1. Introduction
Tuberculosis (TB) is one of the leading causes of illness and death worldwide, posing a significant burden on global public health [
1]. Caused by Mycobacterium tuberculosis (MTB), TB is a chronic infectious disease that can be categorized into pulmonary TB and extrapulmonary TB. Extrapulmonary TB refers to MTB infections occurring outside the lung parenchyma and can affect almost every organ system. Among these, lymph node tuberculosis (LNTB) is the most common form [
2]. LNTB, representing 30% to 55% of extrapulmonary TB cases, is among the most frequent forms of TB outside the lungs [
3]. Cervical lymph nodes are the most frequently affected sites. Patients often present with systemic TB symptoms, such as prolonged low-grade fever and night sweats. As the disease progresses, it may lead to complications, including suppurative infections, chronic sinus tract formation, and skin necrosis [
4,
5]. These conditions are particularly severe in immunocompromised individuals, where the disease can progress rapidly and become life-threatening. Despite the relatively high incidence of LNTB, the molecular mechanisms underlying its pathogenesis remain poorly understood, which has significantly hindered the advancement of effective diagnostic and therapeutic strategies.
The human body hosts a vast population of commensal bacteria, collectively known as the microbiota, which maintain a dynamic interplay with the immune system and play a crucial role in maintaining health. Some studies [
6] have demonstrated strong associations between gut microbiota and both infectious and non-infectious diseases. Specifically, alterations in the gut microbial community have been observed in pulmonary tuberculosis (PTB) patients, and dysbiosis of the gut microbiota has been shown to increase host susceptibility to MTB infection. Evidence [
7] suggests that MTB infection leads to gut microbiota imbalances, characterized by shifts in the abundance of key bacterial taxa, including reductions in short-chain fatty acid (SCFA)-producing bacteria such as
Ruminococcus and
Bifidobacterium. Additionally, gut-derived metabolites, such as indole propionic acid, have been reported to inhibit MTB growth [
8].
The advent of omics technologies has provided powerful tools for investigating disease-related disruptions in metabolic homeostasis. These methods have shed light on disease mechanisms across a range of conditions, including cardiovascular disease, cancer disease, and infectious diseases [
9,
10]. In recent years, the application of omics—particularly metabolomics and lipidomics—in TB research has offered novel insights into host–pathogen interactions and the role of host metabolism in disease progression [
11,
12]. Several potential clinical biomarkers have been identified for diagnosis and therapeutic monitoring [
13,
14]. The integration of metabolomics and gut microbiota analysis has emerged as a powerful approach to elucidate disease development, identify critical regulatory factors, uncover associations between the gut microbiome and metabolic products, and explore potential complex molecular networks [
15,
16,
17]. However, the validation of these findings across different TB forms, especially in LNTB, remains insufficient and demands further investigation.
Previous studies on human LNTB have primarily focused on epidemiology, clinical manifestations, pathology, diagnosis, and treatment [
18]. However, investigations into disease progression, underlying regulatory mechanisms, and key molecular drivers remain limited. In the present study, we employed a multi-omics strategy combining metagenomic sequencing of fecal samples and untargeted plasma metabolomics to investigate both distinct and shared features in patients with LNTB. Our goal was to identify potential diagnostic biomarkers for LNTB and to provide a comprehensive and in-depth understanding of the molecular mechanisms underlying this disease. By integrating rigorous functional interpretations with multi-omics data, we aimed to uncover microbial and metabolic alterations associated with the pathophysiology of LNTB, ultimately contributing to improved disease management and therapeutic strategies.
2. Material and Methods
2.1. Clinical Sample Collection
The collection of blood samples, fecal samples, and clinical data was approved by the Ethics Committee of Shenzhen Third People’s Hospital (Approval No. 2025-015). All participants provided informed consent prior to enrollment. The study population was divided into two groups: healthy volunteers (Health group, n = 8) and patients with lymph node tuberculosis (LNTB group, n = 9). All LNTB cases included in this study were newly diagnosed.
Healthy participants were selected based on stable lifestyles and overall good health. They reported a diverse and balanced diet, regular sleep patterns with adequate nightly rest, and no significant addictive behaviors. None had taken antibiotics within the past year or had a history of substance abuse.
Candidate screening and participant recruitment were conducted strictly according to pre-established criteria [
19], which included the following: (i) general physical and health status for the inclusion of healthy volunteers; (ii) diagnostic criteria for TB patients (1); and (iii) age between 18 and 65 years. Exclusion criteria included failure to meet inclusion criteria or the presence of any of the following conditions [
19]: prior use of therapeutic medications (including antibiotics), hepatitis, HIV/AIDS, rheumatic or autoimmune diseases; coexisting malignancies; serious infections within the past two weeks; pregnancy or known allergy; breastfeeding or intent to conceive; participation in other clinical trials within the past three months; legally recognized disabilities (e.g., blindness, deafness, mutism, intellectual disability, or physical disability); suspected or confirmed history of alcohol or substance abuse; and any circumstances likely to interfere with follow-up (e.g., frequent job relocation). Participants deemed by investigators to have poor compliance potential were also excluded.
2.2. Stool Sample Collection and DNA Extraction
Fresh fecal samples were obtained from both healthy individuals and LNTB patients using sterile collection tubes pre-filled with a stool DNA stabilizing reagent, as provided in the PSP® Spin Stool DNA Plus Kit (Stratec Molecular, Berlin, Germany). Immediately after collection, samples were stored at −20 °C to preserve nucleic acid integrity until further processing. For downstream analysis, approximately 200 mg of each fecal specimen was used for metagenomic DNA extraction, following the standard protocol recommended by the manufacturer.
2.3. Metagenomic Sequencing and Data Processing
According to the manufacturer’s instructions (Illumina, San Diego, CA, USA), paired-end sequencing libraries with an average insert size of approximately 350 bp were constructed. Sequencing was performed on the Illumina HiSeq 2500 platform using a paired-end strategy. The raw sequencing data were subsequently processed using the MOCAT2 analysis pipeline [
20] to remove low-quality reads, adapter contamination, and human-derived sequences. Specifically, sequencing reads were first trimmed using a minimum length threshold of 30 bp and a quality score cutoff of 20. Adapter sequences were filtered out using an e-value threshold of 0.01, and potential human DNA contamination was eliminated by aligning reads to the human genome with a 90% sequence identity threshold. Following quality control and filtering procedures, each sample yielded approximately 5 GB of high-quality clean data on average. For functional profiling, the high-quality reads were assembled into contigs, followed by gene prediction and clustering against a reference gene catalog. Taxonomic profiling and relative abundance estimation were performed using MetaPhlAn2 [
21] with default parameters.
2.4. Microbial Diversity and Enterotype Analysis
Alpha diversity, reflecting the microbial richness and evenness within individual samples, was assessed using the Shannon diversity index derived from the species-level taxonomic profiles. To evaluate inter-sample variation in microbial composition (beta diversity), non-metric multidimensional scaling (NMDS) analysis was performed based on the Bray–Curtis distance matrix, calculated from the relative abundance of bacterial genera. This approach allowed for the visualization of overall microbial community structure and differences across sample groups.
2.5. Plasma Metabolite Extraction for Untargeted Metabolomics
The extraction of plasma metabolites was performed as follows: 50 μL of plasma from each patient was thawed on ice for approximately 30 min to minimize the degradation of thermolabile metabolites and vortexed gently for 10 s to ensure homogeneity. Then, 150 μL of pre-chilled methanol (−20 °C) containing six internal standards—Acetyl-L-carnitine-(N-methyl-d3), L-phenyl-d5-alanine, L-tryptophan-(indole-d5), leucine enkephalin, SM(d18:1/15:0)-d9, and cholic acid-d5—was added to each sample to monitor extraction efficiency and correct for analytical variation. The mixture was vigorously vortexed for 30 s, followed by centrifugation at 14,000 rcf for 2 min at 4 °C to remove precipitated proteins and debris. Next, 150 μL of the supernatant was transferred to a clean tube and evaporated to dryness under a gentle stream of nitrogen at room temperature. The dried residue was reconstituted in 200 μL of 50% methanol (v/v), briefly vortexed, and centrifuged again at 14,000 rcf for 2 min at 4 °C. Finally, 100 μL of the clarified supernatant was collected and transferred to autosampler vials for subsequent untargeted metabolomics analysis.
2.6. Differential Metabolites Analysis
After statistical analysis, the untargeted LC–MS/MS data were further processed to identify differential metabolites across phenotypic groups. Student’s t-test was initially applied to detect statistically significant differences in metabolite levels between groups. The resulting p-values were adjusted for multiple comparisons using the Benjamini–Hochberg false discovery rate (FDR) correction to control for type I error. To further investigate the metabolic distinctions among groups, orthogonal partial least squares discriminant analysis (OPLS-DA), a supervised multivariate method, was performed using the metaX software (version 2.0.0) package. This approach enabled the identification of variables contributing most significantly to group separation. Variable importance in projection (VIP) scores were calculated within the OPLS-DA model, and metabolites with VIP scores greater than 1.0 were considered to be the most relevant features for discriminating between groups.
2.7. Ethics Statement
All fecal and plasma samples were obtained from the Department of Laboratory Medicine at Shenzhen Third People’s Hospital and were fully anonymized prior to analysis. The study protocol, including the use of these biological specimens, was reviewed and approved by the Institutional Ethics Committee of Shenzhen Third People’s Hospital (Approval No. 2025-015).
2.8. Statistical Analysis
Statistical analyses were conducted using GraphPad Prism software (version 9.5.1). Data were analyzed with Mann–Whitney U test, and p values were corrected using the Benjamini–Hochberg method. The results represent the means ± SD of three independent experiments unless specified otherwise. p values < 0.05 were considered statistically significant. False discovery rate controls were set at 10% (q < 0.1) using the Benjamini–Hochberg procedure. Details for all statistical tests can be found in the figure legends.
3. Results
3.1. The Gut Microbiota Composition Is Significantly Altered in LNTB Patients
In the present study, a total of eight healthy volunteers (Health group) and nine patients with LNTB were enrolled in this study. Relevant clinical data were statistically analyzed in
Supplementary Tables S1–S5. Comparative analysis revealed that diagnostic indicators were significantly higher in the LNTB group than in the healthy controls (
Tables S1–S5).
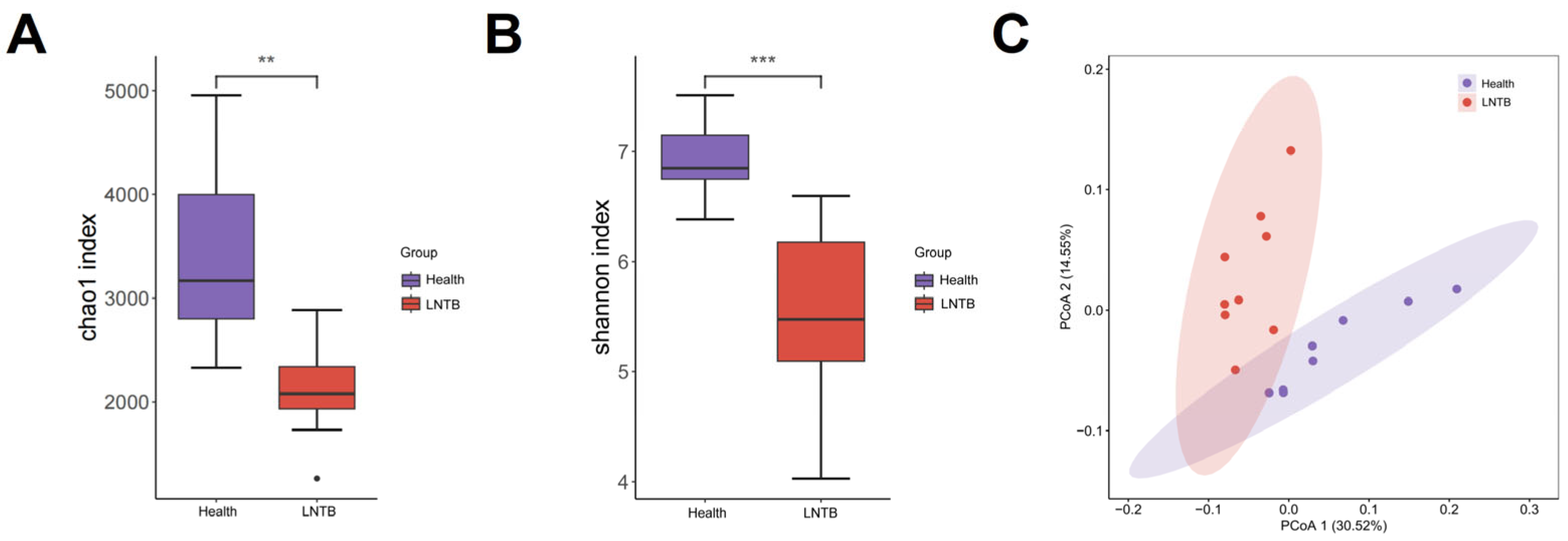
To investigate the gut microbiota composition between the Health and LNTB groups, metagenomic sequencing was performed in the present study. In terms of microbial richness and diversity, both the Chao1 and Shannon indices showed a significant reduction in alpha diversity in the LNTB group compared to healthy controls (
p < 0.01;
Figure 1A,B). These findings suggest that the gut microbial community was markedly altered by LNTB, potentially leading to disruptions in microbial homeostasis and decreased microbial diversity. Beta diversity was assessed by calculating the weighted UniFrac phylogenetic distances based on all ASVs. Principal coordinate analysis (PCA) revealed that the differences in community composition between groups were primarily driven by changes in the gut microbiota of LNTB patients (
Figure 1C).
These results revealed that the gut microbial composition in LNTB patients was significantly different from that of healthy individuals.
3.2. The Composition of Microbiota in LNTB Is Distinct from Healthy Control
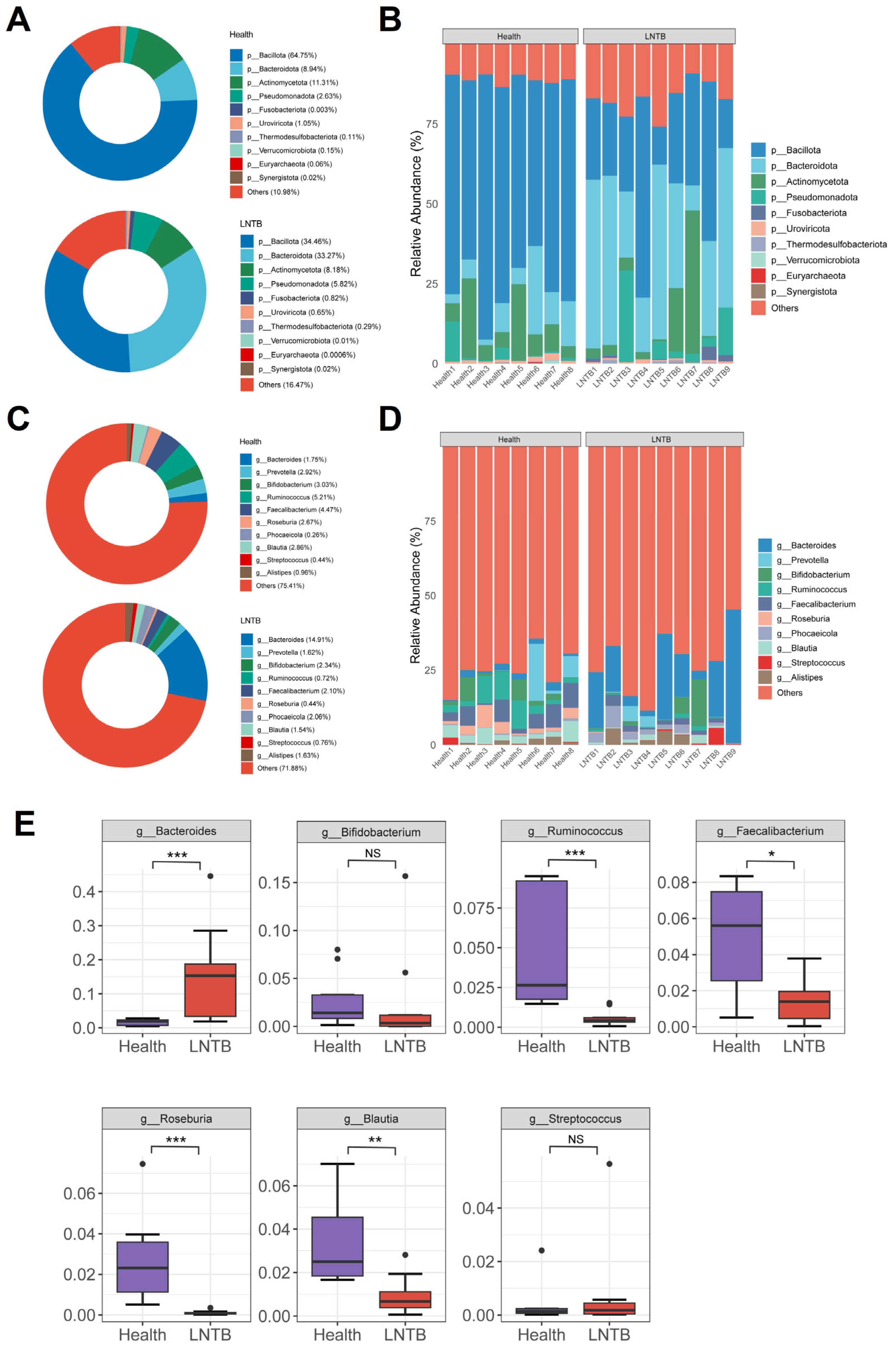
The human gut microbiota is predominantly composed of the following major phyla:
Bacillota,
Bacteroidetes,
Actinobacteria and
Proteobacteria, whose abundance dynamics are closely associated with the host’s physiological state [
22]. In the present study, the relative abundances of
Bacillota and
Bacteroidetes in the healthy group were 64.75% and 8.94%, respectively, while in patients with LNTB, they were 34.46% and 33.27%, respectively (
Figure 2A,B). Notably, the abundance of
Actinobacteria was significantly higher in the LNTB group than that in the healthy group (
Figure 2A).
At the genus level, compared with healthy individuals, the LNTB group exhibited varying degrees of dysbiosis involving several genera, including
Bacteroides,
Prevotella,
Bifidobacterium,
Ruminococcus,
Faecalibacterium,
Roseburia,
Phocaeicola,
Blautia,
Streptococcus, and
Alistipes (
Figure 2C,D). Among these genera,
Ruminococcus,
Faecalibacterium,
Roseburia, and
Blautia were significantly decreased in the LNTB group compared to the healthy group, as illustrated in
Figure 2E. The microbial composition at the species level was shown in
Figure S1. It is worth noting that these genera are known producers of SCFAs, such as butyrate, which play key roles in maintaining intestinal health and immune function.
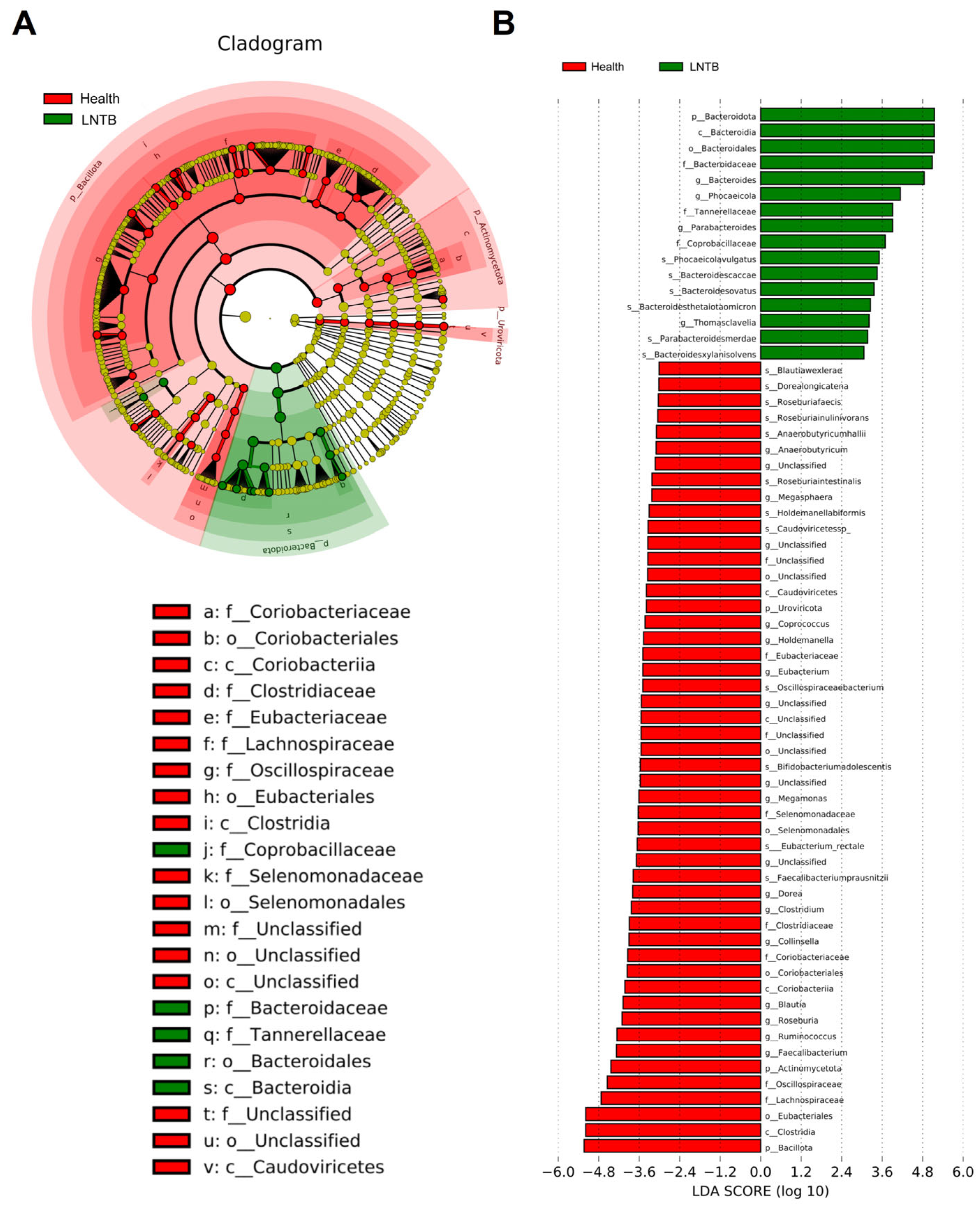
3.3. Differential Microbial Biomarkers Were Identified with LEfSe
To identify differentially distributed fecal microbiota, we conducted Metastat and linear discriminant analysis (LDA) effect size (LEfSe) analyses. These approaches provide valuable insights into the microbial differences between the LNTB and healthy groups. According to the LDA scores (
Figure 3A,B), several taxa were enriched in the healthy group. At the species level,
Blautiawexlerae,
Faecalibacteriumprausnitzii,
Eubacterium_rectale,
Roseburiafaecis,
Roseburia inulinivorans,
Anaerobutyricumhallii, showed significantly higher abundance in healthy individuals. At the genus level,
Faecalibacterium,
Roseburia,
Eubacterium,
Anaerobutyricum,
Clostridium, and
Butyricicoccus were also more abundant in the healthy group. The specific associations between microbial genera and species and different types of SCFAs (including butyrate, acetate, and propionate producers) were illustrated in
Figure S2. And the heatmaps of genus- and species-level differential analyses based on fecal metagenomic data from LNTB patients and healthy controls were shown in
Figures S3 and S4.
These taxa are well-known key SCFA-producing bacteria, particularly involved in butyrate synthesis, which is closely associated with maintaining colonic health and immune homeostasis. Subsequently, the microbial biomarkers identified by LEfSe would be incorporated into a network analysis to explore associations between gut microbiota and plasma metabolomic profiles.
3.4. KEGG Analysis of Gut Microbiota in LNTB Patients and Healthy Individuals
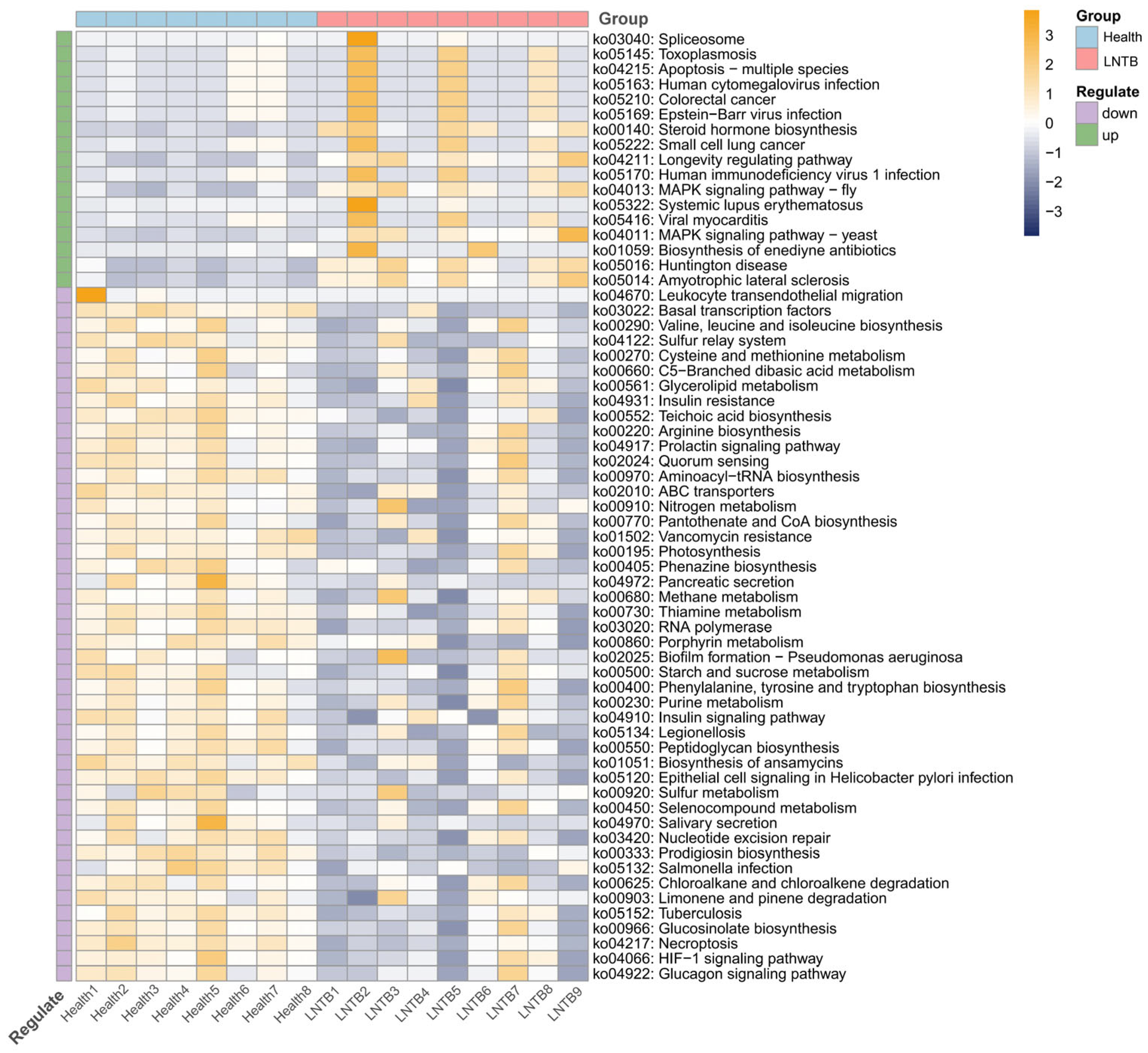
In the present study, functional annotation of the metagenomic microbiota was performed to investigate the potential functions of the gut microbiome and its interactions with the human host. As shown in
Figure 4, compared to healthy individuals, several signaling pathways—including those associated with colorectal cancer and small cell lung cancer—were found to be upregulated in the LNTB group. In contrast, pathways related to the biosynthesis of valine, leucine, and isoleucine, as well as the insulin and glucagon signaling pathways, were significantly downregulated.
The biosynthesis of valine, leucine, and isoleucine has been reported to be closely associated with SCFA production, while insulin and glucagon signaling pathways are known to be critical for host metabolic regulation [
23,
24,
25]. SCFAs are considered important metabolic products of the gut microbiota and have been shown to play key physiological roles. These include the maintenance of intestinal mucosal barrier integrity, inhibition of tumor cell proliferation and differentiation, suppression of intestinal inflammation, and stabilization of the gut microenvironment.
Therefore, the observed alterations suggest that SCFA biosynthesis and host metabolic processes may be disrupted in LNTB patients through functional changes in the gut microbiota.
3.5. SCFA-Related Metabolic Dysregulation in LNTB
Our results revealed that gut microbiota dysbiosis occurred in the LNTB group. Such compositional alterations provide a foundation for further functional characterization through virulence factor databases (e.g., VFDB) and pathway enrichment analyses. As shown in the VFDB results (
Figure S5), the dysbiotic gut microbiota may contribute to immune evasion and other host-related pathological processes.
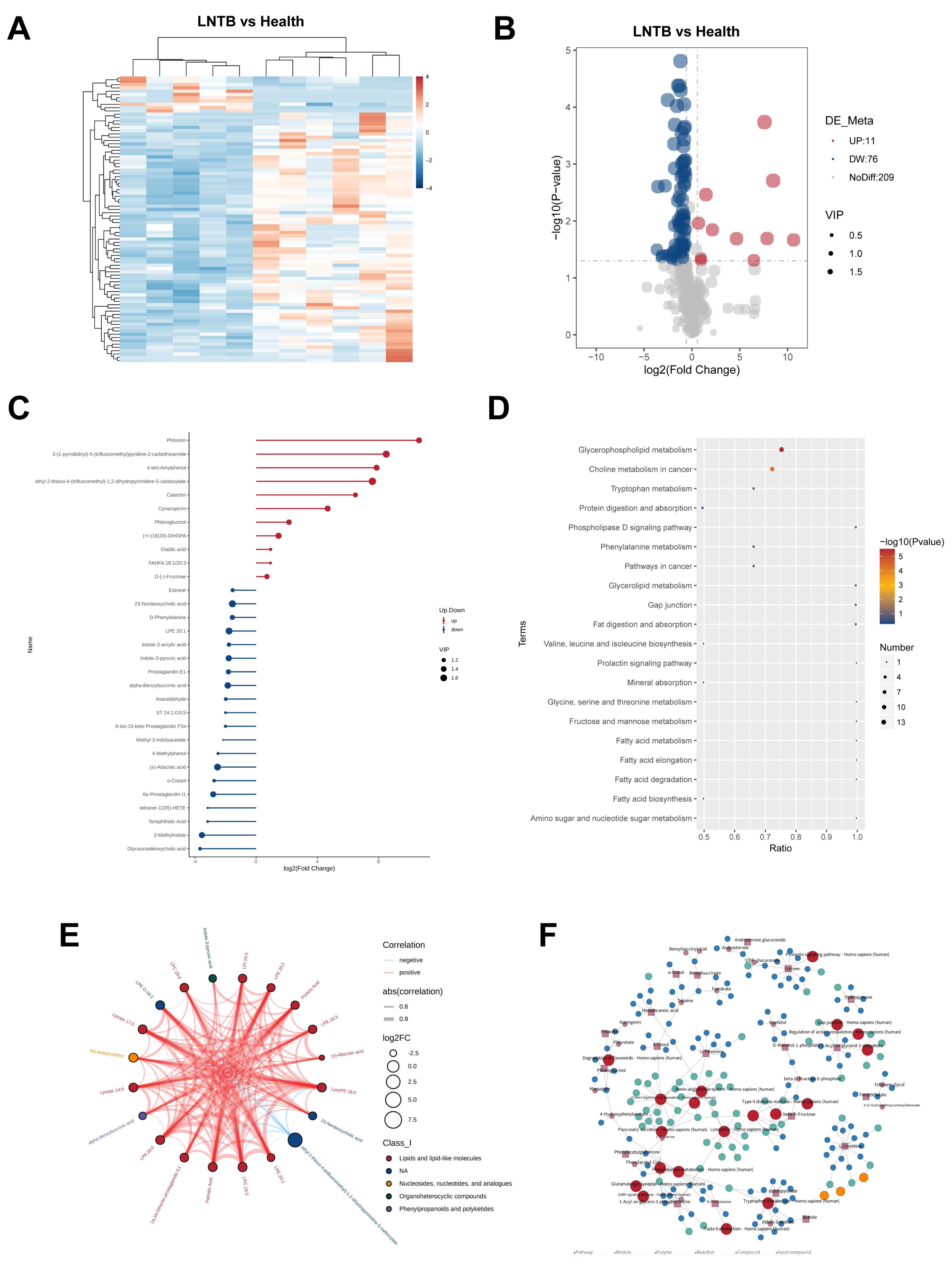
To further investigate alterations in the plasma metabolic profiles of LNTB patients compared to healthy individuals, untargeted plasma metabolomics analysis was conducted. As shown in the heatmap of differentially expressed metabolites (
Figure 5A), a total of 87 significantly altered metabolites were identified in the negative ion mode in the LNTB group, with 11 metabolites being upregulated and 76 downregulated compared to the healthy controls. The corresponding volcano plot is shown in
Figure 5B.
Among these, 30 significantly different metabolites were identified between the LNTB and healthy groups, including D-(–-fructose, α-benzylsuccinic acid, fatty acid-hydroxy fatty acid esters, tetranor-12(R)-HETE, glycochenodeoxycholic acid, and 23-deoxycholic acid, as shown in
Figure 5C. These metabolites have been associated with either SCFA synthesis or inflammatory processes.
In addition, pathway enrichment analysis based on the KEGG database revealed a total of 20 significantly altered pathways, as shown in
Figure 5D. These pathways included the biosynthesis of valine, leucine, and isoleucine; fructose and mannose metabolism; fatty acid metabolism; choline metabolism in cancer; and other cancer-related signaling pathways.
Figure 5E presents a chord diagram illustrating the correlation coefficients among differential metabolites, while
Figure 5F displays a KEGG-based regulatory network highlighting the intersections and functional relationships among the affected metabolic pathways.
These findings suggest that SCFA biosynthesis is indeed disrupted in patients with LNTB, and that multiple metabolic pathways involved in energy production and inflammation are affected.
3.6. Microbial Signatures Associated with Altered Plasma Metabolites in LNTB Patients
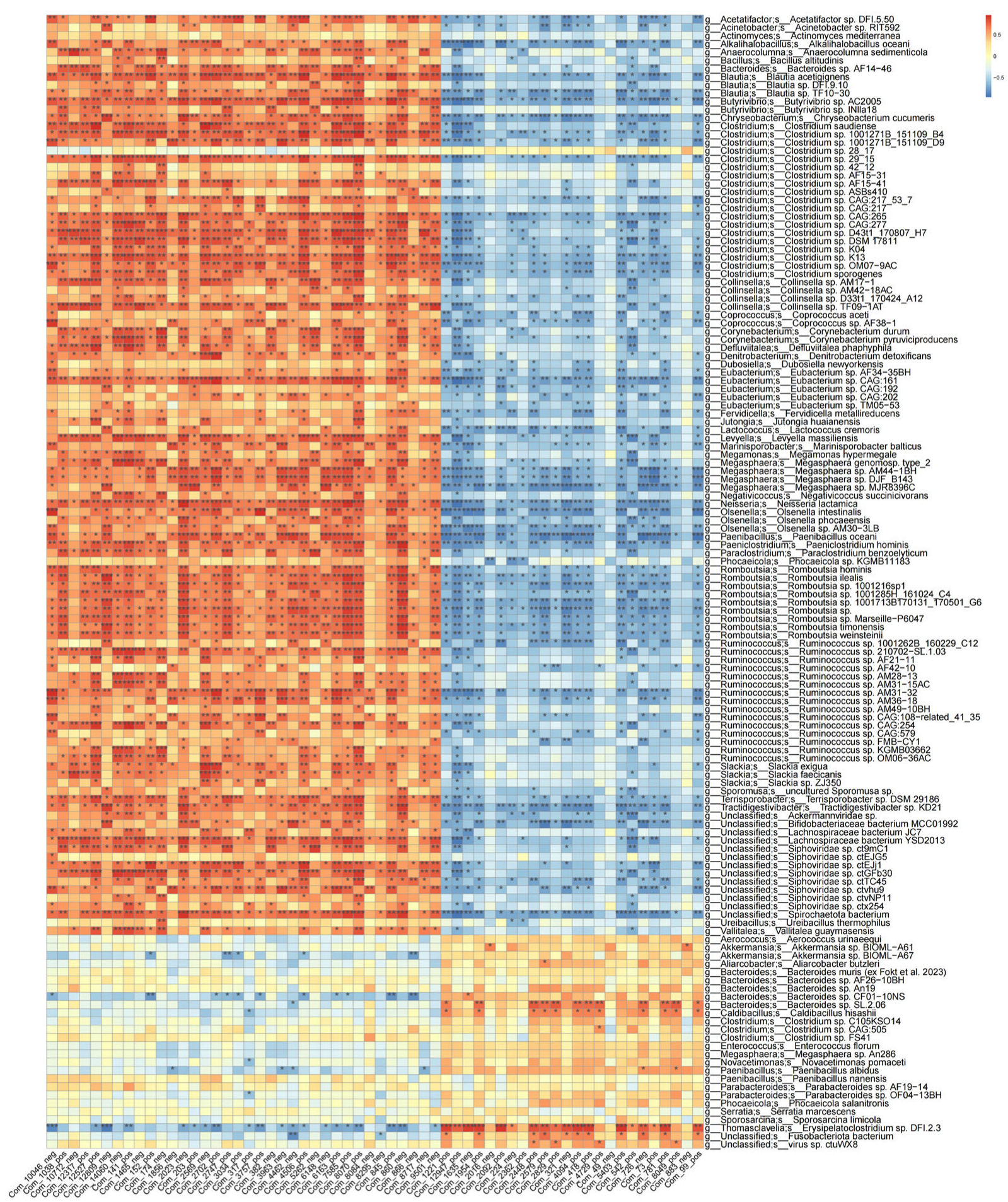
Multi-omics data were utilized to comprehensively characterize the fecal microbiota and plasma metabolite profiles from multiple perspectives. The observed positive or negative correlations between microbial taxa and metabolites may indicate that specific microbial species mediate the production or consumption of corresponding metabolites, or that certain metabolites promote or inhibit the growth of associated microbes.
To investigate these associations, large-scale correlation analyses were performed between the differentially abundant fecal genera and plasma metabolites (
Figure 6). Using Spearman correlation analysis, the significant associations (
p < 0.05) were identified in the LNTB and healthy groups.
The correlation analysis between differential fecal genera and plasma metabolites revealed that several taxa, including Blautia (species: Blautia acidifaciens), Butyricicoccus AC2005 (species: AC2005 AF38-1), Coprococcus (species: AF38-1), Ruminococcus, Bacteroides, and Clostridium (species: C105K04), were significantly correlated with plasma metabolites, such as α-benzylbutyric acid, 2-({2-[(3-methyl-5-pyridazinyl)amino]-2-oxoethyl}thio)acetic acid, 2-{2-[(1-methyl-1H-pyrazol-5-yl)amino]-2-oxoethoxy}acetic acid, and diethyl-2-(3-chloroanilino)succinic acid.
3.7. Functional Pathway–Metabolite Interactions Reveal Microbial Contributions to Host Metabolism in LNTB
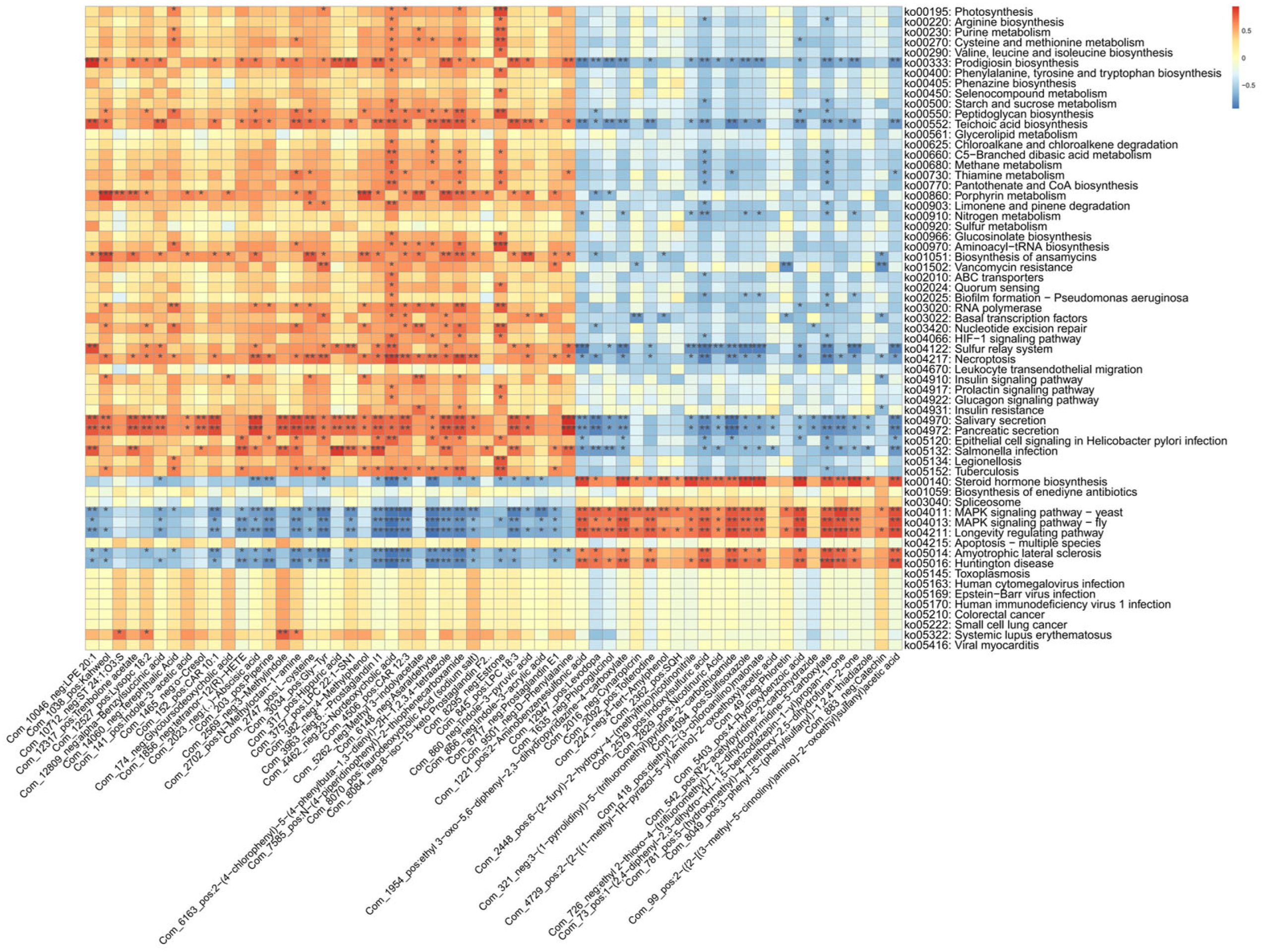
To further investigate these associations, large-scale correlation analyses were also performed between gut microbiota KEGG pathways and plasma metabolites (
Figure 7).
The correlation analysis between gut microbiota KEGG pathways and plasma metabolites revealed that the valine, leucine, and isoleucine biosynthesis pathway was positively correlated with 23-deoxycholic acid and estrone. Additionally, the insulin signaling, glucagon signaling, and diabetes-related pathways also showed positive associations with 23-deoxycholic acid and estrone. Notably, 23-deoxycholic acid has been implicated in the development of diseases, such as colorectal cancer and liver cancer, potentially due to its ability to induce oxidative stress and inflammation within the gut. Meanwhile, the valine, leucine, and isoleucine biosynthesis pathway has been closely associated with the synthesis of SCFAs.
Collectively, these findings highlight the existence of a complex host-microbiota-metabolite interaction network in the context of lymph node tuberculosis.
4. Discussion
Lymph node tuberculosis (LNTB) remains a serious threat to human health, and the identification of potential biomarkers, along with the elucidation of underlying pathogenic mechanisms, is of great significance for improving clinical diagnosis and therapeutic interventions. Many research studies [
7,
26] have demonstrated that alterations in the composition and function of the gut microbiota are closely associated with infectious and immune-related diseases. Therefore, characterizing the taxonomic and functional features of the gut microbiota in LNTB patients, as well as exploring the interactions between gut microbiota and plasma metabolites, is of considerable scientific and clinical value. In the present study, we identified specific gut microbial dysbiosis and plasma metabolic imbalances, and further investigated their functional alterations and interrelationships. These associations underscore the crucial role of host–microbiota–metabolite interactions in the pathogenesis and progression of LNTB.
Until now, multi-omics analyses of the gut microbiota and metabolism in LNTB remain largely unexplored. In the present study, we demonstrated that patients with LNTB exhibit significant alterations in gut microbiota composition compared to healthy individuals. Notably, there was a marked reduction in the relative abundance of SCFA-producing genera, including
Butyricicoccus,
Faecalibacterium,
Blautia,
Roseburia, and
Ruminococcus. In addition, metabolomic profiling revealed 87 significantly altered metabolites in the LNTB group compared to controls, of which 11 were upregulated and 76 were downregulated. Previous studies [
7] have reported that pulmonary tuberculosis (PTB) and anti-tuberculosis treatment can significantly disrupt the gut microbiota. Compared with healthy controls,
Mycobacterium tuberculosis (M.tb) infection has been associated with a slight reduction in gut microbial α-diversity, primarily linked to changes in the abundance of
Bacteroidota [
27]. The phylum
Bacteroidota is the second most dominant bacterial group in the healthy human colon, accounting for approximately 23% of the gut microbiota [
28]. Alterations in
Bacteroidota have also been shown to significantly influence host metabolism and energy homeostasis [
29]. In the present study, we similarly observed a modest decrease in α-diversity and alterations in
Bacteroidota abundance in patients with LNTB compared to healthy individuals. Consistent with previous findings in PTB, our results further support the notion that M.tb infection leads to notable changes in gut microbial diversity and composition, thereby reinforcing the reliability and biological relevance of our observations. Notably, unlike previously reported findings, the proportion of
Bacteroidota was more substantially increased in the gut microbiota of LNTB patients, suggesting that
Bacteroidota may be more sensitive to LNTB-associated changes. Several research studies [
30,
31] have reported a significant reduction in
Bacillota in patients with tuberculosis, which may be associated with immune dysregulation induced by
Mycobacterium tuberculosis infection. Consistent with these findings, our study also revealed a marked decrease in the abundance of
Bacillota in individuals with LNTB.
However, compared with previous studies [
32] on pulmonary tuberculosis (PTB), the dysbiosis of butyrate- and propionate-producing genera in the gut microbiota of LNTB patients shows notable differences. Prior research has demonstrated significant alterations in both the taxonomic composition and functional characteristics of the gut microbiota in patients with active TB compared to healthy controls. Specifically, genera, such as
Prevotella and
Bifidobacterium, were found to be more abundant in healthy individuals, whereas TB patients exhibited increased levels of SCFA-producing genera, including
Faecalibacterium,
Roseburia,
Eubacterium, and
Phascolarctobacterium, which are known producers of butyrate and propionate [
32]. In contrast, the present study revealed a significant reduction in key butyrate-producing genera in the fecal samples of LNTB patients, including
Butyricicoccus,
Coprococcus,
Blautia,
Roseburia, and
Ruminococcus. These findings suggest that SCFA-associated microbial alterations in LNTB may follow a different pattern from those observed in PTB, potentially reflecting distinct host–microbiota interactions in extrapulmonary TB.
Blaak et al. [
33] have revealed that SCFAs, including acetate, propionate, and butyrate, are key microbial metabolites derived from the fermentation of indigestible carbohydrates and play a vital role in maintaining gut and metabolic health [
34]. SCFAs support intestinal integrity by regulating luminal pH, stimulating mucus production, fueling epithelial cells, and modulating immune responses. Systemically, they influence appetite, energy balance, glucose homeostasis, and immunity. Among them, butyrate exerts strong anti-inflammatory effects and modulates immune signaling via the NF-κB pathway. In active pulmonary tuberculosis, intestinal butyrate levels were found to be fivefold lower than in healthy controls [
35], accompanied by reduced abundance of SCFA-producing bacteria and related metabolic pathways [
36,
37]. This disruption may compromise mucosal defenses and promote pathogenic colonization, contributing to impaired immune function. Consistently, our metabolomic data revealed a dysregulation of SCFA-related metabolites in the plasma of patients with LNTB. Notably, metabolomic analysis in the present study revealed abnormal levels of several metabolites in the peripheral blood of patients with LNTB, including D-(-)-fructose, α-benzylsuccinic acid, fatty acid-hydroxy fatty acid esters, tetranor-12(R)-HETE, glycoursodeoxycholic acid, and 23-deoxycholic acid-metabolites, that are closely associated with SCFA biosynthesis. In addition, several key metabolic pathways were found to be suppressed in LNTB patients, including valine, leucine, and isoleucine biosynthesis, fructose and mannose metabolism, and fatty acid metabolism pathways. These alterations further support the notion that SCFA biosynthesis is disrupted in LNTB. Moreover, integrative metagenomic and metabolomic analyses indicated that the biosynthesis pathway of branched-chain amino acids (valine, leucine, and isoleucine) was positively correlated with the production of 23-deoxycholic acid and estrone. Significant changes were also observed in the abundance of SCFA-associated bacterial genera, including
Blautia (acetate-producing),
Butyricimonas (AC2005),
Faecalicoccus (AF38-1),
Ruminococcus, and
Clostridium sp. C105K04. Dysregulation of these taxa was closely linked to the observed alterations in SCFA-related metabolites, further illustrating the disruption of SCFA metabolism and its association with gut microbiota imbalance in LNTB patients. These findings suggest that dysbiosis of specific SCFA-associated microbial populations may influence key physiological processes, such as appetite regulation, energy expenditure, glucose homeostasis, and immune modulation, in individuals with LNTB. This highlights the potential importance of targeting the gut microbiota and SCFA metabolism in the clinical management and therapeutic strategies for LNTB.
Previous studies [
37] have reported that
Clostridium species play an important role in maintaining intestinal homeostasis, protecting the mucosal barrier, and promoting host metabolism. These bacteria are capable of producing SCFAs through the fermentation of dietary fibers. During anti-tuberculosis treatment, the gut microbiota is involved in the metabolic processing of drugs. As treatment progresses and host immunity improves, the abundance of
Clostridium has been observed to increase significantly, coinciding with clinical improvement in tuberculosis patients.
Prevotella, a genus whose primary fermentation products include lactic acid, isovaleric acid, and acetic acid, has also been implicated in SCFA metabolism. Among these, acetate is the most abundant SCFA in the peripheral circulation. Therefore, a reduction in
Prevotella may lead to decreased acetate production, contributing to an overall reduction in SCFA levels [
32]. LNTB remains a challenging condition to treat, often requiring prolonged therapeutic regimens. In this study, we report for the first time that several butyrate-producing species—including
Faecalibacterium prausnitzii,
Butyricicoccus pullicaecorum, and
Eubacterium rectale—were significantly more abundant in healthy controls compared to LNTB patients. At the genus level, SCFA-associated taxa, such as
Butyricicoccus,
Faecalicoccus,
Blautia,
Roseburia, and
Ruminococcus, were also found in higher abundance in healthy individuals. These findings suggest that SCFA-producing bacterial populations may play a beneficial role in modulating host immunity and could potentially serve as targets for future microbiota-based therapeutic strategies for LNTB. Additionally, these microbial taxa may hold promise as non-invasive biomarkers for the diagnosis or monitoring of lymph node tuberculosis.
We acknowledge that further research is needed to address several limitations of the present study. First, although we are the first to characterize gut microbiota dysbiosis in patients with LNTB in our present acknowledgment, the diagnostic performance of the identified combined biomarker panel requires independent validation in larger and geographically diverse cohorts. This is essential to prevent potential overestimation of diagnostic accuracy due to the limited sample size and single-center design of our study. Moreover, given the current scarcity of related studies, many of the observed associations require more robust confirmation through expanded sample collections and experimental validation, including in vivo animal models. According to previous studies [
38,
39,
40], antibiotics—especially broad-spectrum antibiotics—can eliminate a large number of gut microorganisms, including beneficial bacteria, leading to a significant reduction in species richness and diversity of the gut microbiota. To avoid this confounding effect, the present study only included patients who were newly diagnosed with lymph node tuberculosis and had not received any prior medication. Although this strict inclusion criterion limited the number of samples collected, the low heterogeneity among the included samples ensured their validity. In future studies, we plan to expand the sample size to enhance the robustness of our findings. Moreover, while gut microbiota holds promise as a potential diagnostic tool for LNTB, microbiota-based diagnostic approaches must undergo further large-scale investigations to evaluate their accuracy and specificity before clinical implementation can be considered.



 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}