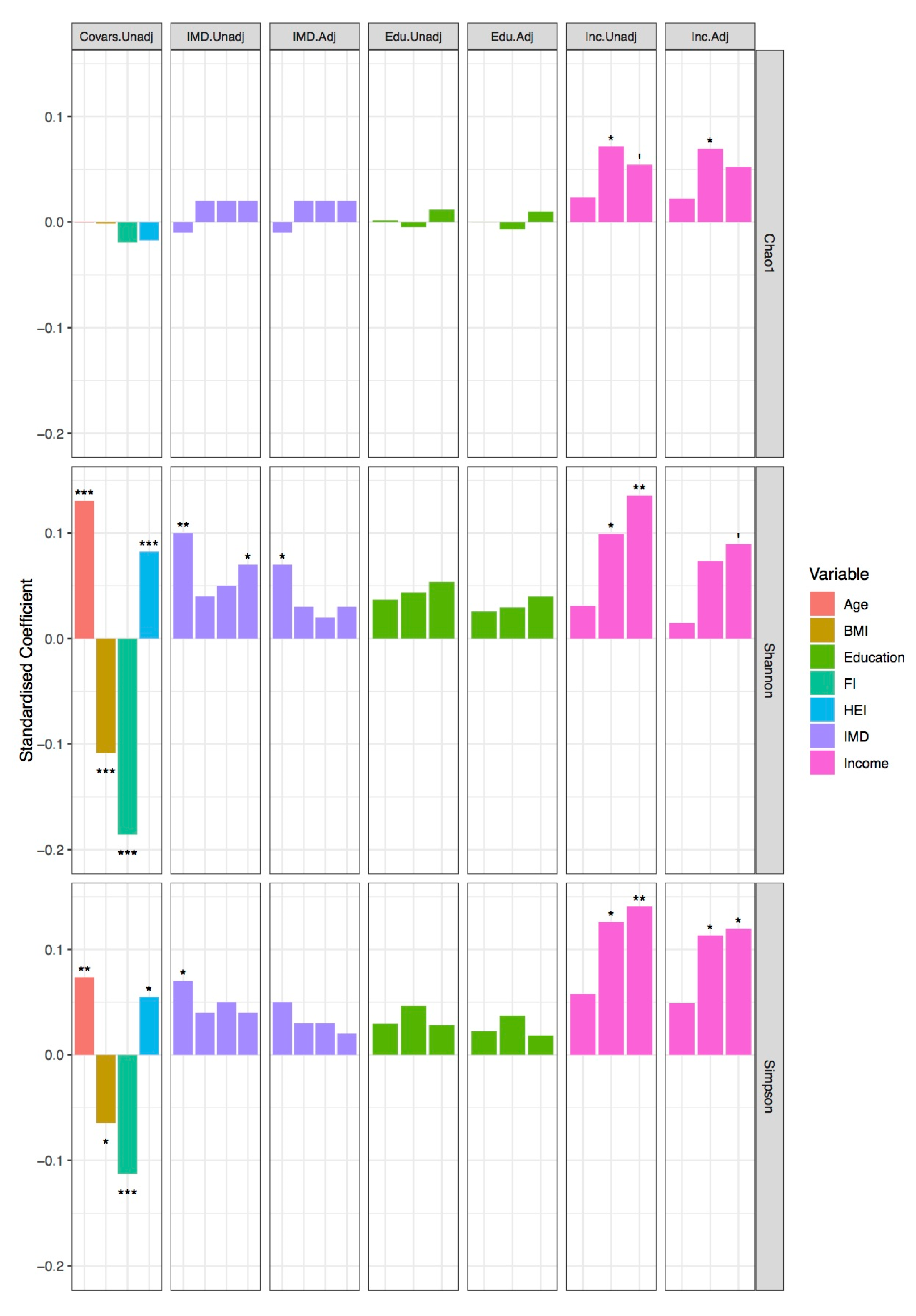
{kind=link}
{kind=link}
| Group | n | %MZ | (%Female) | Age μ | BMI μ | HEI μ | FI μ | |
|---|---|---|---|---|---|---|---|---|
| IMD | Q1 | 336 | 57 | 89 | 60.35 | 26.9 | 59.53 | 0.19 |
| Q2 | 333 | 58 | 91 | 61.37 | 25.52 | 61.09 | 0.18 | |
| Q3 | 334 | 55 | 90 | 61.35 | 26.3 | 60.22 | 0.19 | |
| Q4 | 334 | 59 | 90 | 62.57 | 25.38 | 60.46 | 0.19 | |
| Q5 | 335 | 54 | 93 | 63.7 | 25.53 | 60.46 | 0.18 | |
| Total | 1672 | 56 | 91 | 61.89 | 25.92 | 60.33 | 0.19 | |
| Education | Q1 | 224 | 50 | 91 | 68.92 | 27.18 | 58.92 | 0.24 |
| Q2 | 336 | 52 | 94 | 62.33 | 26.02 | 60.38 | 0.19 | |
| Q3 | 486 | 57 | 89 | 61.2 | 25.81 | 60.32 | 0.19 | |
| Q4 | 359 | 65 | 83 | 55.99 | 24.93 | 61.3 | 0.18 | |
| Total | 1426 | 57 | 89 | 61.48 | 25.87 | 60.34 | 0.2 | |
| Income | Q1 | 139 | 52 | 97 | 67.22 | 26.38 | 58.83 | 0.24 |
| Q2 | 203 | 45 | 93 | 64.62 | 26.32 | 59.55 | 0.21 | |
| Q3 | 310 | 51 | 97 | 62.11 | 26.01 | 59.99 | 0.2 | |
| Q4 | 147 | 52 | 86 | 60.53 | 25 | 61.72 | 0.16 | |
| Total | 799 | 50 | 92 | 63.35 | 25.97 | 59.99 | 0.2 |
| Assigned Taxa | # | i. IMD | ii. IMD Adj | iii. Education | iv. Ed. Adj | v. Income | vi. Inc. Adj | Category | Health Associations |
|---|---|---|---|---|---|---|---|---|---|
| Akkermansia muciniphila (s) | 1 | − | − | A | Disrupts obesity-associated host metabolism [58]. Associated with reduction of inflammation [53]. | ||||
| Anaerofustis (g) | 1 | − | − | D | Decreases where soluble maize fiber used as supplement in adolescents [59,60]. Positively associated with infection in rabbit models [41]. | ||||
| Anaerostipes (g) | 1 | + | A | Butyrate producers that co-occur with other beneficial microbes [61]. Suggestion that excessive short chain fatty acids (SCFA) production promotes gastrointestinal symptoms associated with Rett syndrome [62]. | |||||
| Bacteroides (g) Bacteroides coprophilus (s) | 6 | − | + | + | + | + | B | Member of core microbiome [63]. Degrade dietary polysaccharides (glycans) generating beneficial SCFA [64]. Depletion associated with irritable bowel disorder IBD [65]. | |
| Barnesiellaceae (f) | 4 | + | + | C | Associated with the mucosal microbiota in patients with primary sclerosis cholangitis (PSC) [66] and potentially Parkinson’s disease [67]. Negatively associated with bacteraemia [68]. | ||||
| Blautia (g) Blautia producta (s) | 4 | − | − | +/− | D | Converts plant lignan precursors to enterolactone [69] which may explain its negative association with cancers, such as colorectal cancer (CRC) [70]. Species in genera have been linked with both obese and lean [71]. | |||
| Christensenellaceae (f) | 2 | − | A | Implicated in human longevity [72] and better represented in lean and older individuals [73,74]. | |||||
| Clostridiales (o) Clostridiaceae (f) Clostridium (g) | 13 | +/− | − | +/− | +/− | D | Decreased abundance correlates with inflammatory bowel disease [75] and colorectal cancer [76]. Some members associated with promotion of obesity [69]. | ||
| 3 | 4 | − | + | C | Phyla contains Escherichia coli and opportunistic pathogen Klebsiella pneumoniae. Associated with the development of ulcerative colitis [77]. Bacterially produced β-lactamases are responsible for pyogenic liver abscess [78]. | ||||
| Eubacterium dolichum (s) | 2 | − | − | C | Previously identified within this cohort as being associated with health deficit and higher visceral fat mass [3,79]. Associated with the “obese” gut microbiota [80,81]. | ||||
| Faecalibacterium prausnitzii (s) | 1 | + | + | + | A | Key butyrate producer to the colonic epithelium [82]. Negatively associated with pathogenesis of Crohn’s disease, inflammatory bowel disease, and prostate cancer [52,82,83,84]. Proposed mechanism of action is via production of the anti-inflammatory 15 kDa protein [52]. | |||
| Lachnospiraceae (f) Lachnospira (g) | 11 | − | +/− | +/− | + | − | − | D | Murine models observe improvement to colonization resistance [85]. Induce hyperglycemia in obese mice [86] and negatively associated with resistant starch [87]. Implicated in deficit of caspase-1 which is suggested as having a protective effect in modulation of gut microbiota–brain pathways [88]. |
| Prevotella (g) Prevotella copri (s) Prevotella stercorea (s) | 5 | +/− | + | + | + | B | Reduced in obese patients compared to healthy controls [89]. P. copri has been inferred in the pathogenesis of rheumatoid arthritis [90] and P. stercorea has been observed as associated with carcinoma-in-adenoma [91]. | ||
| RF39 (o) | 3 | +/− | D | Correlates with E. coli under certain dietary conditions in bovine models [92]. | |||||
| Rikenellaceae (f) | 2 | + | + | − | A | Lower abundances associated with lean subjects [74]; depleted in patients with chronic HIV [93]. Target for microbial intervention in obesity management [94]. | |||
| Rumminococcaceae (f) Ruminococcus (g) Ruminococcus gnavus (s) | 11 | +/− | +/− | +/− | +/− | − | D | Dominant and prevalent members of the non-individual specific gut microbiota [55,95]. Associated with diets high in resistant starch [96]. R. gnavus observed to be enriched in Crohn’s disease (CD) patients [97]. Keystone member of the mucus associated microbiome [98]. | |
| S24-7 (f) | 3 | − | + | D | Mouse models suggest role in collagen induced arthritis [99,100]—although the cited papers note different directions of effect. | ||||
| Streptophyta (o) Streptococcus (g) Streptococcus anginosus (s) | 4 | − | − | − | C | S. anginosus a feature of negatively health-associated community assemblages [54]. Increased in colorectal cancer patients [101] and individuals suffering from non-alcoholic fatty liver disease [102]. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bowyer, R.C.E.; Jackson, M.A.; Le Roy, C.I.; Ni Lochlainn, M.; Spector, T.D.; Dowd, J.B.; Steves, C.J. Socioeconomic Status and the Gut Microbiome: A TwinsUK Cohort Study. Microorganisms 2019, 7, 17. https://doi.org/10.3390/microorganisms7010017
Bowyer RCE, Jackson MA, Le Roy CI, Ni Lochlainn M, Spector TD, Dowd JB, Steves CJ. Socioeconomic Status and the Gut Microbiome: A TwinsUK Cohort Study. Microorganisms. 2019; 7(1):17. https://doi.org/10.3390/microorganisms7010017
Chicago/Turabian StyleBowyer, Ruth C. E., Matthew A. Jackson, Caroline I. Le Roy, Mary Ni Lochlainn, Tim D. Spector, Jennifer B. Dowd, and Claire J. Steves. 2019. "Socioeconomic Status and the Gut Microbiome: A TwinsUK Cohort Study" Microorganisms 7, no. 1: 17. https://doi.org/10.3390/microorganisms7010017
APA StyleBowyer, R. C. E., Jackson, M. A., Le Roy, C. I., Ni Lochlainn, M., Spector, T. D., Dowd, J. B., & Steves, C. J. (2019). Socioeconomic Status and the Gut Microbiome: A TwinsUK Cohort Study. Microorganisms, 7(1), 17. https://doi.org/10.3390/microorganisms7010017