
Meta-Transcriptomic Response to Copper Corrosion in Drinking Water Biofilms


Abstract
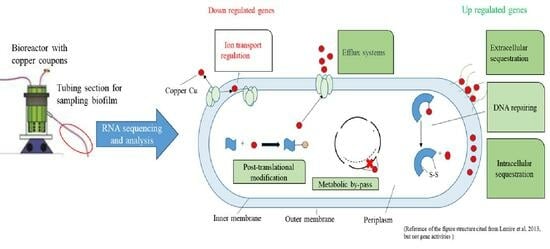
1. Introduction
2. Materials and Methods
2.1. Experiment and Sample Collection
2.2. Total Biofilm RNA Isolation
2.3. cDNA Synthesis, Library Construction, and Sequencing
2.4. Sequence Data Analyses
2.5. Global Gene Expression Classification
2.6. Taxonomic Analysis and Statistical Analysis of the mRNA Sequences
2.7. RT-qPCR Validations
3. Results
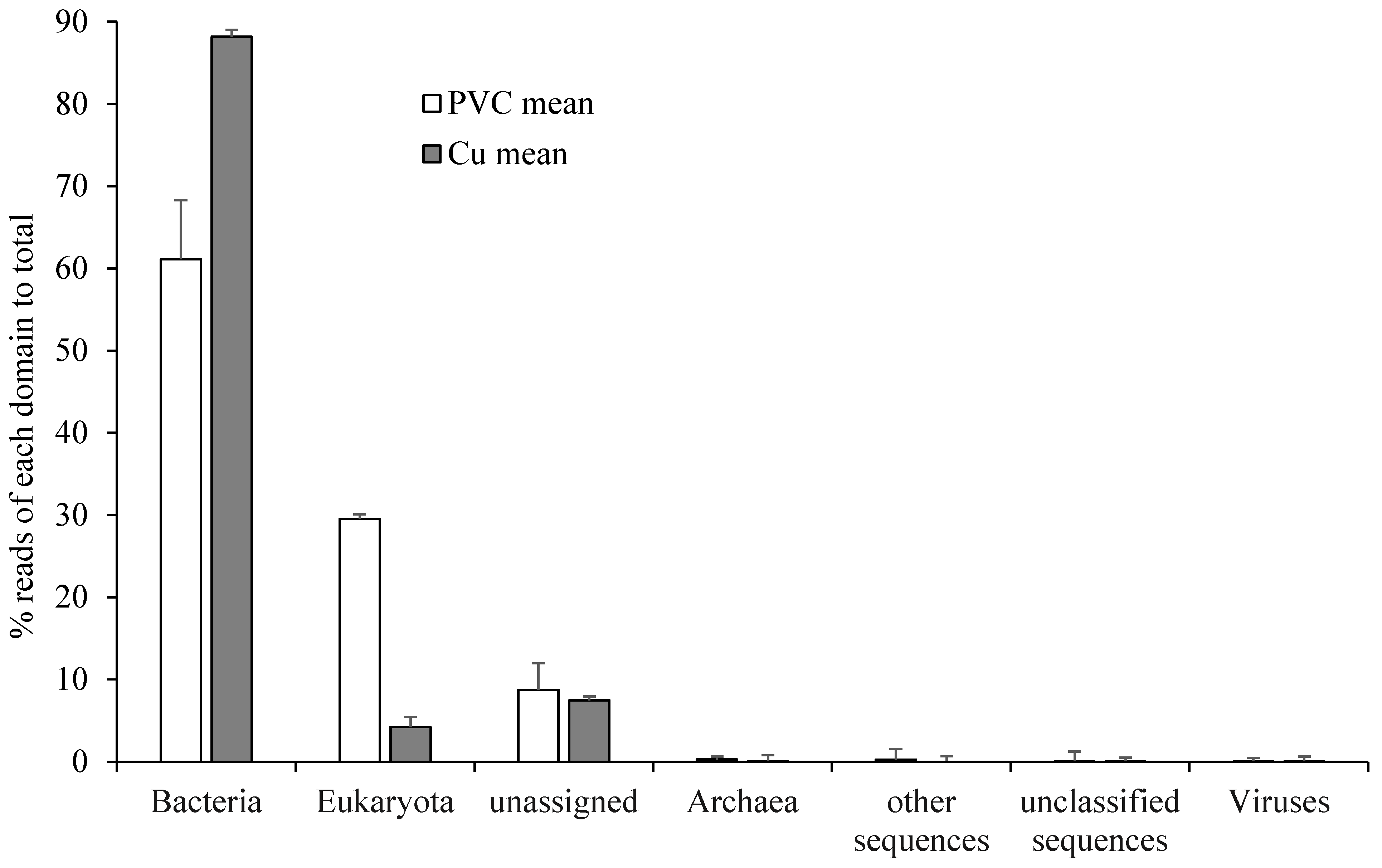
3.1. Retrieved RNA Sequences
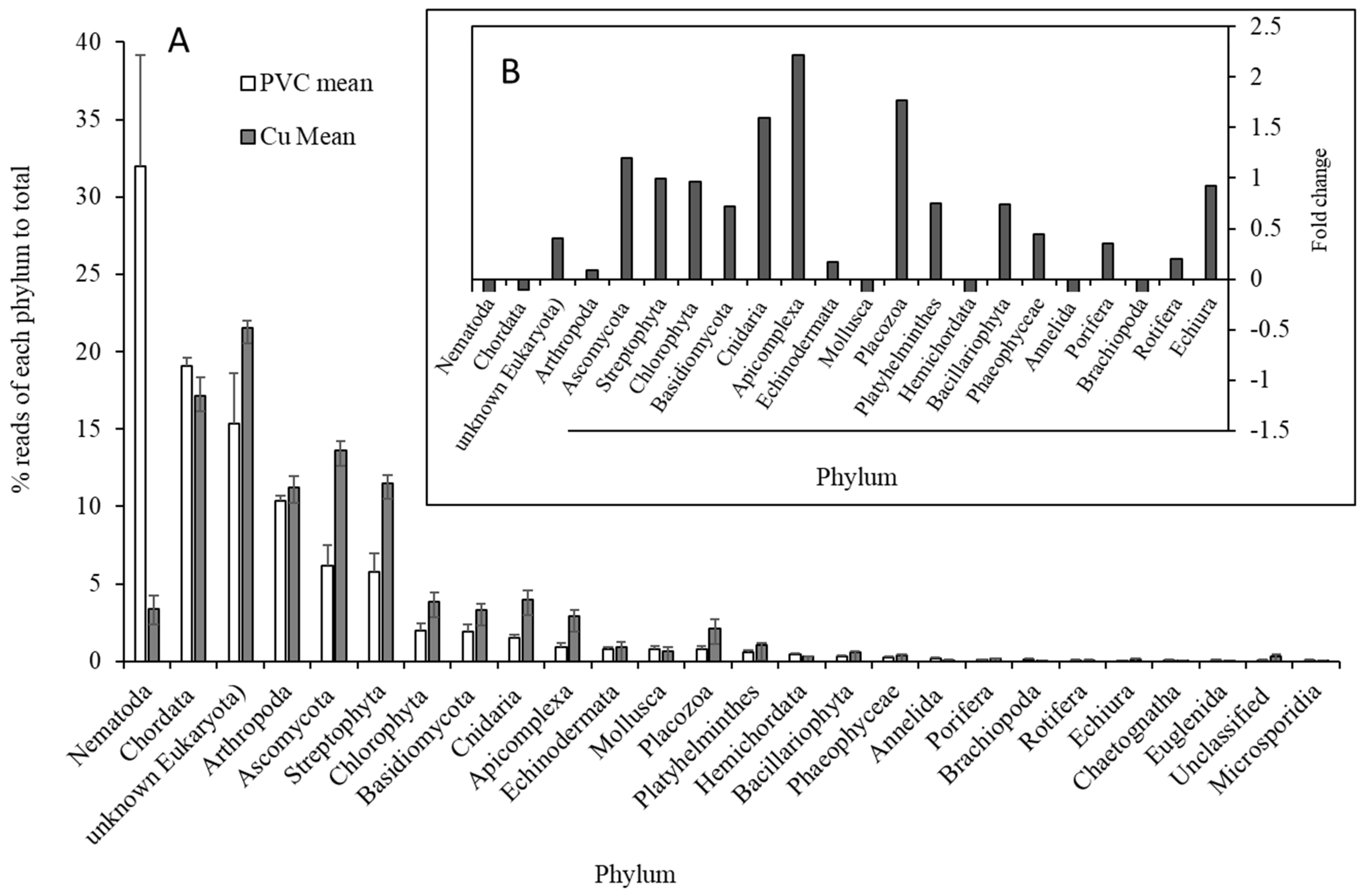
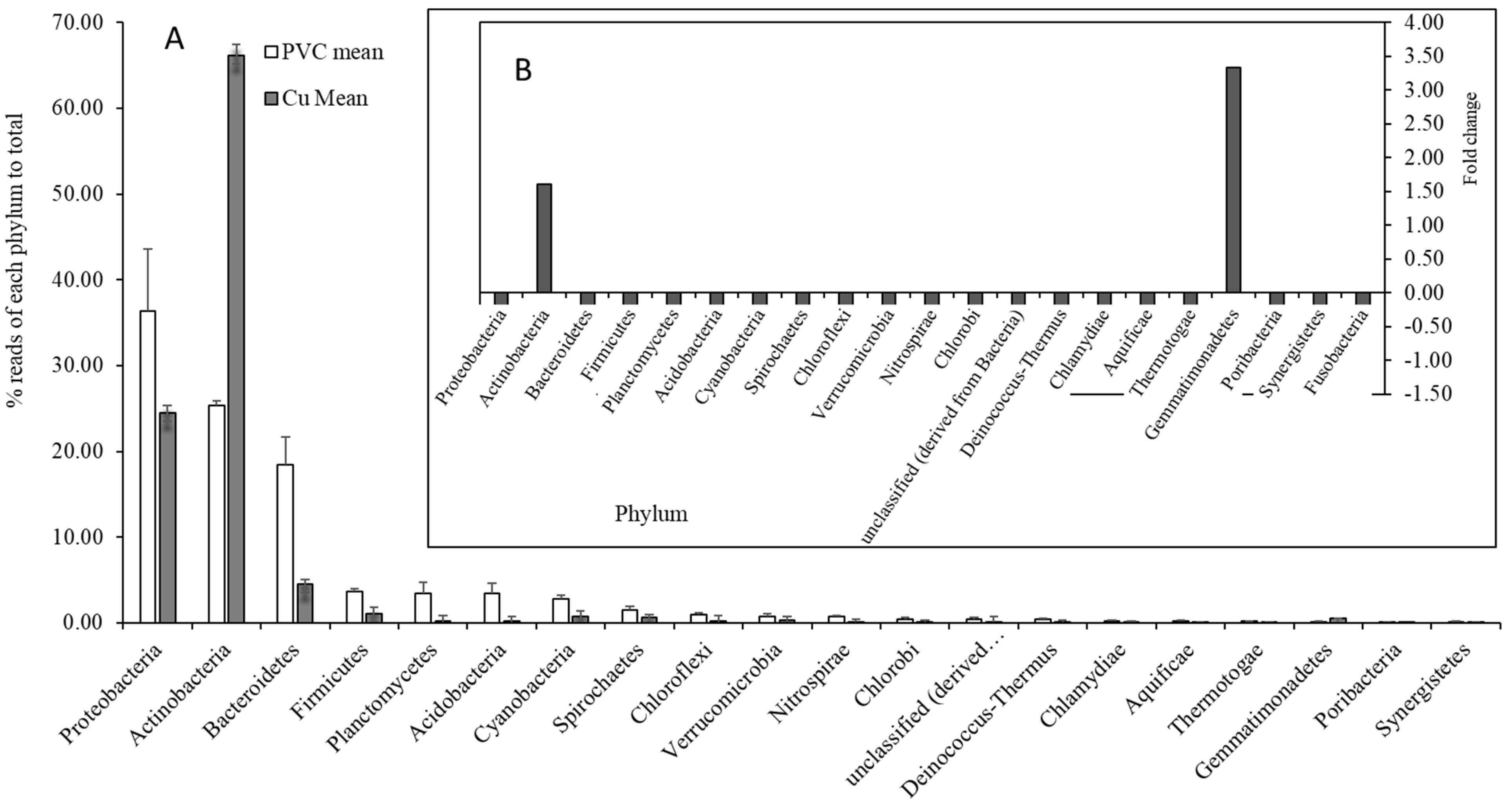
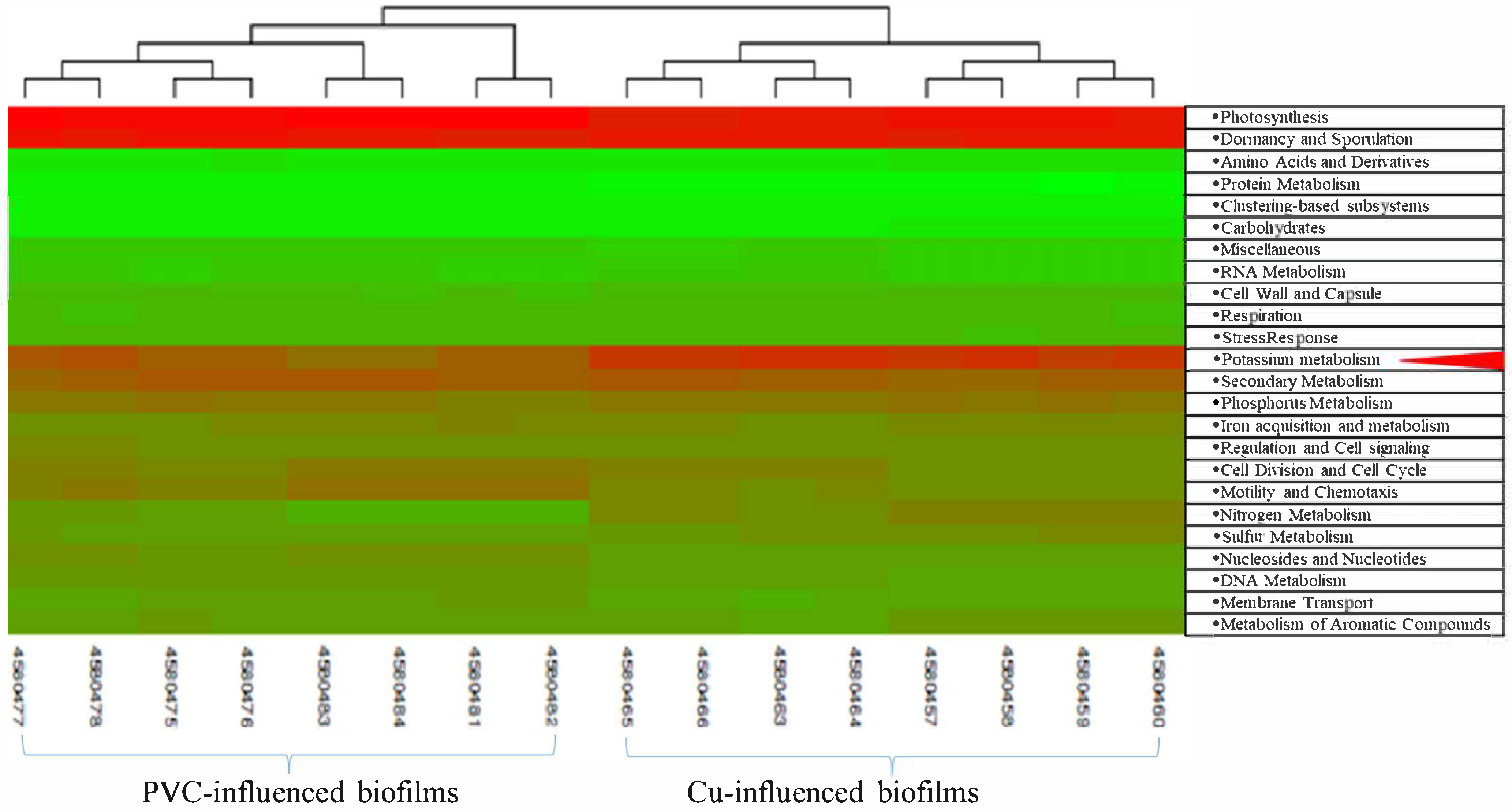
3.2. Comparisons of Microbial Community Between PVC- and Cu-Influenced Biofilms
3.3. Functional Waterborne Pathogens
3.4. Significant Regulated Functional Genes
3.5. Validation of Some Functional Genes
4. Discussion
4.1. The Impact on the Functional Microbial Community
4.2. Impact of Copper on Microbial Organism Functions
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ashbolt, N.J. Environmental (saprozoic) pathogens of engineered water systems: Understanding their ecology for risk assessment and management. Pathogens 2015, 4, 390–405. [Google Scholar] [CrossRef] [PubMed]
- Prest, E.I.; Hammes, F.; Van Loosdrecht, M.C.; Vrouwenvelder, J.S. Biological stability of drinking water: Controlling factors, methods, and challenges. Front. Microbiol. 2016, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Galarce, C.; Fischer, D.; Díez, B.; Vargas, I.T.; Pizarro, G.E. Dynamics of biocorrosion in copper pipes under actual drinking water conditions. Water 2020, 12, 1036. [Google Scholar] [CrossRef]
- Cullom, A.; Spencer, M.S.; Williams, M.D.; Falkinham, J.O., III; Brown, C.; Edwards, M.A.; Pruden, A. Premise plumbing pipe materials and in-building disinfectants shape the potential for proliferation of pathogens and antibiotic resistance genes. Environ. Sci. Technol. 2023, 57, 21382–21394. [Google Scholar] [CrossRef]
- Van der Kooij, D.; Veenendaal, H.R.; Scheffer, W.J. Biofilm formation and multiplication of Legionella in a model warm water system with pipes of copper, stainless steel and cross-linked polyethylene. Water Res. 2005, 39, 2789–2798. [Google Scholar] [CrossRef]
- Doğruöz, N.; Gungor, N.D.; Minnnos, B.; Sungur, E.I.; Çotuk, A. Biofilm formation on copper and galvanized steel surfaces in a cooling-water system. Eur. J. Biol. 2009, 68, 105–111. [Google Scholar]
- Yu, J.; Kim, D.; Lee, T. Microbial diversity in biofilms on water distribution pipes of different materials. Water Sci. Technol. 2010, 61, 163–171. [Google Scholar] [CrossRef]
- Jang, H.J.; Choi, Y.J.; Ka, J.O. Effects of diverse water pipe materials on bacterial communities and water quality in the annular reactor. J. Microbiol. Biotechnol. 2011, 21, 115–123. [Google Scholar] [CrossRef]
- Morvay, A.; Decun, M.; Scurtu, M.; Sala, C.; Morar, A.; Sarandan, M. Biofilm formation on materials commonly used in household drinking water systems. Water Sci. Technol. Water Supply 2011, 11, 252–257. [Google Scholar] [CrossRef]
- Lehtola, M.J.; Miettinen, I.T.; Lampola, T.; Hirvonen, A.; Vartiainen, T.; Martikainen, P.J. Pipeline materials modify the effectiveness of disinfectants in drinking water distribution systems. Water Res. 2005, 39, 1962–1971. [Google Scholar] [CrossRef]
- Buse, H.Y.; Lu, J.; Lu, X.; Mou, X.; Ashbolt, N.J. Microbial diversities (16S and 18S rRNA gene pyrosequencing) and environmental pathogens within drinking water biofilms grown on the common premise plumbing materials unplasticized polyvinylchloride and copper. FEMS Microbiol. Ecol. 2014, 88, 280–295. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Buse, H.; Gomez-Alvarez, V.; Struewing, I.; Santo Domingo, J.; Ashbolt, N.J. Impact of drinking water conditions and copper materials on downstream biofilm microbial communities and Legionella pneumophila colonization. J. Appl. Microbiol. 2014, 117, 905–918. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.K.; Weerasekara, A.; Bond, P.L.; Weynberg, K.D.; Guo, J. Characterizing the premise plumbing microbiome in both water and biofilms of a 50-year-old building. Sci. Total Environ. 2021, 798, 149225. [Google Scholar] [CrossRef] [PubMed]
- van der Kooij, D.; Veenendaal, H.R.; Italiaander, R. Corroding copper and steel exposed to intermittently flowing tap water promote biofilm formation and growth of Legionella pneumophila. Water Res. 2020, 183, 115951. [Google Scholar] [CrossRef]
- Buse, H.Y.; Ji, P.; Gomez-Alvarez, V.; Pruden, A.; Edwards, M.A.; Ashbolt, N.J. Effect of temperature and colonization of Legionella pneumophila and Vermamoeba vermiformis on bacterial community composition of copper drinking water biofilms. Microb. Biotechnol. 2017, 10, 773–788. [Google Scholar] [CrossRef]
- Inkinen, J.; Jayaprakash, B.; Ahonen, M.; Pitkänen, T.; Mäkinen, R.; Pursiainen, A.; Santo Domingo, J.W.; Salonen, H.; Elk, M.; Keinänen-Toivola, M.M. Bacterial community changes in copper and PEX drinking water pipeline biofilms under extra disinfection and magnetic water treatment. J. Appl. Microbiol. 2018, 124, 611–624. [Google Scholar] [CrossRef]
- Gomez-Alvarez, V.; Revetta, R.P.; Santo Domingo, J.W. Metagenomic analyses of drinking water receiving different disinfection treatments. Appl. Environ. Microbiol. 2012, 78, 6095–6102. [Google Scholar] [CrossRef]
- Buse, H.Y.; Lu, J.; Struewing, I.T.; Ashbolt, N.J. Eukaryotic diversity in premise drinking water using 18S rDNA sequencing: Implications for health risks. Environ. Sci. Pollut. Res. 2013, 20, 6351–6366. [Google Scholar] [CrossRef]
- Revetta, R.; Gomez-Alvarez, V.; Gerke, T.; Santo Domingo, J.; Ashbolt, N. Changes in bacterial composition of biofilm in a metropolitan drinking water distribution system. J. Appl. Microbiol. 2016, 121, 294–305. [Google Scholar] [CrossRef]
- Proctor, C.R.; Reimann, M.; Vriens, B.; Hammes, F. Biofilms in shower hoses. Water Res. 2018, 131, 274–286. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59. [Google Scholar] [CrossRef] [PubMed]
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; et al. The metagenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinform. 2008, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Törnqvist, L.; Vartia, P.; Vartia, Y.O. How should relative changes be measured? Am. Stat. 1985, 39, 43–46. [Google Scholar]
- Hemmerling, C.; Labrosse, A.; Ruess, L.; Steinert, M. Legionella pneumophila and free-living nematodes: Environmental co-occurrence and trophic link. Microorganisms 2023, 11, 738. [Google Scholar] [CrossRef]
- Chadha, R.; Grover, M.; Sharma, A.; Lakshmy, A.; Deb, M.; Kumar, A.; Mehta, G. An outbreak of post-surgical wound infections due to Mycobacterium abscessus. Pediatr. Surg. Int. 1998, 13, 406–410. [Google Scholar] [CrossRef]
- Lowry, P.W.; Sague, C.M.B.; Bland, L.A.; Aguero, S.M.; Arduino, M.J.; Minuth, A.N.; Murray, R.A.; Swenson, J.M.; Jarvis, W.R. Mycobacterium chelonae infection among patients receiving high-flux dialysis in a hemodialysis clinic in California. J. Infect. Dis. 1990, 161, 85–90. [Google Scholar] [CrossRef]
- Falkinham, J.O. Current epidemiologic trends of the nontuberculous mycobacteria (NTM). Curr. Environ. Health Rep. 2016, 3, 161–167. [Google Scholar] [CrossRef]
- Buse, H.Y.; Schoen, M.E.; Ashbolt, N.J. Legionellae in engineered systems and use of quantitative microbial risk assessment to predict exposure. Water Res. 2012, 46, 921–933. [Google Scholar] [CrossRef]
- Raoult, D.; Audic, S.; Robert, C.; Abergel, C.; Renesto, P.; Ogata, H.; La Scola, B.; Suzan, M.; Claverie, J.-M. The 1.2-megabase genome sequence of Mimivirus. Science 2004, 306, 1344–1350. [Google Scholar] [CrossRef]
- La Scola, B.; Marrie, T.J.; Auffray, J.-P.; Raoult, D. Mimivirus in pneumonia patients. Emerg. Infect. Dis. 2005, 11, 449. [Google Scholar] [CrossRef]
- Inkinen, J.; Jayaprakash, B.; Siponen, S.; Hokajärvi, A.-M.; Pursiainen, A.; Ikonen, J.; Ryzhikov, I.; Täubel, M.; Kauppinen, A.; Paananen, J. Active eukaryotes in drinking water distribution systems of ground and surface waterworks. Microbiome 2019, 7, 99. [Google Scholar] [CrossRef] [PubMed]
- Schenk, J.; Höss, S.; Brinke, M.; Kleinbölting, N.; Brüchner-Hüttemann, H.; Traunspurger, W. Nematodes as bioindicators of polluted sediments using metabarcoding and microscopic taxonomy. Environ. Int. 2020, 143, 105922. [Google Scholar] [CrossRef] [PubMed]
- ASTM E2172-22; Standard Guide for Conducting Laboratory Soil Toxicity Tests with the Nematode Caenorhabditis Elegans. ASTM: West Conshohocken, PA, USA, 2022.
- Boyd, W.A.; Williams, P.L. Comparison of the sensitivity of three nematode species to copper and their utility in aquatic and soil toxicity tests. Environ. Toxicol. Chem. 2003, 22, 2768–2774. [Google Scholar] [CrossRef] [PubMed]
- Harada, H.; Kurauchi, M.; Hayashi, R.; Eki, T. Shortened lifespan of nematode Caenorhabditis elegans after prolonged exposure to heavy metals and detergents. Ecotoxicol. Environ. Saf. 2007, 66, 378–383. [Google Scholar] [CrossRef]
- Lytle, D.A.; Liggett, J. Impact of water quality on chlorine demand of corroding copper. Water Res. 2016, 92, 11–21. [Google Scholar] [CrossRef]
- van Lieverloo, J.H.M.; van der Kooij, D.; Hoogenboezem, W. Invertebrates and protozoa (free-living) in drinking water distribution systems. In Encyclopedia of Environmental Microbiology; Bitton, G., Ed.; John Wiley & Sons: New York, NY, USA, 2002; pp. 1718–1733. [Google Scholar] [CrossRef]
- Borella, P.; Guerrieri, E.; Marchesi, I.; Bondi, M.; Messi, P. Water ecology of Legionella and protozoan: Environmental and public health perspectives. Biotechnol. Annu. Rev. 2005, 11, 355–380. [Google Scholar]
- Lin, Y.-s.E.; Vidic, R.D.; Stout, J.E.; Yu, V.L. Negative effect of high pH on biocidal efficacy of copper and silver ions in controlling Legionella pneumophila. Appl. Environ. Microbiol. 2002, 68, 2711–2715. [Google Scholar] [CrossRef]
- Ladomersky, E.; Petris, M.J. Copper tolerance and virulence in bacteria. Metallomics 2015, 7, 957–964. [Google Scholar] [CrossRef]
- Gaetke, L.M.; Chow, C.K. Copper toxicity, oxidative stress, and antioxidant nutrients. Toxicology 2003, 189, 147–163. [Google Scholar] [CrossRef]
- Macomber, L.; Imlay, J.A. The iron-sulfur clusters of dehydratases are primary intracellular targets of copper toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 8344–8349. [Google Scholar] [CrossRef]
- Dupont, C.L.; Grass, G.; Rensing, C.J.M. Copper toxicity and the origin of bacterial resistance—New insights and applications. Metallomics 2011, 3, 1109–1118. [Google Scholar] [CrossRef] [PubMed]
- Quintana, J.; Novoa-Aponte, L.; Argüello, J.M. Copper homeostasis networks in the bacterium Pseudomonas aeruginosa. J. Biol. Chem. 2017, 292, 15691–15704. [Google Scholar] [CrossRef] [PubMed]
- Everaert, C.; Luypaert, M.; Maag, J.L.; Cheng, Q.X.; Dinger, M.E.; Hellemans, J.; Mestdagh, P. Benchmarking of RNA-sequencing analysis workflows using whole-transcriptome RT-qPCR expression data. Sci. Rep. 2017, 7, 1559. [Google Scholar] [CrossRef] [PubMed]
- Stautz, J.; Hellmich, Y.; Fuss, M.F.; Silberberg, J.M.; Devlin, J.R.; Stockbridge, R.B.; Hänelt, I. Molecular mechanisms for bacterial potassium homeostasis. J. Mol. Biol. 2021, 433, 166968. [Google Scholar] [CrossRef]
- Solioz, M.; Abicht, H.K.; Mermod, M.; Mancini, S. Response of Gram-positive bacteria to copper stress. JBIC J. Biol. Inorg. Chem. 2010, 15, 3. [Google Scholar] [CrossRef]
- Argüello, J.M.; Eren, E.; González-Guerrero, M. The structure and function of heavy metal transport P 1B-ATPases. Biometals 2007, 20, 233–248. [Google Scholar] [CrossRef]
- González-Guerrero, M.; Argüello, J.M. Mechanism of Cu+-transporting ATPases: Soluble Cu+ chaperones directly transfer Cu+ to transmembrane transport sites. Proc. Natl. Acad. Sci. USA 2008, 105, 5992–5997. [Google Scholar] [CrossRef]
- Jung, K.; Altendorf, K. Towards an understanding of the molecular mechanisms of stimulus perception and signal transduction by the KdpD/KdpE system of Escherichia coli. J. Mol. Microbiol. Biotechnol. 2002, 4, 223–228. [Google Scholar]
- Cholo, M.C.; van Rensburg, E.J.; Osman, A.G.; Anderson, R. Expression of the genes encoding the Trk and Kdp potassium transport systems of Mycobacterium tuberculosis during growth in vitro. BioMed Res. Int. 2015, 2015, 608682. [Google Scholar] [CrossRef]
- Chen, Y.; Yang, F.; Sun, Z.; Wang, Q.; Mi, K.; Deng, H. Proteomic analysis of drug-resistant mycobacteria: Co-evolution of copper and INH resistance. PLoS ONE 2015, 10, e0127788. [Google Scholar] [CrossRef]
- Chaturvedi, K.S.; Hung, C.S.; Crowley, J.R.; Stapleton, A.E.; Henderson, J.P. The siderophore yersiniabactin binds copper to protect pathogens during infection. Nat. Chem. Biol. 2012, 8, 731. [Google Scholar] [CrossRef] [PubMed]
- Bush, M.J.; Tschowri, N.; Schlimpert, S.; Flärdh, K.; Buttner, M.J. c-di-GMP signalling and the regulation of developmental transitions in streptomycetes. Nat. Rev. Microbiol. 2015, 13, 749–760. [Google Scholar] [CrossRef] [PubMed]
- Khan, F.Z.; Palmer, K.; Dillon, N. Siderophores mediate antibiotic resistance. Nat. Microbiol. 2024, 9, 587–588. [Google Scholar] [CrossRef] [PubMed]
- Roberts, S.A.; Weichsel, A.; Grass, G.; Thakali, K.; Hazzard, J.T.; Tollin, G.; Rensing, C.; Montfort, W.R. Crystal structure and electron transfer kinetics of CueO, a multicopper oxidase required for copper homeostasis in Escherichia coli. Proc. Natl. Acad. Sci. USA 2002, 99, 2766–2771. [Google Scholar] [CrossRef]
- Rowland, J.L.; Niederweis, M. Resistance mechanisms of Mycobacterium tuberculosis against phagosomal copper overload. Tuberculosis 2012, 92, 202–210. [Google Scholar] [CrossRef]
- Bodet, C.; Sahr, T.; Dupuy, M.; Buchrieser, C.; Héchard, Y. Legionella pneumophila transcriptional response to chlorine treatment. Water Res. 2012, 46, 808–816. [Google Scholar] [CrossRef]
- O’Connor, A.; McClean, S. The role of universal stress proteins in bacterial infections. Curr. Med. Chem. 2017, 24, 3970–3979. [Google Scholar] [CrossRef]
- Takahashi, N.; Sato, T.; Yamada, T. Metabolic pathways for cytotoxic end product formation from glutamate-and aspartate-containing peptides by Porphyromonas gingivalis. J. Bacteriol. 2000, 182, 4704–4710. [Google Scholar] [CrossRef]
- Johnson, D.C.; Dean, D.R.; Smith, A.D.; Johnson, M.K. Structure, function, and formation of biological iron-sulfur clusters. Annu. Rev. Biochem. 2005, 74, 247–281. [Google Scholar] [CrossRef]
- Paul, V.D.; Lill, R. Biogenesis of cytosolic and nuclear iron–sulfur proteins and their role in genome stability. Biochim. Et Biophys. Acta-Mol. Cell Res. 2015, 1853, 1528–1539. [Google Scholar] [CrossRef]
- Avery, S.V.; Howlett, N.G.; Radice, S. Copper toxicity towards Saccharomyces cerevisiae: Dependence on plasma membrane fatty acid composition. Appl. Environ. Microbiol. 1996, 62, 3960–3966. [Google Scholar] [CrossRef] [PubMed]
- Portevin, D.; de Sousa-D’Auria, C.; Houssin, C.; Grimaldi, C.; Chami, M.; Daffé, M.; Guilhot, C. A polyketide synthase catalyzes the last condensation step of mycolic acid biosynthesis in mycobacteria and related organisms. Proc. Natl. Acad. Sci. USA 2004, 101, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Armitige, L.Y.; Jagannath, C.; Wanger, A.R.; Norris, S.J. Disruption of the genes encoding antigen 85A and antigen 85B of Mycobacterium tuberculosis H37Rv: Effect on growth in culture and in macrophages. Infect. Immun. 2000, 68, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Linton, D.; Owen, R.J.; Stanley, J. Rapid identification by PCR of the genus Campylobacter and of five Campylobacter species enteropathogenic for man and animals. Res. Microbiol. 1996, 147, 707–718. [Google Scholar] [CrossRef]
- Lund, M.; Nordentoft, S.; Pedersen, K.; Madsen, M. Detection of Campylobacter spp. in chicken fecal samples by real-time PCR. J. Clin. Microbiol. 2004, 42, 5125–5132. [Google Scholar] [CrossRef]
- González-Escalona, N.; Hammack, T.S.; Russell, M.; Jacobson, A.P.; De Jesús, A.J.; Brown, E.W.; Lampel, K.A. Detection of live Salmonella sp. cells in produce by a TaqMan-based quantitative reverse transcriptase real-time PCR targeting invA mRNA. Appl. Environ. Microbiol. 2009, 75, 3714–3720. [Google Scholar] [CrossRef]
- Bruijnesteijn van Coppenraet, E.S.; Lindeboom, J.A.; Prins, J.M.; Peeters, M.F.; Claas, E.C.J.; Kuijper, E.J. Real-time PCR assay using fine-needle aspirates and tissue biopsy specimens for rapid diagnosis of mycobacterial lymphadenitis in children. J. Clin. Microbiol. 2004, 42, 2644. [Google Scholar] [CrossRef]
- Riviere, D.; Szczebara, F.M.; Berjeaud, J.M.; Frere, J.; Hechard, Y. Development of a real-time PCR assay for quantification of Acanthamoeba trophozoites and cysts. J. Microbiol. Methods 2006, 64, 78–83. [Google Scholar] [CrossRef]
- Kuiper, M.W.; Valster, R.M.; Wullings, B.A.; Boonstra, H.; Smidt, H.; van der Kooij, D. Quantitative detection of the free-living amoeba Hartmannella vermiformis in surface water by using real-time PCR. Appl. Environ. Microbiol. 2006, 72, 5750–5756. [Google Scholar] [CrossRef]
- Schroeder, J.M.; Booton, G.C.; Hay, J.; Niszl, I.A.; Seal, D.V.; Markus, M.B.; Fuerst, P.A.; Byers, T.J. Use of subgenic 18S ribosomal DNA PCR and sequencing for genes and genotype identification of acanthamoebae from human with keratitis and sewage sludge. J. Clin. Microbiol. 2001, 39, 1903–1911. [Google Scholar] [CrossRef]
- Hadfield, S.J.; Robinson, G.; Elwin, K.; Chalmers, R.M. Detection and differentiation of Cryptosporidium spp. in human clinical samples by use of realtime PCR. J. Clin. Microbiol. 2011, 49, 918–924. [Google Scholar] [CrossRef] [PubMed]
- Guy, R.A.; Payment, P.; Krull, U.J.; Horgen, P.A. Real-time PCR for quantification of Giardia and Cryptosporidium in environmental water samples and sewage. Appl. Environ. Microbiol. 2003, 69, 5178–5185. [Google Scholar] [CrossRef] [PubMed]
- Qvarnstrom, Y.; Visvesvara, G.S.; Sriram, R.; da Silva, A.J. Multiplex real-time PCR assay for simultaneous detection of Acanthamoeba spp.; Balamuthia mandrillaris, and Naegleria fowleri. J. Clin. Microbiol. 2006, 44, 3589–3595. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genus | Sequence Fold Change 1 | ***: p ≤ 0.001 | PVC Mean | StDev | Cu Mean | StDev | Reference Species Mainly Hit | RT-qPCR Fold Change 2 | **: p ≤ 0.01 |
|---|---|---|---|---|---|---|---|---|---|
| Acanthamoeba | −0.86 | *** | 0.54 | 0.259 | 0.075 | 0.035 | A. castellanii, A. culbertsoni, A. healyi, A. polyphaga | ||
| Campylobacter | −0.60 | *** | 0.03 | 0.01 | 0.01 | 0.00 | C. jejuni, C. hominis, C. Concisus, C. Curvus, C. fetus, C. lari, C. rectus, C. coli, C. showae | ||
| Cryptosporidium | −0.67 | *** | 0.06 | 0.01 | 0.02 | 0.01 | C. parvum, C. hominis, C. muris | ||
| Escherichia | −0.29 | 0.11 | 0.05 | 0.08 | 0.06 | E. coli | |||
| Giardia | −0.69 | *** | 0.01 | 0.00 | 0.00 | 0.00 | G. intestinalis | ||
| Vermamoeba | −0.38 | 0.32 | 0.22 | 0.20 | 0.21 | V. vermiformis | 0.81 | ** | |
| Legionella | −0.91 | *** | 0.55 | 0.11 | 0.05 | 0.02 | L. pneumophila, L. longbeachae, L. drancourtii | 0.91 | ** |
| Mimivirus | −0.86 | *** | 0.00 | 0.00 | 0.00 | 0.00 | A. polyphaga mimivirus | ||
| Mycobacterium | 3.97 | *** | 10.25 | 3.38 | 50.94 | 4.81 | M. abscessus, M. avium, M. bovis, M. chelonae, M. gilvum, M. marinum, M. smegmatis, C. tuberculosis, C. smegmatis, M. gilvum, C. marinum, M. parascrofulaceum, M. microti, | 3.06 | ** |
| Naegleria | −0.76 | *** | 0.17 | 0.04 | 0.04 | 0.02 | N. gruberi, N. fowleri | ||
| Pseudomonas | −0.23 | 1.05 | 0.53 | 0.80 | 0.45 | P. Fluorescens, P. aeruginosa, | |||
| Salmonella | −0.30 | *** | 0.03 | 0.01 | 0.02 | 0.01 | S. enterica, S. bongori |
| Phylum | Class/Order/Family/Species | Fold Change | *: p ≤ 0.05, **: p ≤ 0.01, ***: p ≤ 0.001 | PVC Mean | PVC StDev | Cu Mean | Cu StDev |
|---|---|---|---|---|---|---|---|
| Actinomycetota (Actinobacteria) | Mycobacteriaceae | 0.09 | *** | 74.34 | 1.20 | 80.98 | 0.63 |
| M. abscessus | 0.03 | *** | 39.13 | 0.42 | 40.38 | 0.73 | |
| M. bovis | 0.10 | ** | 3.18 | 0.19 | 3.51 | 0.07 | |
| M. microti | 0.22 | *** | 1.53 | 0.13 | 1.87 | 0.07 | |
| M. smegmatis | 0.06 | ** | 6.16 | 0.13 | 6.56 | 0.25 | |
| M. tuberculosis | 0.08 | ** | 4.50 | 0.15 | 4.88 | 0.13 | |
| M. vanbaalenii | 0.16 | *** | 4.02 | 0.27 | 4.66 | 0.20 | |
| Pseudomonadota (Proteobacteria) | 0.54 | *** | 48.98 | 0.98 | 75.48 | 2.70 | |
| Rhodobacterales | 1.87 | *** | 8.77 | 0.58 | 25.20 | 2.79 | |
| Rhodobacteraceae | 0.23 | *** | 77.05 | 1.16 | 94.77 | 0.86 | |
| Rhodobacter (sp. SW2) | 2.77 | *** | 12.35 | 0.69 | 46.54 | 1.97 | |
| Sphingomonadales | 0.79 | *** | 11.10 | 0.62 | 19.88 | 4.95 | |
| Bacteroidota | Flavobacteriia | 1.55 | *** | 17.67 | 1.83 | 45.14 | 5.81 |
| Bacteroidota | Sphingobacteriia | 0.55 | * | 13.71 | 1.07 | 21.30 | 7.01 |
| 1st Category | 2nd-Level Category | Fold Change (Total (Cu%-uPVC%))/Total uPVC% | *: p ≤ 0.05, **: p ≤ 0.01, ***: p ≤ 0.001 | Major Genes Upregulated | Major Genes Downregulated |
|---|---|---|---|---|---|
| Potassium-Associated Metabolism | Potassium homeostasis | 3.29 | *** | Potassium-transporting ATPase: A (6.5-fold), B (3.2-fold), and C (2.5-fold) chain | ATP/GTP-binding site motif A, binding-protein-dependent transport systems’ inner membrane: FKBP-type peptidyl-prolyl cis-trans isomerase; potassium uptake protein: TrkH; potassium voltage-gated channel subfamily; TrkA-N: potassium; glutathione-regulated potassium–efflux system ATP-binding protein: KefA, KefC, KdpE, Kup system |
| CBSS-83332.1.peg.3803 (antigen proteins) | 2.81 | *** | Propionyl-CoA activated by K+ carboxylase beta chain (0.5), probable polyketide synthase (0.6) | Antigen 85-A and -C precursor (85A and 85C) | |
| Universal stress protein family | 1.80 | *** | Universal stress protein family: the universal stress protein (USP) domain is a superfamily of conserved genes that can be found in bacteria, archaea, fungi, protozoa, and plant proteins | ||
| CBSS-196620.1.peg.2477 (1–5%) | 1.75 | *** | Copper-translocating P-type ATPase | Delta-1-pyrroline-5-carboxylate dehydrogenase, ferrous iron transport protein B, maltose O-acetyltransferase | |
| Nitrate and nitrite ammonification | 1.58 | *** | Nitrite reductase [NAD(P)H] subunits, nitrate/nitrite transporter | Respiratory nitrate reductase alpha, beta, delta, and gamma chain | |
| Ammonia assimilation | 1.51 | *** | uridylyltransferase: Nucleotidyl transferase is a component of the repair pathway for single-nucleotide base excision repair. This repair mechanism begins when a single nucleotide is recognized by DNA glycosylase as incorrectly matched or has been mutated in some way (UV light, chemical mutagen, etc.), and is removed. Later, a nucleotidyl tranferase is used to fill in the gap with the correct base, using the template strand as the reference. | Glutamate synthas: This enzyme belongs to the family of oxidoreductases, specifically those acting on the CH-NH2 group of donors with NAD+ or NADP+ as acceptor. This enzyme participates in glutamate metabolism and nitrogen metabolism. It has 5 cofactors: FAD, iron, FMN, sulfur, and iron–sulfur. | |
| WhiB and WhiB-type regulatory proteins | 1.32 | ** | Sporulation regulatory protein WhiD and WhiB family transcriptional regulator | ||
| Bacterial cyanide production and tolerance mechanisms | 1.18 | *** | Thiosulfate sulfurtransferase | Formate dehydrogenase O alpha, beta, and gama subunit | |
| Heme and hemin uptake and utilization systems in Gram-positive bacteria | 1.12 | *** | Iron-dependent repressor | Heme-degrading monoxygenase | |
| Allophanate hydrolase 2 and biotin carboxylase cluster | 0.92 | *** | Allophanate hydrolase 2 subunit 1 (EC 3.5.1.54) | Biotin carboxylase | |
| Soluble cytochromes and functionally related electron carriers | 0.76 | *** | Ferredoxin | Cytochrome | |
| Heme and hemin uptake and utilization systems in Gram-negative bacteria | 0.55 | * | Electron transfer flavoprotein, beta subunit | Ferric siderophore transport system, periplasmic binding protein TonB, outer membrane receptor proteins, heme iron utilization protein | |
| RNA modification cluster | 0.43 | *** | Inner membrane protein translocase component YidC, long form | Inner membrane protein translocase component YidC, short form OxaI-like (YidC) | |
| Amino Acids and Derivatives | Glutamine, glutamate, aspartate, asparagine; ammonia assimilation | 0.71 | *** | Glutamine synthetases: plays an essential role in the metabolism of nitrogen | Aspartate aminotransferase, glutamate, and aspartate uptake in bacteria; glutamate dehydrogenases |
| Cell Wall and Capsule | Major component of cell wall of mycobacteria: mycolic acids, but not Gram-negative and -positive cell wall components and capsular and extracellular polysaccharides | 0.94 | *** | Antibiotics and resistance: Acyl carrier protein, linoleoyl-CoA desaturase | 3-oxoacyl-[acyl-carrier protein] reductase and synthase |
| Fatty Acids, Lipids, and Isoprenoids | Fatty acids (biosynthesis) and phospholipids | 0.34 | *** | Fatty acids: membrane; phospholipids: membrane and oxidant (fab genes) | Possibly associated with disruption of plasma membrane integrity; triacylglycerols |
| Stress Response | Heat shock | 0.37 | ** | Heat shock DNA cluster | Oxidative stress: glutathione reductase to reduce oxidized glutathione within cells, indicating cellular toxicity by Cu. |
| Oxidative stress | 0.2 | *** | Upregulation of oxidative stress response glutaredoxins, glutathione analogs: mycothiol and CoA-disulfide reductase | Down: some glutathione redox cycles | |
| Sulfur Metabolism | Inorganic sulfur assimilation | 0.45 | *** | Ferredoxin–sulfite reductase, sulfate adenylyltransferase, sulfate transporter | |
| Virulence, Disease, and Defense | Adhesion and resistance to antibiotics and toxic compounds | 0.78 | *** | Copper homeostasis (copper-translocating P-type ATPase, copper chaperone, copper resistance protein C precursor), BlaR1 family regulatory sensor transducer disambiguation, erythromycin resistance | Ferredoxin–sulfite reductase, sulfate- and thiosulfate-binding protein CysP |
| Miscellaneous | Iron–sulfur cluster assembly protein | *** | Iron–sulfur cluster assembly protein SufA, B, D, R (0.28–1.92) | Cysteine desulfurase and chaperone protein HscB and HscA | |
| Nitrogen Metabolism | Nitrate/nitrite transporter Nitrite reductase [NAD(P)H] large subunit Respiratory nitrate reductase alpha chain | 0.74 0.50 −3.54 | ** ** *** |
| Primers—> Downstream of: | MAC 1 ceoBf2/r2 | MAC 1 ceo Bf1/r1 | MAC 2 acp1 F1/R1 | 85A 3 F5/R5 | 85C 3 F3/R3 | LP 4 kupB |
|---|---|---|---|---|---|---|
| PVC: mean (Ct) | 25.28 | 25.02 | 18.92 | 30.08 | 27.04 | 32.99 |
| PVC: variance (Ct) | 0.98 | 0.83 | 1.42 | 0.58 | 0.67 | 0.59 |
| Cu: mean (Ct) | 24.43 | 24.16 | 16.86 | 30.98 | 28.62 | 35.78 |
| Cu: variance (Ct) | 0.59 | 0.49 | 0.59 | 0.82 | 0.87 | 0.27 |
| P (t-test, n = 8) | 0.06 | 0.02 | 0.01 | 0.06 | 0.00 | 0.00 |
| Fold change (2∆Ct) | 1.80 | 1.82 | 4.18 | 0.54 | 0.33 | 0.15 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, J.; Struewing, I.; Ashbolt, N.J. Meta-Transcriptomic Response to Copper Corrosion in Drinking Water Biofilms. Microorganisms 2025, 13, 1528. https://doi.org/10.3390/microorganisms13071528
Lu J, Struewing I, Ashbolt NJ. Meta-Transcriptomic Response to Copper Corrosion in Drinking Water Biofilms. Microorganisms. 2025; 13(7):1528. https://doi.org/10.3390/microorganisms13071528
Chicago/Turabian StyleLu, Jingrang, Ian Struewing, and Nicholas J. Ashbolt. 2025. "Meta-Transcriptomic Response to Copper Corrosion in Drinking Water Biofilms" Microorganisms 13, no. 7: 1528. https://doi.org/10.3390/microorganisms13071528
APA StyleLu, J., Struewing, I., & Ashbolt, N. J. (2025). Meta-Transcriptomic Response to Copper Corrosion in Drinking Water Biofilms. Microorganisms, 13(7), 1528. https://doi.org/10.3390/microorganisms13071528