
Impact of Gut Microbiome on Gut Permeability in Liver and Gut Diseases


Abstract
1. Introduction
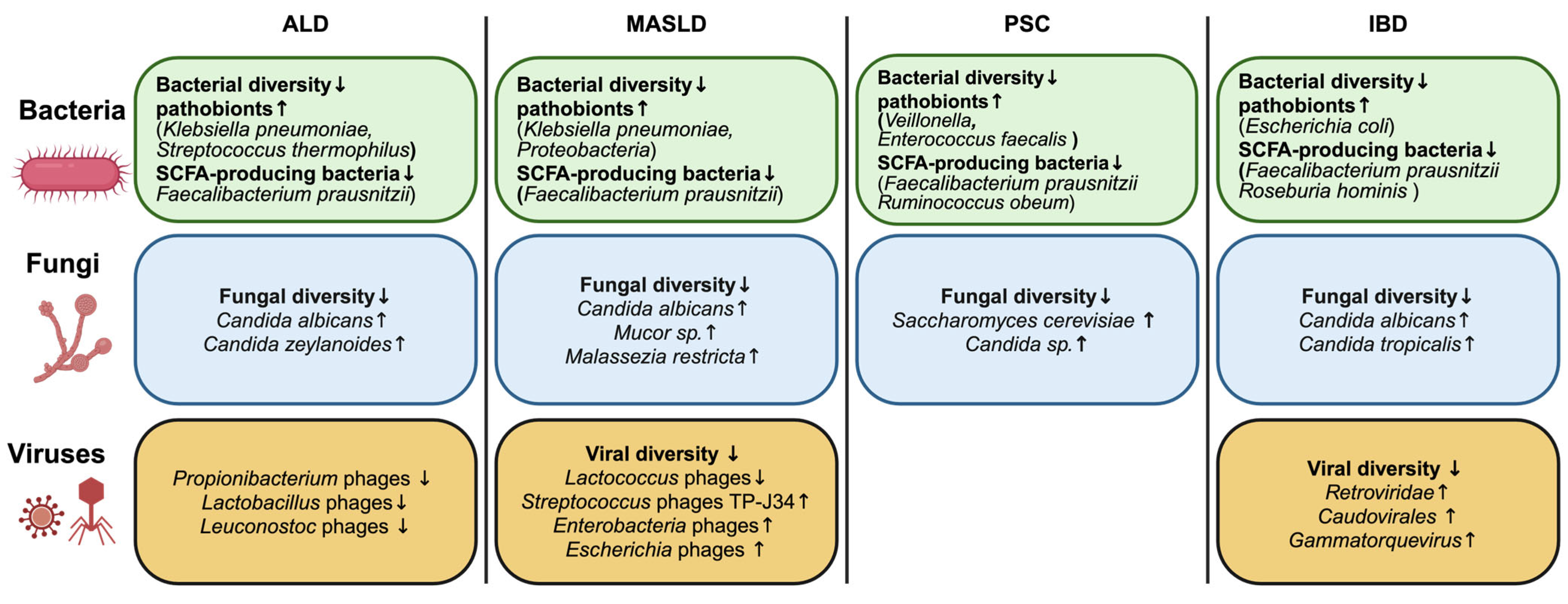
2. Changes in the Intestinal Microbiome in Liver and Intestinal Diseases
2.1. Alcohol-Associated Liver Disease
2.2. Metabolic Dysfunction-Associated Steatotic Liver Disease
2.3. Cholestatic Liver Disease
2.4. Inflammatory Bowel Disease
2.5. Common Findings
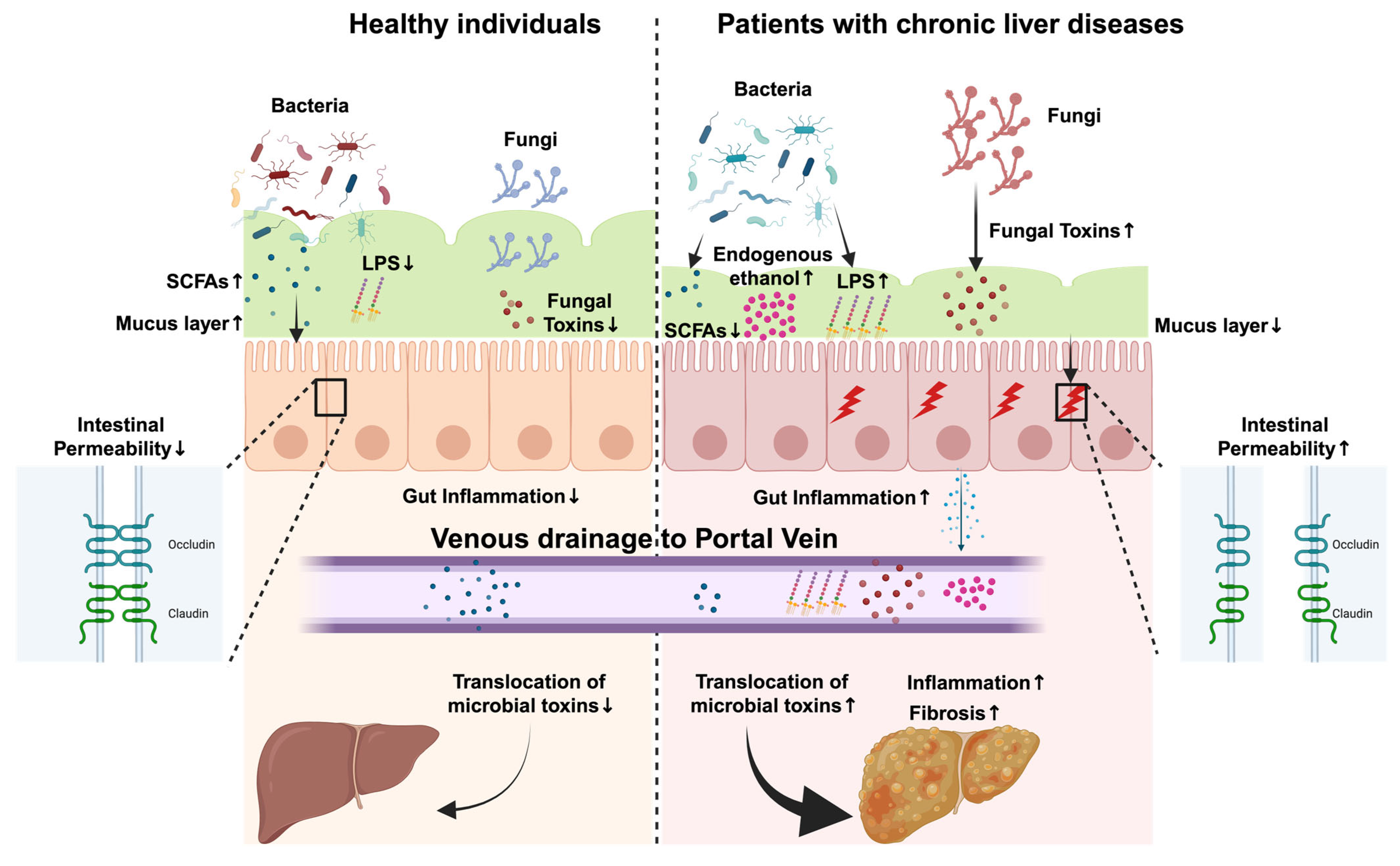
3. Role of Microbial Metabolites and Toxins in Liver and Gut Diseases
3.1. Short-Chain Fatty Acids
3.2. Lipopolysaccharides
3.3. Fungal Toxins
3.4. Endogenous Ethanol
3.5. Bile Acids
3.6. Indoles
4. Therapeutic Interventions to Alleviate Liver and Gut Diseases
4.1. Untargeted Microbiome-Based Therapeutic Approaches
4.1.1. Fecal Microbiota Transplantation
4.1.2. Prebiotics, Probiotics, Synbiotics, and Postbiotics
4.1.3. Fungi-Focused Interventions
4.2. Targeted Microbiome-Based Therapeutic Approaches
4.2.1. Farnesoid X Receptor (FXR) Agonists
4.2.2. Bioengineered Bacteria
4.2.3. Precision Editing Against Specific Gut Pathobionts
4.2.4. Phage Therapy

{kind=link}
{kind=link}
| Type of Intervention | Disease Type | Intervention Details | Outcome | Reference |
|---|---|---|---|---|
| Fecal microbiota transplantion | MASLD | RCT in 21 adult patients with MASLD; follow-up at 2 weeks, 6 weeks, and 6 months post-FMT. |
| Craven et al. [159] |
| MASLD | RCT in 75 adult patients with MASLD, who were divided into non-FMT (28 patients) and FMT (47 patients) groups; follow-up after 1 month. |
| Xue et al. [160] | |
| Probiotics | ALD | Meta-analysis of 9 randomized controlled trials from 2008 to 2023 (n = 639) |
| Xiong et al. [174] |
| Probiotics, Prebiotics, and Synbiotics | MASLD | Meta-analysis of 34 randomized controlled trials until March 2024 (n = 12,682) |
| Pan et al. [180] |
| Probiotics | PSC–IBD | 14 adult patients with PSC–IBD treated with probiotics comprising six strains for 3 months |
| Vleggaar et al. [181] |
| FXR agonists | MASLD | Randomized clinical trial in adult patients with non-cirrhotic MASH; obeticholic acid (25 mg daily) or the placebo was given orally for 72 weeks. |
| Neuschwander-Tetri et al. [205] |
| PSC | Randomized clinical trial in 76 adult patients with PSC with a placebo or 2 doses of obeticholic acid once daily for 24 weeks; followed by a 2-year, long-term safety extension. |
| Kowdley et al. [207] | |
| ALD | Evaluation of the FXR agonist fexaramine in mice with chronic alcohol-induced liver disease. |
| Hartmann et al. [214] | |
| Bioengineered bacteria | ALD | Bioengineered Lactobacillus reuteri strain in mice with chronic alcohol-induced liver disease. |
| Hendrikx, T., et al. [155] |
| ALD | Bioengineered Escherichia coli Nissle 1917 strain (EcN-Ahr) in mice with chronic alcohol-induced liver disease. |
| Kouno et al. [218] | |
| Precision editing of the gut microbiome | IBD | Inhibition of microbial respiratory pathways by tungstate in a murine model of colitis. |
| Zhu et al. [219] |
| Fungi-focused interventions | ALD | Oral administration of the antifungal amphotericin B in alcohol-induced liver disease. |
| Yang et al. [120] |
| MASH | Oral administration of amphotericin B in Western-diet-fed germ-free mice (transplanted with cells from patients with steatohepatitis). |
| Demir et al. [32] | |
| IBD | Oral fluconazole therapy (200 mg daily for 3 weeks) in 68 Candida-positive adult patients with UC; follow-up after 4 weeks. |
| Jena et al. [193] | |
| Phage therapy | ALD | Bacteriophage treatment to target cytolysin-positive Enterococcus faecalis in murine model of alcohol-induced steatohepatitis. |
| Duan et al. [220] |
| MASLD | Phage therapy against alcohol-producing Klebsiella pneumoniae in MASLD. |
| Gan et al. [221] | |
| PSC | Phage cocktail treatment in gnotobiotic mice (transplanted with Klebsiella pneumoniae). |
| Ichikawa et al. [222] |
5. Existing Challenges and Future Directions
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Moon, A.M.; Singal, A.G.; Tapper, E.B. Contemporary Epidemiology of Chronic Liver Disease and Cirrhosis. Clin. Gastroenterol. Hepatol. 2020, 18, 2650–2666. [Google Scholar] [CrossRef] [PubMed]
- Adolph, T.E.; Meyer, M.; Jukic, A.; Tilg, H. Heavy arch: From inflammatory bowel diseases to metabolic disorders. Gut 2024, 73, 1376–1387. [Google Scholar] [CrossRef]
- Devarbhavi, H.; Asrani, S.K.; Arab, J.P.; Nartey, Y.A.; Pose, E.; Kamath, P.S. Global burden of liver disease: 2023 update. J. Hepatol. 2023, 79, 516–537. [Google Scholar] [CrossRef]
- Curtin, S.C.; Tejada-Vera, B.; Bastian, B.A. Deaths: Leading Causes for 2020. Natl. Vital. Stat. Rep. 2023, 72, 1–115. [Google Scholar]
- Yarur, A.J.; Czul, F.; Levy, C. Hepatobiliary manifestations of inflammatory bowel disease. Inflamm. Bowel Dis. 2014, 20, 1655–1667. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Li, Z.; Liu, S.; Zhang, D. Global, regional and national burden of inflammatory bowel disease in 204 countries and territories from 1990 to 2019: A systematic analysis based on the Global Burden of Disease Study 2019. BMJ Open 2023, 13, e065186. [Google Scholar] [CrossRef]
- Gaspar, R.; Branco, C.C.; Macedo, G. Liver manifestations and complications in inflammatory bowel disease: A review. World J. Hepatol. 2021, 13, 1956–1967. [Google Scholar] [CrossRef]
- Palmela, C.; Peerani, F.; Castaneda, D.; Torres, J.; Itzkowitz, S.H. Inflammatory Bowel Disease and Primary Sclerosing Cholangitis: A Review of the Phenotype and Associated Specific Features. Gut Liver 2018, 12, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Loftus, E.V.; Harewood, G.C.; Loftus, C.G.; Tremaine, W.J.; Harmsen, W.S.; Zinsmeister, A.R.; Jewell, D.A.; Sandborn, W.J. PSC-IBD: A unique form of inflammatory bowel disease associated with primary sclerosing cholangitis. Gut 2005, 54, 91–96. [Google Scholar] [CrossRef]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef]
- Patrignani, P.; Tacconelli, S.; Bruno, A. Gut microbiota, host gene expression, and aging. J. Clin. Gastroenterol. 2014, 48 (Suppl. S1), S28–S31. [Google Scholar] [CrossRef] [PubMed]
- Voreades, N.; Kozil, A.; Weir, T.L. Diet and the development of the human intestinal microbiome. Front. Microbiol. 2014, 5, 494. [Google Scholar] [CrossRef] [PubMed]
- Saha, P.; Bose, D.; Stebliankin, V.; Cickovski, T.; Seth, R.K.; Porter, D.E.; Brooks, B.W.; Mathee, K.; Narasimhan, G.; Colwell, R.; et al. Prior exposure to microcystin alters host gut resistome and is associated with dysregulated immune homeostasis in translatable mouse models. Sci. Rep. 2022, 12, 11516. [Google Scholar] [CrossRef] [PubMed]
- Zuniga-Chaves, I.; Eggers, S.; Kates, A.E.; Safdar, N.; Suen, G.; Malecki, K.M.C. Neighborhood socioeconomic status is associated with low diversity gut microbiomes and multi-drug resistant microorganism colonization. NPJ Biofilms Microbiomes 2023, 9, 61. [Google Scholar] [CrossRef] [PubMed]
- Jandhyala, S.M.; Talukdar, R.; Subramanyam, C.; Vuyyuru, H.; Sasikala, M.; Nageshwar Reddy, D. Role of the normal gut microbiota. World J. Gastroenterol. 2015, 21, 8787–8803. [Google Scholar] [CrossRef]
- Hartmann, P.; Schnabl, B. Risk factors for progression of and treatment options for NAFLD in children. Clin. Liver Dis. 2018, 11, 11–15. [Google Scholar] [CrossRef]
- Hartmann, P.; Schnabl, B. New Developments in Microbiome in Alcohol-Associated and Nonalcoholic Fatty Liver Disease. Semin. Liver Dis. 2021, 41, 87–102. [Google Scholar] [CrossRef]
- Kaufmann, B.; Seyfried, N.; Hartmann, D.; Hartmann, P. Probiotics, prebiotics, and synbiotics in nonalcoholic fatty liver disease and alcohol-associated liver disease. Am. J. Physiol. Gastrointest. Liver Physiol. 2023, 325, G42–G61. [Google Scholar] [CrossRef]
- Mutlu, E.A.; Gillevet, P.M.; Rangwala, H.; Sikaroodi, M.; Naqvi, A.; Engen, P.A.; Kwasny, M.; Lau, C.K.; Keshavarzian, A. Colonic microbiome is altered in alcoholism. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G966–G978. [Google Scholar] [CrossRef]
- Dubinkina, V.B.; Tyakht, A.V.; Odintsova, V.Y.; Yarygin, K.S.; Kovarsky, B.A.; Pavlenko, A.V.; Ischenko, D.S.; Popenko, A.S.; Alexeev, D.G.; Taraskina, A.Y.; et al. Links of gut microbiota composition with alcohol dependence syndrome and alcoholic liver disease. Microbiome 2017, 5, 141. [Google Scholar] [CrossRef]
- Litwinowicz, K.; Gamian, A. Microbiome Alterations in Alcohol Use Disorder and Alcoholic Liver Disease. Int. J. Mol. Sci. 2023, 24, 2461. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, P.; Schnabl, B. Fungal infections and the fungal microbiome in hepatobiliary disorders. J. Hepatol. 2023, 78, 836–851. [Google Scholar] [CrossRef] [PubMed]
- Lang, S.; Duan, Y.; Liu, J.; Torralba, M.G.; Kuelbs, C.; Ventura-Cots, M.; Abraldes, J.G.; Bosques-Padilla, F.; Verna, E.C.; Brown, R.S.; et al. Intestinal Fungal Dysbiosis and Systemic Immune Response to Fungi in Patients with Alcoholic Hepatitis. Hepatology 2020, 71, 522–538. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, P.; Lang, S.; Zeng, S.; Duan, Y.; Zhang, X.; Wang, Y.; Bondareva, M.; Kruglov, A.; Fouts, D.E.; Stärkel, P.; et al. Dynamic Changes of the Fungal Microbiome in Alcohol Use Disorder. Front. Physiol. 2021, 12, 699253. [Google Scholar] [CrossRef]
- Hsu, C.L.; Zhang, X.; Jiang, L.; Lang, S.; Hartmann, P.; Pride, D.; Fouts, D.E.; Stärkel, P.; Schnabl, B. Intestinal virome in patients with alcohol use disorder and after abstinence. Hepatol. Commun. 2022, 6, 2058–2069. [Google Scholar] [CrossRef] [PubMed]
- Rinella, M.E.; Lazarus, J.V.; Ratziu, V.; Francque, S.M.; Sanyal, A.J.; Kanwal, F.; Romero, D.; Abdelmalek, M.F.; Anstee, Q.M.; Arab, J.P.; et al. A multisociety Delphi consensus statement on new fatty liver disease nomenclature. Ann. Hepatol. 2024, 29, 101133. [Google Scholar] [CrossRef]
- Del Chierico, F.; Nobili, V.; Vernocchi, P.; Russo, A.; De Stefanis, C.; Gnani, D.; Furlanello, C.; Zandonà, A.; Paci, P.; Capuani, G.; et al. Gut microbiota profiling of pediatric nonalcoholic fatty liver disease and obese patients unveiled by an integrated meta-omics-based approach. Hepatology 2017, 65, 451–464. [Google Scholar] [CrossRef]
- Wang, B.; Jiang, X.; Cao, M.; Ge, J.; Bao, Q.; Tang, L.; Chen, Y.; Li, L. Altered Fecal Microbiota Correlates with Liver Biochemistry in Nonobese Patients with Non-alcoholic Fatty Liver Disease. Sci. Rep. 2016, 6, 32002. [Google Scholar] [CrossRef] [PubMed]
- Schwimmer, J.B.; Johnson, J.S.; Angeles, J.E.; Behling, C.; Belt, P.H.; Borecki, I.; Bross, C.; Durelle, J.; Goyal, N.P.; Hamilton, G.; et al. Microbiome Signatures Associated with Steatohepatitis and Moderate to Severe Fibrosis in Children with Nonalcoholic Fatty Liver Disease. Gastroenterology 2019, 157, 1109–1122. [Google Scholar] [CrossRef]
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2017, 25, 1054–1062.e1055. [Google Scholar] [CrossRef]
- Yang, C.; Wu, J.; Yang, L.; Hu, Q.; Li, L.; Yang, Y.; Hu, J.; Pan, D.; Zhao, Q. Altered gut microbial profile accompanied by abnormal short chain fatty acid metabolism exacerbates nonalcoholic fatty liver disease progression. Sci. Rep. 2024, 14, 22385. [Google Scholar] [CrossRef] [PubMed]
- Demir, M.; Lang, S.; Hartmann, P.; Duan, Y.; Martin, A.; Miyamoto, Y.; Bondareva, M.; Zhang, X.; Wang, Y.; Kasper, P.; et al. The fecal mycobiome in non-alcoholic fatty liver disease. J. Hepatol. 2022, 76, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Viebahn, G.; Hartmann, P.; Lang, S.; Demir, M.; Zhang, X.; Fouts, D.E.; Stärkel, P.; Schnabl, B. Fungal signature differentiates alcohol-associated liver disease from nonalcoholic fatty liver disease. Gut Microbes 2024, 16, 2307586. [Google Scholar] [CrossRef]
- Lang, S.; Demir, M.; Martin, A.; Jiang, L.; Zhang, X.; Duan, Y.; Gao, B.; Wisplinghoff, H.; Kasper, P.; Roderburg, C.; et al. Intestinal Virome Signature Associated with Severity of Nonalcoholic Fatty Liver Disease. Gastroenterology 2020, 159, 1839–1852. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of cholestatic liver diseases. J. Hepatol. 2009, 51, 237–267. [Google Scholar] [CrossRef]
- Manns, M.P.; Bergquist, A.; Karlsen, T.H.; Levy, C.; Muir, A.J.; Ponsioen, C.; Trauner, M.; Wong, G.; Younossi, Z.M. Primary sclerosing cholangitis. Nat. Rev. Dis. Primers 2025, 11, 17. [Google Scholar] [CrossRef]
- Kummen, M.; Holm, K.; Anmarkrud, J.A.; Nygård, S.; Vesterhus, M.; Høivik, M.L.; Trøseid, M.; Marschall, H.U.; Schrumpf, E.; Moum, B.; et al. The gut microbial profile in patients with primary sclerosing cholangitis is distinct from patients with ulcerative colitis without biliary disease and healthy controls. Gut 2017, 66, 611–619. [Google Scholar] [CrossRef]
- Bajer, L.; Kverka, M.; Kostovcik, M.; Macinga, P.; Dvorak, J.; Stehlikova, Z.; Brezina, J.; Wohl, P.; Spicak, J.; Drastich, P. Distinct gut microbiota profiles in patients with primary sclerosing cholangitis and ulcerative colitis. World J. Gastroenterol. 2017, 23, 4548–4558. [Google Scholar] [CrossRef]
- Sabino, J.; Vieira-Silva, S.; Machiels, K.; Joossens, M.; Falony, G.; Ballet, V.; Ferrante, M.; Van Assche, G.; Van der Merwe, S.; Vermeire, S.; et al. Primary sclerosing cholangitis is characterised by intestinal dysbiosis independent from IBD. Gut 2016, 65, 1681–1689. [Google Scholar] [CrossRef]
- Rühlemann, M.; Liwinski, T.; Heinsen, F.A.; Bang, C.; Zenouzi, R.; Kummen, M.; Thingholm, L.; Tempel, M.; Lieb, W.; Karlsen, T.; et al. Consistent alterations in faecal microbiomes of patients with primary sclerosing cholangitis independent of associated colitis. Aliment. Pharmacol. Ther. 2019, 50, 580–589. [Google Scholar] [CrossRef]
- Iwasawa, K.; Suda, W.; Tsunoda, T.; Oikawa-Kawamoto, M.; Umetsu, S.; Inui, A.; Fujisawa, T.; Morita, H.; Sogo, T.; Hattori, M. Characterisation of the faecal microbiota in Japanese patients with paediatric-onset primary sclerosing cholangitis. Gut 2017, 66, 1344–1346. [Google Scholar] [CrossRef] [PubMed]
- Özdirik, B.; Berger, H.; Tonetti, F.R.; Cabré, N.; Treichel, N.; Clavel, T.; Tacke, F.; Sigal, M.; Schnabl, B. Faecal Cytolysin is Associated with Worse Survival in Patients with Primary Sclerosing Cholangitis. Liver Int. 2025, 45, e16181. [Google Scholar] [CrossRef] [PubMed]
- Lemoinne, S.; Kemgang, A.; Ben Belkacem, K.; Straube, M.; Jegou, S.; Corpechot, C.; Chazouillères, O.; Housset, C.; Sokol, H.; Network, S.-A.I. Fungi participate in the dysbiosis of gut microbiota in patients with primary sclerosing cholangitis. Gut 2020, 69, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Rühlemann, M.C.; Solovjeva, M.E.L.; Zenouzi, R.; Liwinski, T.; Kummen, M.; Lieb, W.; Hov, J.R.; Schramm, C.; Franke, A.; Bang, C. Gut mycobiome of primary sclerosing cholangitis patients is characterised by an increase of. Gut 2020, 69, 1890–1892. [Google Scholar] [CrossRef]
- Del Chierico, F.; Cardile, S.; Baldelli, V.; Alterio, T.; Reddel, S.; Bramuzzo, M.; Knafelz, D.; Lega, S.; Bracci, F.; Torre, G.; et al. Characterization of the Gut Microbiota and Mycobiota in Italian Pediatric Patients with Primary Sclerosing Cholangitis and Ulcerative Colitis. Inflamm. Bowel Dis. 2024, 30, 529–537. [Google Scholar] [CrossRef]
- Glassner, K.L.; Abraham, B.P.; Quigley, E.M.M. The microbiome and inflammatory bowel disease. J. Allergy Clin. Immunol. 2020, 145, 16–27. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef]
- Nagalingam, N.A.; Lynch, S.V. Role of the microbiota in inflammatory bowel diseases. Inflamm. Bowel Dis. 2012, 18, 968–984. [Google Scholar] [CrossRef]
- Lucas López, R.; Grande Burgos, M.J.; Gálvez, A.; Pérez Pulido, R. The human gastrointestinal tract and oral microbiota in inflammatory bowel disease: A state of the science review. APMIS J. Pathol. Microbiol. Immunol. 2017, 125, 3–10. [Google Scholar] [CrossRef]
- Pascal, V.; Pozuelo, M.; Borruel, N.; Casellas, F.; Campos, D.; Santiago, A.; Martinez, X.; Varela, E.; Sarrabayrouse, G.; Machiels, K.; et al. A microbial signature for Crohn’s disease. Gut 2017, 66, 813–822. [Google Scholar] [CrossRef]
- Ma, H.Q.; Yu, T.T.; Zhao, X.J.; Zhang, Y.; Zhang, H.J. Fecal microbial dysbiosis in Chinese patients with inflammatory bowel disease. World J. Gastroenterol. 2018, 24, 1464–1477. [Google Scholar] [CrossRef]
- Teofani, A.; Marafini, I.; Laudisi, F.; Pietrucci, D.; Salvatori, S.; Unida, V.; Biocca, S.; Monteleone, G.; Desideri, A. Intestinal Taxa Abundance and Diversity in Inflammatory Bowel Disease Patients: An Analysis including Covariates and Confounders. Nutrients 2022, 14, 260. [Google Scholar] [CrossRef] [PubMed]
- Halfvarson, J.; Brislawn, C.J.; Lamendella, R.; Vázquez-Baeza, Y.; Walters, W.A.; Bramer, L.M.; D’Amato, M.; Bonfiglio, F.; McDonald, D.; Gonzalez, A.; et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat. Microbiol. 2017, 2, 17004. [Google Scholar] [CrossRef]
- Lloyd-Price, J.; Arze, C.; Ananthakrishnan, A.N.; Schirmer, M.; Avila-Pacheco, J.; Poon, T.W.; Andrews, E.; Ajami, N.J.; Bonham, K.S.; Brislawn, C.J.; et al. Multi-omics of the gut microbial ecosystem in inflammatory bowel diseases. Nature 2019, 569, 655–662. [Google Scholar] [CrossRef]
- Chehoud, C.; Albenberg, L.G.; Judge, C.; Hoffmann, C.; Grunberg, S.; Bittinger, K.; Baldassano, R.N.; Lewis, J.D.; Bushman, F.D.; Wu, G.D. Fungal Signature in the Gut Microbiota of Pediatric Patients with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2015, 21, 1948–1956. [Google Scholar] [CrossRef]
- Hoarau, G.; Mukherjee, P.K.; Gower-Rousseau, C.; Hager, C.; Chandra, J.; Retuerto, M.A.; Neut, C.; Vermeire, S.; Clemente, J.; Colombel, J.F.; et al. Bacteriome and Mycobiome Interactions Underscore Microbial Dysbiosis in Familial Crohn’s Disease. mBio 2016, 7, e01250-16. [Google Scholar] [CrossRef] [PubMed]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal microbiota dysbiosis in IBD. Gut 2017, 66, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef]
- Jangi, S.; Hsia, K.; Zhao, N.; Kumamoto, C.A.; Friedman, S.; Singh, S.; Michaud, D.S. Dynamics of the Gut Mycobiome in Patients with Ulcerative Colitis. Clin. Gastroenterol. Hepatol. 2024, 22, 821–830.e827. [Google Scholar] [CrossRef]
- Ungaro, F.; Massimino, L.; D’Alessio, S.; Danese, S. The gut virome in inflammatory bowel disease pathogenesis: From metagenomics to novel therapeutic approaches. United Eur. Gastroenterol. J. 2019, 7, 999–1007. [Google Scholar] [CrossRef]
- Tun, H.M.; Peng, Y.; Massimino, L.; Sin, Z.Y.; Parigi, T.L.; Facoetti, A.; Rahman, S.; Danese, S.; Ungaro, F. Gut virome in inflammatory bowel disease and beyond. Gut 2024, 73, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Clooney, A.G.; Sutton, T.D.S.; Shkoporov, A.N.; Holohan, R.K.; Daly, K.M.; O’Regan, O.; Ryan, F.J.; Draper, L.A.; Plevy, S.E.; Ross, R.P.; et al. Whole-Virome Analysis Sheds Light on Viral Dark Matter in Inflammatory Bowel Disease. Cell Host Microbe 2019, 26, 764–778.e765. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Brocal, V.; García-López, R.; Nos, P.; Beltrán, B.; Moret, I.; Moya, A. Metagenomic Analysis of Crohn’s Disease Patients Identifies Changes in the Virome and Microbiome Related to Disease Status and Therapy, and Detects Potential Interactions and Biomarkers. Inflamm. Bowel Dis. 2015, 21, 2515–2532. [Google Scholar] [CrossRef]
- Fernandes, M.A.; Verstraete, S.G.; Phan, T.G.; Deng, X.; Stekol, E.; LaMere, B.; Lynch, S.V.; Heyman, M.B.; Delwart, E. Enteric Virome and Bacterial Microbiota in Children with Ulcerative Colitis and Crohn Disease. J. Pediatr. Gastroenterol. Nutr. 2019, 68, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Zuo, T.; Lu, X.J.; Zhang, Y.; Cheung, C.P.; Lam, S.; Zhang, F.; Tang, W.; Ching, J.Y.L.; Zhao, R.; Chan, P.K.S.; et al. Gut mucosal virome alterations in ulcerative colitis. Gut 2019, 68, 1169–1179. [Google Scholar] [CrossRef]
- Cao, Z.; Fan, D.; Sun, Y.; Huang, Z.; Li, Y.; Su, R.; Zhang, F.; Li, Q.; Yang, H.; Miao, Y.; et al. The gut ileal mucosal virome is disturbed in patients with Crohn’s disease and exacerbates intestinal inflammation in mice. Nat. Commun. 2024, 15, 1638. [Google Scholar] [CrossRef]
- Li, R.; Mao, Z.; Ye, X.; Zuo, T. Human Gut Microbiome and Liver Diseases: From Correlation to Causation. Microorganisms 2021, 9, 1017. [Google Scholar] [CrossRef]
- Wang, L.; Cao, Z.M.; Zhang, L.L.; Li, J.M.; Lv, W.L. The Role of Gut Microbiota in Some Liver Diseases: From an Immunological Perspective. Front. Immunol. 2022, 13, 923599. [Google Scholar] [CrossRef]
- Hsu, C.L.; Duan, Y.; Fouts, D.E.; Schnabl, B. Intestinal virome and therapeutic potential of bacteriophages in liver disease. J. Hepatol. 2021, 75, 1465–1475. [Google Scholar] [CrossRef]
- An, L.; Wirth, U.; Koch, D.; Schirren, M.; Drefs, M.; Koliogiannis, D.; Nieß, H.; Andrassy, J.; Guba, M.; Bazhin, A.V.; et al. The Role of Gut-Derived Lipopolysaccharides and the Intestinal Barrier in Fatty Liver Diseases. J. Gastrointest. Surg. 2022, 26, 671–683. [Google Scholar] [CrossRef]
- Pant, K.; Venugopal, S.K.; Lorenzo Pisarello, M.J.; Gradilone, S.A. The Role of Gut Microbiome-Derived Short-Chain Fatty Acid Butyrate in Hepatobiliary Diseases. Am. J. Pathol. 2023, 193, 1455–1467. [Google Scholar] [CrossRef] [PubMed]
- Visekruna, A.; Luu, M. The Role of Short-Chain Fatty Acids and Bile Acids in Intestinal and Liver Function, Inflammation, and Carcinogenesis. Front. Cell Dev. Biol. 2021, 9, 703218. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Luo, X.; Tang, J.; Mo, Q.; Zhong, H.; Zhang, H.; Feng, F. A bridge for short-chain fatty acids to affect inflammatory bowel disease, type 1 diabetes, and non-alcoholic fatty liver disease positively: By changing gut barrier. Eur. J. Nutr. 2021, 60, 2317–2330. [Google Scholar] [CrossRef] [PubMed]
- Meijnikman, A.S.; Nieuwdorp, M.; Schnabl, B. Endogenous ethanol production in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2024, 21, 556–571. [Google Scholar] [CrossRef]
- Jirillo, E.; Caccavo, D.; Magrone, T.; Piccigallo, E.; Amati, L.; Lembo, A.; Kalis, C.; Gumenscheimer, M. The role of the liver in the response to LPS: Experimental and clinical findings. J. Endotoxin Res. 2002, 8, 319–327. [Google Scholar] [CrossRef]
- Männistö, V.; Färkkilä, M.; Pussinen, P.; Jula, A.; Männistö, S.; Lundqvist, A.; Valsta, L.; Salomaa, V.; Perola, M.; Åberg, F. Serum lipopolysaccharides predict advanced liver disease in the general population. JHEP Rep. 2019, 1, 345–352. [Google Scholar] [CrossRef]
- Jaeger, J.W.; Brandt, A.; Gui, W.; Yergaliyev, T.; Hernández-Arriaga, A.; Muthu, M.M.; Edlund, K.; Elashy, A.; Molinaro, A.; Möckel, D.; et al. Microbiota modulation by dietary oat beta-glucan prevents steatotic liver disease progression. JHEP Rep. 2024, 6, 100987. [Google Scholar] [CrossRef]
- Farias E Silva, K.; Nanini, H.F.; Cascabulho, C.M.; Rosas, S.L.B.; Santana, P.T.; Carneiro, A.J.V.; Anaissie, E.; Nucci, M.; de Souza, H.S.P. Serum 1,3-beta-D-glucan as a noninvasive test to predict histologic activity in patients with inflammatory bowel disease. World J. Gastroenterol. 2021, 27, 866–885. [Google Scholar] [CrossRef]
- Chu, H.; Duan, Y.; Lang, S.; Jiang, L.; Wang, Y.; Llorente, C.; Liu, J.; Mogavero, S.; Bosques-Padilla, F.; Abraldes, J.G.; et al. The Candida albicans exotoxin candidalysin promotes alcohol-associated liver disease. J. Hepatol. 2020, 72, 391–400. [Google Scholar] [CrossRef]
- den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef]
- Cummings, J.H.; Pomare, E.W.; Branch, W.J.; Naylor, C.P.; Macfarlane, G.T. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 1987, 28, 1221–1227. [Google Scholar] [CrossRef] [PubMed]
- Donohoe, D.R.; Garge, N.; Zhang, X.; Sun, W.; O’Connell, T.M.; Bunger, M.K.; Bultman, S.J. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011, 13, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Roediger, W.E. Utilization of nutrients by isolated epithelial cells of the rat colon. Gastroenterology 1982, 83, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.H.; Park, J.; Kim, M. Gut microbiota-derived short-chain Fatty acids, T cells, and inflammation. Immune Netw. 2014, 14, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Lin, J.; Zhang, C.; Gao, H.; Lu, H.; Gao, X.; Zhu, R.; Li, Z.; Li, M.; Liu, Z. Microbiota metabolite butyrate constrains neutrophil functions and ameliorates mucosal inflammation in inflammatory bowel disease. Gut Microbes 2021, 13, 1968257. [Google Scholar] [CrossRef]
- Singh, V.; Lee, G.; Son, H.; Koh, H.; Kim, E.S.; Unno, T.; Shin, J.H. Butyrate producers, “The Sentinel of Gut”: Their intestinal significance with and beyond butyrate, and prospective use as microbial therapeutics. Front. Microbiol. 2022, 13, 1103836. [Google Scholar] [CrossRef]
- Rey, F.E.; Faith, J.J.; Bain, J.; Muehlbauer, M.J.; Stevens, R.D.; Newgard, C.B.; Gordon, J.I. Dissecting the in vivo metabolic potential of two human gut acetogens. J. Biol. Chem. 2010, 285, 22082–22090. [Google Scholar] [CrossRef]
- Hosseini, E.; Grootaert, C.; Verstraete, W.; Van de Wiele, T. Propionate as a health-promoting microbial metabolite in the human gut. Nutr. Rev. 2011, 69, 245–258. [Google Scholar] [CrossRef]
- Cao, X.; Zolnikova, O.; Maslennikov, R.; Reshetova, M.; Poluektova, E.; Bogacheva, A.; Zharkova, M.; Ivashkin, V. Differences in Fecal Short-Chain Fatty Acids between Alcoholic Fatty Liver-Induced Cirrhosis and Non-alcoholic (Metabolic-Associated) Fatty Liver-Induced Cirrhosis. Metabolites 2023, 13, 859. [Google Scholar] [CrossRef]
- Pohl, K.; Moodley, P.; Dhanda, A. The effect of increasing intestinal short-chain fatty acid concentration on gut permeability and liver injury in the context of liver disease: A systematic review. J. Gastroenterol. Hepatol. 2022, 37, 1498–1506. [Google Scholar] [CrossRef]
- Cresci, G.A.; Bush, K.; Nagy, L.E. Tributyrin supplementation protects mice from acute ethanol-induced gut injury. Alcohol Clin. Exp. Res. 2014, 38, 1489–1501. [Google Scholar] [CrossRef] [PubMed]
- Cresci, G.A.; Glueck, B.; McMullen, M.R.; Xin, W.; Allende, D.; Nagy, L.E. Prophylactic tributyrin treatment mitigates chronic-binge ethanol-induced intestinal barrier and liver injury. J. Gastroenterol. Hepatol. 2017, 32, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Zhang, R.; Mu, Y.; Song, Y.; Hao, N.; Wei, Y.; Wang, Q.; Mackay, C.R. Propionate Ameliorates Alcohol-Induced Liver Injury in Mice via the Gut-Liver Axis: Focus on the Improvement of Intestinal Permeability. J. Agric. Food Chem. 2022, 70, 6084–6096. [Google Scholar] [CrossRef] [PubMed]
- Jin, C.J.; Sellmann, C.; Engstler, A.J.; Ziegenhardt, D.; Bergheim, I. Supplementation of sodium butyrate protects mice from the development of non-alcoholic steatohepatitis (NASH). Br. J. Nutr. 2015, 114, 1745–1755. [Google Scholar] [CrossRef]
- Jin, C.J.; Engstler, A.J.; Sellmann, C.; Ziegenhardt, D.; Landmann, M.; Kanuri, G.; Lounis, H.; Schröder, M.; Vetter, W.; Bergheim, I. Sodium butyrate protects mice from the development of the early signs of non-alcoholic fatty liver disease: Role of melatonin and lipid peroxidation. Br. J. Nutr. 2016, 116, 1682–1693. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; He, M.; Yi, X.; Lu, X.; Zhu, M.; Xue, M.; Tang, Y.; Zhu, Y. Short-chain fatty acids in nonalcoholic fatty liver disease: New prospects for short-chain fatty acids as therapeutic targets. Heliyon 2024, 10, e26991. [Google Scholar] [CrossRef]
- Luther, J.; Garber, J.J.; Khalili, H.; Dave, M.; Bale, S.S.; Jindal, R.; Motola, D.L.; Luther, S.; Bohr, S.; Jeoung, S.W.; et al. Hepatic Injury in Nonalcoholic Steatohepatitis Contributes to Altered Intestinal Permeability. Cell Mol. Gastroenterol. Hepatol. 2015, 1, 222–232. [Google Scholar] [CrossRef]
- Dai, X.; Hou, H.; Zhang, W.; Liu, T.; Li, Y.; Wang, S.; Wang, B.; Cao, H. Microbial Metabolites: Critical Regulators in NAFLD. Front. Microbiol. 2020, 11, 567654. [Google Scholar] [CrossRef]
- Wang, P.; Li, T.; Niu, C.; Sun, S.; Liu, D. ROS-activated MAPK/ERK pathway regulates crosstalk between Nrf2 and Hif-1α to promote IL-17D expression protecting the intestinal epithelial barrier under hyperoxia. Int. Immunopharmacol. 2023, 116, 109763. [Google Scholar] [CrossRef]
- Yan, H.; Ajuwon, K.M. Butyrate modifies intestinal barrier function in IPEC-J2 cells through a selective upregulation of tight junction proteins and activation of the Akt signaling pathway. PLoS ONE 2017, 12, e0179586. [Google Scholar] [CrossRef]
- Macia, L.; Tan, J.; Vieira, A.T.; Leach, K.; Stanley, D.; Luong, S.; Maruya, M.; Ian McKenzie, C.; Hijikata, A.; Wong, C.; et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat. Commun. 2015, 6, 6734. [Google Scholar] [CrossRef] [PubMed]
- Skelly, A.N.; Sato, Y.; Kearney, S.; Honda, K. Mining the microbiota for microbial and metabolite-based immunotherapies. Nat. Rev. Immunol. 2019, 19, 305–323. [Google Scholar] [CrossRef]
- Endo, H.; Niioka, M.; Kobayashi, N.; Tanaka, M.; Watanabe, T. Butyrate-producing probiotics reduce nonalcoholic fatty liver disease progression in rats: New insight into the probiotics for the gut-liver axis. PLoS ONE 2013, 8, e63388. [Google Scholar] [CrossRef]
- Little, R.; Wine, E.; Kamath, B.M.; Griffiths, A.M.; Ricciuto, A. Gut microbiome in primary sclerosing cholangitis: A review. World J. Gastroenterol. 2020, 26, 2768–2780. [Google Scholar] [CrossRef] [PubMed]
- Özdirik, B.; Schnabl, B. Microbial Players in Primary Sclerosing Cholangitis: Current Evidence and Concepts. Cell Mol. Gastroenterol. Hepatol. 2024, 17, 423–438. [Google Scholar] [CrossRef]
- Parada Venegas, D.; De la Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef]
- Zhuang, X.; Li, T.; Li, M.; Huang, S.; Qiu, Y.; Feng, R.; Zhang, S.; Chen, M.; Xiong, L.; Zeng, Z. Systematic Review and Meta-analysis: Short-Chain Fatty Acid Characterization in Patients with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2019, 25, 1751–1763. [Google Scholar] [CrossRef]
- Fujimoto, M.; Uemura, M.; Nakatani, Y.; Tsujita, S.; Hoppo, K.; Tamagawa, T.; Kitano, H.; Kikukawa, M.; Ann, T.; Ishii, Y.; et al. Plasma endotoxin and serum cytokine levels in patients with alcoholic hepatitis: Relation to severity of liver disturbance. Alcohol. Clin. Exp. Res. 2000, 24, 48S–54S. [Google Scholar] [CrossRef]
- Schäfer, C.; Parlesak, A.; Schütt, C.; Bode, J.C.; Bode, C. Concentrations of lipopolysaccharide-binding protein, bactericidal/permeability-increasing protein, soluble CD14 and plasma lipids in relation to endotoxaemia in patients with alcoholic liver disease. Alcohol Alcohol. 2002, 37, 81–86. [Google Scholar] [CrossRef]
- Parlesak, A.; Schäfer, C.; Schütz, T.; Bode, J.C.; Bode, C. Increased intestinal permeability to macromolecules and endotoxemia in patients with chronic alcohol abuse in different stages of alcohol-induced liver disease. J. Hepatol. 2000, 32, 742–747. [Google Scholar] [CrossRef]
- Mathurin, P.; Deng, Q.G.; Keshavarzian, A.; Choudhary, S.; Holmes, E.W.; Tsukamoto, H. Exacerbation of alcoholic liver injury by enteral endotoxin in rats. Hepatology 2000, 32, 1008–1017. [Google Scholar] [CrossRef] [PubMed]
- Purohit, V.; Bode, J.C.; Bode, C.; Brenner, D.A.; Choudhry, M.A.; Hamilton, F.; Kang, Y.J.; Keshavarzian, A.; Rao, R.; Sartor, R.B.; et al. Alcohol, intestinal bacterial growth, intestinal permeability to endotoxin, and medical consequences: Summary of a symposium. Alcohol 2008, 42, 349–361. [Google Scholar] [CrossRef]
- Uesugi, T.; Froh, M.; Arteel, G.E.; Bradford, B.U.; Thurman, R.G. Toll-like receptor 4 is involved in the mechanism of early alcohol-induced liver injury in mice. Hepatology 2001, 34, 101–108. [Google Scholar] [CrossRef]
- Carpino, G.; Del Ben, M.; Pastori, D.; Carnevale, R.; Baratta, F.; Overi, D.; Francis, H.; Cardinale, V.; Onori, P.; Safarikia, S.; et al. Increased Liver Localization of Lipopolysaccharides in Human and Experimental NAFLD. Hepatology 2020, 72, 470–485. [Google Scholar] [CrossRef]
- Fei, N.; Bruneau, A.; Zhang, X.; Wang, R.; Wang, J.; Rabot, S.; Gérard, P.; Zhao, L. Endotoxin Producers Overgrowing in Human Gut Microbiota as the Causative Agents for Nonalcoholic Fatty Liver Disease. mBio 2020, 11, e03263-19. [Google Scholar] [CrossRef] [PubMed]
- Roh, Y.S.; Seki, E. Toll-like receptors in alcoholic liver disease, non-alcoholic steatohepatitis and carcinogenesis. J. Gastroenterol. Hepatol. 2013, 28 (Suppl. S1), 38–42. [Google Scholar] [CrossRef] [PubMed]
- Pastor Rojo, O.; López San Román, A.; Albéniz Arbizu, E.; de la Hera Martínez, A.; Ripoll Sevillano, E.; Albillos Martínez, A. Serum lipopolysaccharide-binding protein in endotoxemic patients with inflammatory bowel disease. Inflamm. Bowel Dis. 2007, 13, 269–277. [Google Scholar] [CrossRef]
- Gardiner, K.R.; Halliday, M.I.; Barclay, G.R.; Milne, L.; Brown, D.; Stephens, S.; Maxwell, R.J.; Rowlands, B.J. Significance of systemic endotoxaemia in inflammatory bowel disease. Gut 1995, 36, 897–901. [Google Scholar] [CrossRef]
- Ma, T.Y.; Iwamoto, G.K.; Hoa, N.T.; Akotia, V.; Pedram, A.; Boivin, M.A.; Said, H.M. TNF-alpha-induced increase in intestinal epithelial tight junction permeability requires NF-kappa B activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G367–G376. [Google Scholar] [CrossRef]
- Yang, A.M.; Inamine, T.; Hochrath, K.; Chen, P.; Wang, L.; Llorente, C.; Bluemel, S.; Hartmann, P.; Xu, J.; Koyama, Y.; et al. Intestinal fungi contribute to development of alcoholic liver disease. J. Clin. Investig. 2017, 127, 2829–2841. [Google Scholar] [CrossRef]
- Zeng, S.; Hartmann, P.; Park, M.; Duan, Y.; Lang, S.; Llorente, C.; Wang, Y.; Cabré, N.; Fouts, D.E.; Bacher, P.; et al. Malassezia restricta promotes alcohol-induced liver injury. Hepatol. Commun. 2023, 7, e0029. [Google Scholar] [CrossRef] [PubMed]
- Kasper, L.; König, A.; Koenig, P.A.; Gresnigt, M.S.; Westman, J.; Drummond, R.A.; Lionakis, M.S.; Groß, O.; Ruland, J.; Naglik, J.R.; et al. The fungal peptide toxin Candidalysin activates the NLRP3 inflammasome and causes cytolysis in mononuclear phagocytes. Nat. Commun. 2018, 9, 4260. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.P.; Willems, H.M.E.; Moyes, D.L.; Shoaie, S.; Barker, K.S.; Tan, S.L.; Palmer, G.E.; Hube, B.; Naglik, J.R.; Peters, B.M. Candidalysin Drives Epithelial Signaling, Neutrophil Recruitment, and Immunopathology at the Vaginal Mucosa. Infect. Immun. 2018, 86, e00645-17. [Google Scholar] [CrossRef]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Cope, K.; Risby, T.; Diehl, A.M. Increased gastrointestinal ethanol production in obese mice: Implications for fatty liver disease pathogenesis. Gastroenterology 2000, 119, 1340–1347. [Google Scholar] [CrossRef]
- Yuan, J.; Chen, C.; Cui, J.; Lu, J.; Yan, C.; Wei, X.; Zhao, X.; Li, N.; Li, S.; Xue, G.; et al. Fatty Liver Disease Caused by High-Alcohol-Producing Klebsiella pneumoniae. Cell Metab. 2019, 30, 675–688.e677. [Google Scholar] [CrossRef]
- Elamin, E.; Jonkers, D.; Juuti-Uusitalo, K.; van Ijzendoorn, S.; Troost, F.; Duimel, H.; Broers, J.; Verheyen, F.; Dekker, J.; Masclee, A. Effects of ethanol and acetaldehyde on tight junction integrity: In vitro study in a three dimensional intestinal epithelial cell culture model. PLoS ONE 2012, 7, e35008. [Google Scholar] [CrossRef]
- Zong, H.; Armoni, M.; Harel, C.; Karnieli, E.; Pessin, J.E. Cytochrome P-450 CYP2E1 knockout mice are protected against high-fat diet-induced obesity and insulin resistance. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E532–E539. [Google Scholar] [CrossRef]
- Ekström, G.; Ingelman-Sundberg, M. Rat liver microsomal NADPH-supported oxidase activity and lipid peroxidation dependent on ethanol-inducible cytochrome P-450 (P-450IIE1). Biochem. Pharmacol. 1989, 38, 1313–1319. [Google Scholar] [CrossRef]
- Collins, S.L.; Stine, J.G.; Bisanz, J.E.; Okafor, C.D.; Patterson, A.D. Bile acids and the gut microbiota: Metabolic interactions and impacts on disease. Nat. Rev. Microbiol. 2023, 21, 236–247. [Google Scholar] [CrossRef]
- Jones, B.V.; Begley, M.; Hill, C.; Gahan, C.G.; Marchesi, J.R. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc. Natl. Acad. Sci. USA 2008, 105, 13580–13585. [Google Scholar] [CrossRef] [PubMed]
- Adams, L.A.; Wang, Z.; Liddle, C.; Melton, P.E.; Ariff, A.; Chandraratna, H.; Tan, J.; Ching, H.; Coulter, S.; de Boer, B.; et al. Bile acids associate with specific gut microbiota, low-level alcohol consumption and liver fibrosis in patients with non-alcoholic fatty liver disease. Liver Int. 2020, 40, 1356–1365. [Google Scholar] [CrossRef] [PubMed]
- Caussy, C.; Hsu, C.; Singh, S.; Bassirian, S.; Kolar, J.; Faulkner, C.; Sinha, N.; Bettencourt, R.; Gara, N.; Valasek, M.A.; et al. Serum bile acid patterns are associated with the presence of NAFLD in twins, and dose-dependent changes with increase in fibrosis stage in patients with biopsy-proven NAFLD. Aliment. Pharmacol. Ther. 2019, 49, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Mousa, O.Y.; Juran, B.D.; McCauley, B.M.; Vesterhus, M.N.; Folseraas, T.; Turgeon, C.T.; Ali, A.H.; Schlicht, E.M.; Atkinson, E.J.; Hu, C.; et al. Bile Acid Profiles in Primary Sclerosing Cholangitis and Their Ability to Predict Hepatic Decompensation. Hepatology 2021, 74, 281–295. [Google Scholar] [CrossRef]
- Shlomai, A.; Halfon, P.; Goldiner, I.; Zelber-Sagi, S.; Halpern, Z.; Oren, R.; Bruck, R. Serum bile acid levels as a predictor for the severity of liver fibrosis in patients with chronic hepatitis C. J. Viral Hepat. 2013, 20, 95–102. [Google Scholar] [CrossRef]
- Yan, L.T.; Wang, L.L.; Yao, J.; Yang, Y.T.; Mao, X.R.; Yue, W.; Mao, Y.W.; Zhou, W.; Chen, Q.F.; Chen, Y.; et al. Total bile acid-to-cholesterol ratio as a novel noninvasive marker for significant liver fibrosis and cirrhosis in patients with non-cholestatic chronic hepatitis B virus infection. Medicine 2020, 99, e19248. [Google Scholar] [CrossRef]
- Parks, D.J.; Blanchard, S.G.; Bledsoe, R.K.; Chandra, G.; Consler, T.G.; Kliewer, S.A.; Stimmel, J.B.; Willson, T.M.; Zavacki, A.M.; Moore, D.D.; et al. Bile acids: Natural ligands for an orphan nuclear receptor. Science 1999, 284, 1365–1368. [Google Scholar] [CrossRef]
- Goodwin, B.; Jones, S.A.; Price, R.R.; Watson, M.A.; McKee, D.D.; Moore, L.B.; Galardi, C.; Wilson, J.G.; Lewis, M.C.; Roth, M.E.; et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol. Cell 2000, 6, 517–526. [Google Scholar] [CrossRef]
- Jiao, N.; Baker, S.S.; Chapa-Rodriguez, A.; Liu, W.; Nugent, C.A.; Tsompana, M.; Mastrandrea, L.; Buck, M.J.; Baker, R.D.; Genco, R.J.; et al. Suppressed hepatic bile acid signalling despite elevated production of primary and secondary bile acids in NAFLD. Gut 2018, 67, 1881–1891. [Google Scholar] [CrossRef]
- Hartmann, P.; Duan, Y.; Miyamoto, Y.; Demir, M.; Lang, S.; Hasa, E.; Stern, P.; Yamashita, D.; Conrad, M.; Eckmann, L.; et al. Colesevelam ameliorates non-alcoholic steatohepatitis and obesity in mice. Hepatol. Int. 2022, 16, 359–370. [Google Scholar] [CrossRef]
- Brandl, K.; Hartmann, P.; Jih, L.J.; Pizzo, D.P.; Argemi, J.; Ventura-Cots, M.; Coulter, S.; Liddle, C.; Ling, L.; Rossi, S.J.; et al. Dysregulation of serum bile acids and FGF19 in alcoholic hepatitis. J. Hepatol. 2018, 69, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Kakiyama, G.; Hylemon, P.B.; Zhou, H.; Pandak, W.M.; Heuman, D.M.; Kang, D.J.; Takei, H.; Nittono, H.; Ridlon, J.M.; Fuchs, M.; et al. Colonic inflammation and secondary bile acids in alcoholic cirrhosis. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G929–G937. [Google Scholar] [CrossRef] [PubMed]
- Hao, H.; Cao, L.; Jiang, C.; Che, Y.; Zhang, S.; Takahashi, S.; Wang, G.; Gonzalez, F.J. Farnesoid X Receptor Regulation of the NLRP3 Inflammasome Underlies Cholestasis-Associated Sepsis. Cell Metab. 2017, 25, 856–867.e855. [Google Scholar] [CrossRef] [PubMed]
- Carino, A.; Biagioli, M.; Marchianò, S.; Scarpelli, P.; Zampella, A.; Limongelli, V.; Fiorucci, S. Disruption of TFGβ-SMAD3 pathway by the nuclear receptor SHP mediates the antifibrotic activities of BAR704, a novel highly selective FXR ligand. Pharmacol. Res. 2018, 131, 17–31. [Google Scholar] [CrossRef]
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Trinath, J.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Wu, L.; et al. Bile acid metabolites control T. Nature 2019, 576, 143–148. [Google Scholar] [CrossRef]
- Wang, L.; Gong, Z.; Zhang, X.; Zhu, F.; Liu, Y.; Jin, C.; Du, X.; Xu, C.; Chen, Y.; Cai, W.; et al. Gut microbial bile acid metabolite skews macrophage polarization and contributes to high-fat diet-induced colonic inflammation. Gut Microbes 2020, 12, 1819155. [Google Scholar] [CrossRef]
- Gao, J.; Xu, K.; Liu, H.; Liu, G.; Bai, M.; Peng, C.; Li, T.; Yin, Y. Impact of the Gut Microbiota on Intestinal Immunity Mediated by Tryptophan Metabolism. Front. Cell Infect. Microbiol. 2018, 8, 13. [Google Scholar] [CrossRef]
- Tennoune, N.; Andriamihaja, M.; Blachier, F. Production of Indole and Indole-Related Compounds by the Intestinal Microbiota and Consequences for the Host: The Good, the Bad, and the Ugly. Microorganisms 2022, 10, 930. [Google Scholar] [CrossRef]
- Hubbard, T.D.; Murray, I.A.; Bisson, W.H.; Lahoti, T.S.; Gowda, K.; Amin, S.G.; Patterson, A.D.; Perdew, G.H. Adaptation of the human aryl hydrocarbon receptor to sense microbiota-derived indoles. Sci. Rep. 2015, 5, 12689. [Google Scholar] [CrossRef]
- Sun, M.; Ma, N.; He, T.; Johnston, L.J.; Ma, X. Tryptophan (Trp) modulates gut homeostasis via aryl hydrocarbon receptor (AhR). Crit. Rev. Food Sci. Nutr. 2020, 60, 1760–1768. [Google Scholar] [CrossRef]
- Powell, D.N.; Swimm, A.; Sonowal, R.; Bretin, A.; Gewirtz, A.T.; Jones, R.M.; Kalman, D. Indoles from the commensal microbiota act via the AHR and IL-10 to tune the cellular composition of the colonic epithelium during aging. Proc. Natl. Acad. Sci. USA 2020, 117, 21519–21526. [Google Scholar] [CrossRef] [PubMed]
- Mawe, G.M.; Hoffman, J.M. Serotonin signalling in the gut--functions, dysfunctions and therapeutic targets. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 473–486. [Google Scholar] [CrossRef]
- Bansal, T.; Alaniz, R.C.; Wood, T.K.; Jayaraman, A. The bacterial signal indole increases epithelial-cell tight-junction resistance and attenuates indicators of inflammation. Proc. Natl. Acad. Sci. USA 2010, 107, 228–233. [Google Scholar] [CrossRef]
- Ma, L.; Li, H.; Hu, J.; Zheng, J.; Zhou, J.; Botchlett, R.; Matthews, D.; Zeng, T.; Chen, L.; Xiao, X.; et al. Indole Alleviates Diet-Induced Hepatic Steatosis and Inflammation in a Manner Involving Myeloid Cell 6-Phosphofructo-2-Kinase/Fructose-2,6-Biphosphatase 3. Hepatology 2020, 72, 1191–1203. [Google Scholar] [CrossRef]
- Hendrikx, T.; Duan, Y.; Wang, Y.; Oh, J.H.; Alexander, L.M.; Huang, W.; Stärkel, P.; Ho, S.B.; Gao, B.; Fiehn, O.; et al. Bacteria engineered to produce IL-22 in intestine induce expression of REG3G to reduce ethanol-induced liver disease in mice. Gut 2019, 68, 1504–1515. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Ji, Y.Y.; Wen, X.L.; Duan, S.L. Fecal microbiota transplantation in the metabolic diseases: Current status and perspectives. World J. Gastroenterol. 2022, 28, 2546–2560. [Google Scholar] [CrossRef]
- Yang, R.; Chen, Z.; Cai, J. Fecal microbiota transplantation: Emerging applications in autoimmune diseases. J. Autoimmun. 2023, 141, 103038. [Google Scholar] [CrossRef] [PubMed]
- Vendrik, K.E.W.; Ooijevaar, R.E.; de Jong, P.R.C.; Laman, J.D.; van Oosten, B.W.; van Hilten, J.J.; Ducarmon, Q.R.; Keller, J.J.; Kuijper, E.J.; Contarino, M.F. Fecal Microbiota Transplantation in Neurological Disorders. Front. Cell Infect. Microbiol. 2020, 10, 98. [Google Scholar] [CrossRef]
- Craven, L.; Rahman, A.; Nair Parvathy, S.; Beaton, M.; Silverman, J.; Qumosani, K.; Hramiak, I.; Hegele, R.; Joy, T.; Meddings, J.; et al. Allogenic Fecal Microbiota Transplantation in Patients with Nonalcoholic Fatty Liver Disease Improves Abnormal Small Intestinal Permeability: A Randomized Control Trial. Am. J. Gastroenterol. 2020, 115, 1055–1065. [Google Scholar] [CrossRef]
- Xue, L.; Deng, Z.; Luo, W.; He, X.; Chen, Y. Effect of Fecal Microbiota Transplantation on Non-Alcoholic Fatty Liver Disease: A Randomized Clinical Trial. Front. Cell Infect. Microbiol. 2022, 12, 759306. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Fagan, A.; Gavis, E.A.; Sterling, R.K.; Gallagher, M.L.; Lee, H.; Matherly, S.C.; Siddiqui, M.S.; Bartels, A.; Mousel, T.; et al. Microbiota transplant for hepatic encephalopathy in cirrhosis: The THEMATIC trial. J. Hepatol. 2025, in press. [CrossRef] [PubMed]
- Allegretti, J.R.; Kassam, Z.; Carrellas, M.; Mullish, B.H.; Marchesi, J.R.; Pechlivanis, A.; Smith, M.; Gerardin, Y.; Timberlake, S.; Pratt, D.S.; et al. Fecal Microbiota Transplantation in Patients with Primary Sclerosing Cholangitis: A Pilot Clinical Trial. Am. J. Gastroenterol. 2019, 114, 1071–1079. [Google Scholar] [CrossRef]
- Moayyedi, P.; Surette, M.G.; Kim, P.T.; Libertucci, J.; Wolfe, M.; Onischi, C.; Armstrong, D.; Marshall, J.K.; Kassam, Z.; Reinisch, W.; et al. Fecal Microbiota Transplantation Induces Remission in Patients with Active Ulcerative Colitis in a Randomized Controlled Trial. Gastroenterology 2015, 149, 102–109.e106. [Google Scholar] [CrossRef]
- Rossen, N.G.; Fuentes, S.; van der Spek, M.J.; Tijssen, J.G.; Hartman, J.H.; Duflou, A.; Löwenberg, M.; van den Brink, G.R.; Mathus-Vliegen, E.M.; de Vos, W.M.; et al. Findings From a Randomized Controlled Trial of Fecal Transplantation for Patients with Ulcerative Colitis. Gastroenterology 2015, 149, 110–118.e114. [Google Scholar] [CrossRef]
- Paramsothy, S.; Kamm, M.A.; Kaakoush, N.O.; Walsh, A.J.; van den Bogaerde, J.; Samuel, D.; Leong, R.W.L.; Connor, S.; Ng, W.; Paramsothy, R.; et al. Multidonor intensive faecal microbiota transplantation for active ulcerative colitis: A randomised placebo-controlled trial. Lancet 2017, 389, 1218–1228. [Google Scholar] [CrossRef] [PubMed]
- Costello, S.P.; Hughes, P.A.; Waters, O.; Bryant, R.V.; Vincent, A.D.; Blatchford, P.; Katsikeros, R.; Makanyanga, J.; Campaniello, M.A.; Mavrangelos, C.; et al. Effect of Fecal Microbiota Transplantation on 8-Week Remission in Patients with Ulcerative Colitis: A Randomized Clinical Trial. JAMA 2019, 321, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Březina, J.; Bajer, L.; Wohl, P.; Ďuricová, D.; Hrabák, P.; Novotný, A.; Koželuhová, J.; Lukáš, M.; Mrázek, J.; Fliegerová, K.O.; et al. Fecal Microbial Transplantation versus Mesalamine Enema for Treatment of Active Left-Sided Ulcerative Colitis-Results of a Randomized Controlled Trial. J. Clin. Med. 2021, 10, 2753. [Google Scholar] [CrossRef]
- Al-Habsi, N.; Al-Khalili, M.; Haque, S.A.; Elias, M.; Olqi, N.A.; Al Uraimi, T. Health Benefits of Prebiotics, Probiotics, Synbiotics, and Postbiotics. Nutrients 2024, 16, 3955. [Google Scholar] [CrossRef]
- Li, H.Y.; Zhou, D.D.; Gan, R.Y.; Huang, S.Y.; Zhao, C.N.; Shang, A.; Xu, X.Y.; Li, H.B. Effects and Mechanisms of Probiotics, Prebiotics, Synbiotics, and Postbiotics on Metabolic Diseases Targeting Gut Microbiota: A Narrative Review. Nutrients 2021, 13, 3211. [Google Scholar] [CrossRef]
- Vieira, A.T.; Teixeira, M.M.; Martins, F.S. The role of probiotics and prebiotics in inducing gut immunity. Front. Immunol. 2013, 4, 445. [Google Scholar] [CrossRef]
- Cani, P.D.; Possemiers, S.; Van de Wiele, T.; Guiot, Y.; Everard, A.; Rottier, O.; Geurts, L.; Naslain, D.; Neyrinck, A.; Lambert, D.M.; et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 2009, 58, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Shukla, P.K.; Meena, A.S.; Manda, B.; Gomes-Solecki, M.; Dietrich, P.; Dragatsis, I.; Rao, R. Lactobacillus plantarum prevents and mitigates alcohol-induced disruption of colonic epithelial tight junctions, endotoxemia, and liver damage by an EGF receptor-dependent mechanism. FASEB J. 2018, 32, fj201800351R. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Ren, T.; Zhou, M.; Cheng, M. The combination of blueberry juice and probiotics reduces apoptosis of alcoholic fatty liver of mice by affecting SIRT1 pathway. Drug Des. Devel Ther. 2016, 10, 1649–1661. [Google Scholar] [CrossRef] [PubMed]
- Xiong, S.Y.; Wu, G.S.; Li, C.; Ma, W.; Luo, H.R. Clinical efficacy of probiotics in the treatment of alcoholic liver disease: A systematic review and meta-analysis. Front. Cell Infect. Microbiol. 2024, 14, 1358063. [Google Scholar] [CrossRef]
- Patel, D.; Desai, C.; Singh, D.; Soppina, V.; Parwani, K.; Patel, F.; Mandal, P. Synbiotic Intervention Ameliorates Oxidative Stress and Gut Permeability in an In Vitro and In Vivo Model of Ethanol-Induced Intestinal Dysbiosis. Biomedicines 2022, 10, 3285. [Google Scholar] [CrossRef]
- Naudin, C.R.; Maner-Smith, K.; Owens, J.A.; Wynn, G.M.; Robinson, B.S.; Matthews, J.D.; Reedy, A.R.; Luo, L.; Wolfarth, A.A.; Darby, T.M.; et al. Lactococcus lactis Subspecies cremoris Elicits Protection Against Metabolic Changes Induced by a Western-Style Diet. Gastroenterology 2020, 159, 639–651.e635. [Google Scholar] [CrossRef]
- Yan, Y.; Liu, C.; Zhao, S.; Wang, X.; Wang, J.; Zhang, H.; Wang, Y.; Zhao, G. Probiotic Bifidobacterium lactis V9 attenuates hepatic steatosis and inflammation in rats with non-alcoholic fatty liver disease. AMB Express 2020, 10, 101. [Google Scholar] [CrossRef]
- Duseja, A.; Acharya, S.K.; Mehta, M.; Chhabra, S.; Rana, S.; Das, A.; Dattagupta, S.; Dhiman, R.K.; Chawla, Y.K.; Shalimar. High potency multistrain probiotic improves liver histology in non-alcoholic fatty liver disease (NAFLD): A randomised, double-blind, proof of concept study. BMJ Open Gastroenterol. 2019, 6, e000315. [Google Scholar] [CrossRef]
- Ahn, S.B.; Jun, D.W.; Kang, B.K.; Lim, J.H.; Lim, S.; Chung, M.J. Randomized, Double-blind, Placebo-controlled Study of a Multispecies Probiotic Mixture in Nonalcoholic Fatty Liver Disease. Sci. Rep. 2019, 9, 5688. [Google Scholar] [CrossRef]
- Pan, Y.; Yang, Y.; Wu, J.; Zhou, H.; Yang, C. Efficacy of probiotics, prebiotics, and synbiotics on liver enzymes, lipid profiles, and inflammation in patients with non-alcoholic fatty liver disease: A systematic review and meta-analysis of randomized controlled trials. BMC Gastroenterol. 2024, 24, 283. [Google Scholar] [CrossRef]
- Vleggaar, F.P.; Monkelbaan, J.F.; van Erpecum, K.J. Probiotics in primary sclerosing cholangitis: A randomized placebo-controlled crossover pilot study. Eur. J. Gastroenterol. Hepatol. 2008, 20, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Ojetti, V.; Saviano, A.; Brigida, M.; Petruzziello, C.; Caronna, M.; Gayani, G.; Franceschi, F. Randomized control trial on the efficacy of Limosilactobacillus reuteri ATCC PTA 4659 in reducing inflammatory markers in acute uncomplicated diverticulitis. Eur. J. Gastroenterol. Hepatol. 2022, 34, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Bamola, V.D.; Dubey, D.; Samanta, P.; Kedia, S.; Ahuja, V.; Madempudi, R.S.; Neelamraju, J.; Chaudhry, R. Role of a probiotic strain in the modulation of gut microbiota and cytokines in inflammatory bowel disease. Anaerobe 2022, 78, 102652. [Google Scholar] [CrossRef] [PubMed]
- Agraib, L.M.; Yamani, M.I.; Tayyem, R.; Abu-Sneineh, A.T.; Rayyan, Y.M. Probiotic supplementation induces remission and changes in the immunoglobulins and inflammatory response in active ulcerative colitis patients: A pilot, randomized, double-blind, placebo-controlled study. Clin. Nutr. ESPEN 2022, 51, 83–91. [Google Scholar] [CrossRef]
- Iheozor-Ejiofor, Z.; Kaur, L.; Gordon, M.; Baines, P.A.; Sinopoulou, V.; Akobeng, A.K. Probiotics for maintenance of remission in ulcerative colitis. Cochrane Database Syst. Rev. 2020, 3, CD007443. [Google Scholar] [CrossRef]
- Matsuoka, K.; Uemura, Y.; Kanai, T.; Kunisaki, R.; Suzuki, Y.; Yokoyama, K.; Yoshimura, N.; Hibi, T. Efficacy of Bifidobacterium breve Fermented Milk in Maintaining Remission of Ulcerative Colitis. Dig. Dis. Sci. 2018, 63, 1910–1919. [Google Scholar] [CrossRef]
- Valcheva, R.; Koleva, P.; Martínez, I.; Walter, J.; Gänzle, M.G.; Dieleman, L.A. Inulin-type fructans improve active ulcerative colitis associated with microbiota changes and increased short-chain fatty acids levels. Gut Microbes 2019, 10, 334–357. [Google Scholar] [CrossRef]
- Kamarli Altun, H.; Akal Yildiz, E.; Akin, M. Impact of Synbiotic Therapy on the Quality of Life in Patients with Mild-to-Moderately Active Ulcerative Colitis. J. Gastrointestin Liver Dis. 2022, 31, 417–423. [Google Scholar] [CrossRef]
- Amiriani, T.; Rajabli, N.; Faghani, M.; Besharat, S.; Roshandel, G.; Akhavan Tabib, A.; Joshaghani, H. Effect of Lactocare® Synbiotic on Disease Severity in Ulcerative Colitis: A Randomized Placebo-Controlled Double-Blind Clinical Trial. Middle East J. Dig. Dis. 2020, 12, 27–33. [Google Scholar] [CrossRef]
- Kavita; Om, H.; Chand, U.; Kushawaha, P.K. Postbiotics: An alternative and innovative intervention for the therapy of inflammatory bowel disease. Microbiol. Res. 2024, 279, 127550. [Google Scholar] [CrossRef]
- Zhang, T.; Zhang, W.; Feng, C.; Kwok, L.Y.; He, Q.; Sun, Z. Stronger gut microbiome modulatory effects by postbiotics than probiotics in a mouse colitis model. NPJ Sci. Food 2022, 6, 53. [Google Scholar] [CrossRef] [PubMed]
- Yin, R.; Wang, T.; Sun, J.; Dai, H.; Zhang, Y.; Liu, N.; Liu, H. Postbiotics From Lactobacillus Johnsonii Activates Gut Innate Immunity to Mitigate Alcohol-Associated Liver Disease. Adv. Sci. 2025, 12, e2405781. [Google Scholar] [CrossRef] [PubMed]
- Jena, A.; Dutta, U.; Shah, J.; Sharma, V.; Prasad, K.K.; Shivaprakash, R.M.; Mandavdhare, H.S.; Samanta, J.; Sharma, P.; Popli, P.; et al. Oral Fluconazole Therapy in Patients with Active Ulcerative Colitis Who Have Detectable Candida in the Stool: A Double-Blind Randomized Placebo-controlled Trial. J. Clin. Gastroenterol. 2022, 56, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Chen, J.; Hollister, K.; Sowers, L.C.; Forman, B.M. Endogenous bile acids are ligands for the nuclear receptor FXR/BAR. Mol. Cell 1999, 3, 543–553. [Google Scholar] [CrossRef]
- Ma, K.; Saha, P.K.; Chan, L.; Moore, D.D. Farnesoid X receptor is essential for normal glucose homeostasis. J. Clin. Investig. 2006, 116, 1102–1109. [Google Scholar] [CrossRef]
- Watanabe, M.; Houten, S.M.; Wang, L.; Moschetta, A.; Mangelsdorf, D.J.; Heyman, R.A.; Moore, D.D.; Auwerx, J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Investig. 2004, 113, 1408–1418. [Google Scholar] [CrossRef]
- Armstrong, L.E.; Guo, G.L. Role of FXR in Liver Inflammation during Nonalcoholic Steatohepatitis. Curr. Pharmacol. Rep. 2017, 3, 92–100. [Google Scholar] [CrossRef]
- Mudaliar, S.; Henry, R.R.; Sanyal, A.J.; Morrow, L.; Marschall, H.U.; Kipnes, M.; Adorini, L.; Sciacca, C.I.; Clopton, P.; Castelloe, E.; et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 2013, 145, 574–582.e571. [Google Scholar] [CrossRef]
- Almeqdadi, M.; Gordon, F.D. Farnesoid X Receptor Agonists: A Promising Therapeutic Strategy for Gastrointestinal Diseases. Gastro Hep Adv. 2024, 3, 344–352. [Google Scholar] [CrossRef]
- Maliha, S.; Guo, G.L. Farnesoid X receptor and fibroblast growth factor 15/19 as pharmacological targets. Liver Res. 2021, 5, 142–150. [Google Scholar] [CrossRef]
- Stofan, M.; Guo, G.L. Bile Acids and FXR: Novel Targets for Liver Diseases. Front. Med. 2020, 7, 544. [Google Scholar] [CrossRef] [PubMed]
- Nevens, F.; Andreone, P.; Mazzella, G.; Strasser, S.I.; Bowlus, C.; Invernizzi, P.; Drenth, J.P.; Pockros, P.J.; Regula, J.; Beuers, U.; et al. A Placebo-Controlled Trial of Obeticholic Acid in Primary Biliary Cholangitis. N. Engl. J. Med. 2016, 375, 631–643. [Google Scholar] [CrossRef]
- Kowdley, K.V.; Luketic, V.; Chapman, R.; Hirschfield, G.M.; Poupon, R.; Schramm, C.; Vincent, C.; Rust, C.; Parés, A.; Mason, A.; et al. A randomized trial of obeticholic acid monotherapy in patients with primary biliary cholangitis. Hepatology 2018, 67, 1890–1902. [Google Scholar] [CrossRef]
- Trauner, M.; Nevens, F.; Shiffman, M.L.; Drenth, J.P.H.; Bowlus, C.L.; Vargas, V.; Andreone, P.; Hirschfield, G.M.; Pencek, R.; Malecha, E.S.; et al. Long-term efficacy and safety of obeticholic acid for patients with primary biliary cholangitis: 3-year results of an international open-label extension study. Lancet Gastroenterol. Hepatol. 2019, 4, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef]
- Kowdley, K.V.; Vuppalanchi, R.; Levy, C.; Floreani, A.; Andreone, P.; LaRusso, N.F.; Shrestha, R.; Trotter, J.; Goldberg, D.; Rushbrook, S.; et al. A randomized, placebo-controlled, phase II study of obeticholic acid for primary sclerosing cholangitis. J. Hepatol. 2020, 73, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Lindor, K.D.; Bowlus, C.L.; Boyer, J.; Levy, C.; Mayo, M. Primary Biliary Cholangitis: 2018 Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology 2019, 69, 394–419. [Google Scholar] [CrossRef]
- Loomba, R.; Noureddin, M.; Kowdley, K.V.; Kohli, A.; Sheikh, A.; Neff, G.; Bhandari, B.R.; Gunn, N.; Caldwell, S.H.; Goodman, Z.; et al. Combination Therapies Including Cilofexor and Firsocostat for Bridging Fibrosis and Cirrhosis Attributable to NASH. Hepatology 2021, 73, 625–643. [Google Scholar] [CrossRef]
- Trauner, M.; Gulamhusein, A.; Hameed, B.; Caldwell, S.; Shiffman, M.L.; Landis, C.; Eksteen, B.; Agarwal, K.; Muir, A.; Rushbrook, S.; et al. The Nonsteroidal Farnesoid X Receptor Agonist Cilofexor (GS-9674) Improves Markers of Cholestasis and Liver Injury in Patients with Primary Sclerosing Cholangitis. Hepatology 2019, 70, 788–801. [Google Scholar] [CrossRef]
- You, M.; Liang, X.; Ajmo, J.M.; Ness, G.C. Involvement of mammalian sirtuin 1 in the action of ethanol in the liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 294, G892–G898. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.M.; Du, K.; You, M.; Ding, W.X. Critical role of FoxO3a in alcohol-induced autophagy and hepatotoxicity. Am. J. Pathol. 2013, 183, 1815–1825. [Google Scholar] [CrossRef] [PubMed]
- Manley, S.; Ni, H.M.; Williams, J.A.; Kong, B.; DiTacchio, L.; Guo, G.; Ding, W.X. Farnesoid X receptor regulates forkhead Box O3a activation in ethanol-induced autophagy and hepatotoxicity. Redox Biol. 2014, 2, 991–1002. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, P.; Hochrath, K.; Horvath, A.; Chen, P.; Seebauer, C.T.; Llorente, C.; Wang, L.; Alnouti, Y.; Fouts, D.E.; Stärkel, P.; et al. Modulation of the intestinal bile acid/farnesoid X receptor/fibroblast growth factor 15 axis improves alcoholic liver disease in mice. Hepatology 2018, 67, 2150–2166. [Google Scholar] [CrossRef]
- Wu, W.; Zhu, B.; Peng, X.; Zhou, M.; Jia, D.; Gu, J. Activation of farnesoid X receptor attenuates hepatic injury in a murine model of alcoholic liver disease. Biochem. Biophys. Res. Commun. 2014, 443, 68–73. [Google Scholar] [CrossRef]
- Lívero, F.A.; Stolf, A.M.; Dreifuss, A.A.; Bastos-Pereira, A.L.; Chicorski, R.; de Oliveira, L.G.; de Souza, C.E.; Fabossi, I.A.; Rabitto, I.S.; Gremski, L.H.; et al. The FXR agonist 6ECDCA reduces hepatic steatosis and oxidative stress induced by ethanol and low-protein diet in mice. Chem. Biol. Interact. 2014, 217, 19–27. [Google Scholar] [CrossRef]
- Gadaleta, R.M.; van Erpecum, K.J.; Oldenburg, B.; Willemsen, E.C.; Renooij, W.; Murzilli, S.; Klomp, L.W.; Siersema, P.D.; Schipper, M.E.; Danese, S.; et al. Farnesoid X receptor activation inhibits inflammation and preserves the intestinal barrier in inflammatory bowel disease. Gut 2011, 60, 463–472. [Google Scholar] [CrossRef]
- Kouno, T.; Zeng, S.; Wang, Y.; Duan, Y.; Lang, S.; Gao, B.; Hartmann, P.; Cabré, N.; Llorente, C.; Galbert, C.; et al. Engineered bacteria producing aryl-hydrocarbon receptor agonists protect against ethanol-induced liver disease in mice. Alcohol Clin. Exp. Res. 2023, 47, 856–867. [Google Scholar] [CrossRef]
- Zhu, W.; Winter, M.G.; Byndloss, M.X.; Spiga, L.; Duerkop, B.A.; Hughes, E.R.; Büttner, L.; de Lima Romão, E.; Behrendt, C.L.; Lopez, C.A.; et al. Precision editing of the gut microbiota ameliorates colitis. Nature 2018, 553, 208–211. [Google Scholar] [CrossRef]
- Duan, Y.; Llorente, C.; Lang, S.; Brandl, K.; Chu, H.; Jiang, L.; White, R.C.; Clarke, T.H.; Nguyen, K.; Torralba, M.; et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature 2019, 575, 505–511. [Google Scholar] [CrossRef]
- Gan, L.; Feng, Y.; Du, B.; Fu, H.; Tian, Z.; Xue, G.; Yan, C.; Cui, X.; Zhang, R.; Cui, J.; et al. Bacteriophage targeting microbiota alleviates non-alcoholic fatty liver disease induced by high alcohol-producing Klebsiella pneumoniae. Nat. Commun. 2023, 14, 3215. [Google Scholar] [CrossRef] [PubMed]
- Ichikawa, M.; Nakamoto, N.; Kredo-Russo, S.; Weinstock, E.; Weiner, I.N.; Khabra, E.; Ben-Ishai, N.; Inbar, D.; Kowalsman, N.; Mordoch, R.; et al. Bacteriophage therapy against pathological Klebsiella pneumoniae ameliorates the course of primary sclerosing cholangitis. Nat. Commun. 2023, 14, 3261. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.L.; Wang, Y.; Duan, Y.; Chu, H.; Hartmann, P.; Llorente, C.; Zhou, R.; Schnabl, B. Differences in Bacterial Translocation and Liver Injury in Ethanol Versus Diet-Induced Liver Disease. Dig. Dis. Sci. 2023, 68, 3059–3069. [Google Scholar] [CrossRef]
- Zeng, S.; Rosati, E.; Saggau, C.; Messner, B.; Chu, H.; Duan, Y.; Hartmann, P.; Wang, Y.; Ma, S.; Huang, W.J.M.; et al. Candida albicans-specific Th17 cell-mediated response contributes to alcohol-associated liver disease. Cell Host Microbe 2023, 31, 389–404.e387. [Google Scholar] [CrossRef]
- Münte, E.; Viebahn, G.; Khurana, A.; Fujiki, J.; Nakamura, T.; Lang, S.; Demir, M.; Schnabl, B.; Hartmann, P. Is Associated with Disease Severity in MASLD but Its Supplementation Does Not Improve Diet-Induced Steatohepatitis in Mice. Microorganisms 2025, 13, 675. [Google Scholar] [CrossRef]
- Forsyth, C.B.; Farhadi, A.; Jakate, S.M.; Tang, Y.; Shaikh, M.; Keshavarzian, A. Lactobacillus GG treatment ameliorates alcohol-induced intestinal oxidative stress, gut leakiness, and liver injury in a rat model of alcoholic steatohepatitis. Alcohol 2009, 43, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Almeida, C.; Oliveira, R.; Baylina, P.; Fernandes, R.; Teixeira, F.G.; Barata, P. Current Trends and Challenges of Fecal Microbiota Transplantation-An Easy Method That Works for All? Biomedicines 2022, 10, 2742. [Google Scholar] [CrossRef]
- DeFilipp, Z.; Bloom, P.P.; Torres Soto, M.; Mansour, M.K.; Sater, M.R.A.; Huntley, M.H.; Turbett, S.; Chung, R.T.; Chen, Y.B.; Hohmann, E.L. Drug-Resistant. N. Engl. J. Med. 2019, 381, 2043–2050. [Google Scholar] [CrossRef]
- Nicco, C.; Paule, A.; Konturek, P.; Edeas, M. From Donor to Patient: Collection, Preparation and Cryopreservation of Fecal Samples for Fecal Microbiota Transplantation. Diseases 2020, 8, 9. [Google Scholar] [CrossRef]
- Lee, C.H.; Steiner, T.; Petrof, E.O.; Smieja, M.; Roscoe, D.; Nematallah, A.; Weese, J.S.; Collins, S.; Moayyedi, P.; Crowther, M.; et al. Frozen vs Fresh Fecal Microbiota Transplantation and Clinical Resolution of Diarrhea in Patients with Recurrent Clostridium difficile Infection: A Randomized Clinical Trial. JAMA 2016, 315, 142–149. [Google Scholar] [CrossRef]
- Freedman, S.B.; Schnadower, D.; Tarr, P.I. The Probiotic Conundrum: Regulatory Confusion, Conflicting Studies, and Safety Concerns. JAMA 2020, 323, 823–824. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Jin, W.; Liu, S.J.; Jiao, Z.; Li, X. Probiotics, prebiotics, and postbiotics in health and disease. MedComm 2023, 4, e420. [Google Scholar] [CrossRef] [PubMed]
- Merenstein, D.; Pot, B.; Leyer, G.; Ouwehand, A.C.; Preidis, G.A.; Elkins, C.A.; Hill, C.; Lewis, Z.T.; Shane, A.L.; Zmora, N.; et al. Emerging issues in probiotic safety: 2023 perspectives. Gut Microbes 2023, 15, 2185034. [Google Scholar] [CrossRef] [PubMed]
- Kothari, D.; Patel, S.; Kim, S.K. Probiotic supplements might not be universally-effective and safe: A review. Biomed. Pharmacother. 2019, 111, 537–547. [Google Scholar] [CrossRef]
- Górski, A.; Międzybrodzki, R.; Borysowski, J.; Dąbrowska, K.; Wierzbicki, P.; Ohams, M.; Korczak-Kowalska, G.; Olszowska-Zaremba, N.; Łusiak-Szelachowska, M.; Kłak, M.; et al. Phage as a modulator of immune responses: Practical implications for phage therapy. Adv. Virus Res. 2012, 83, 41–71. [Google Scholar] [CrossRef]
- Zalewska-Piątek, B. Phage Therapy-Challenges, Opportunities and Future Prospects. Pharmaceuticals 2023, 16, 1638. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saha, P.; Hartmann, P. Impact of Gut Microbiome on Gut Permeability in Liver and Gut Diseases. Microorganisms 2025, 13, 1188. https://doi.org/10.3390/microorganisms13061188
Saha P, Hartmann P. Impact of Gut Microbiome on Gut Permeability in Liver and Gut Diseases. Microorganisms. 2025; 13(6):1188. https://doi.org/10.3390/microorganisms13061188
Chicago/Turabian StyleSaha, Punnag, and Phillipp Hartmann. 2025. "Impact of Gut Microbiome on Gut Permeability in Liver and Gut Diseases" Microorganisms 13, no. 6: 1188. https://doi.org/10.3390/microorganisms13061188
APA StyleSaha, P., & Hartmann, P. (2025). Impact of Gut Microbiome on Gut Permeability in Liver and Gut Diseases. Microorganisms, 13(6), 1188. https://doi.org/10.3390/microorganisms13061188