
Exosome-Modified AAV Gene Therapy Attenuates Autoimmune Hepatitis via Enhanced Regulatory T Cell Targeting and Immune Modulation

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Experimental Design in AIH Mouse Model
2.3. Histology
2.4. Lentivirus Packaging and Stable Cell Line Construction
2.5. Exo-AAV and AAV Preparations
2.6. Flow Cytometry Analysis
2.7. ELISA
2.8. Western Blot Analysis
2.9. Statistical Analysis
3. Results
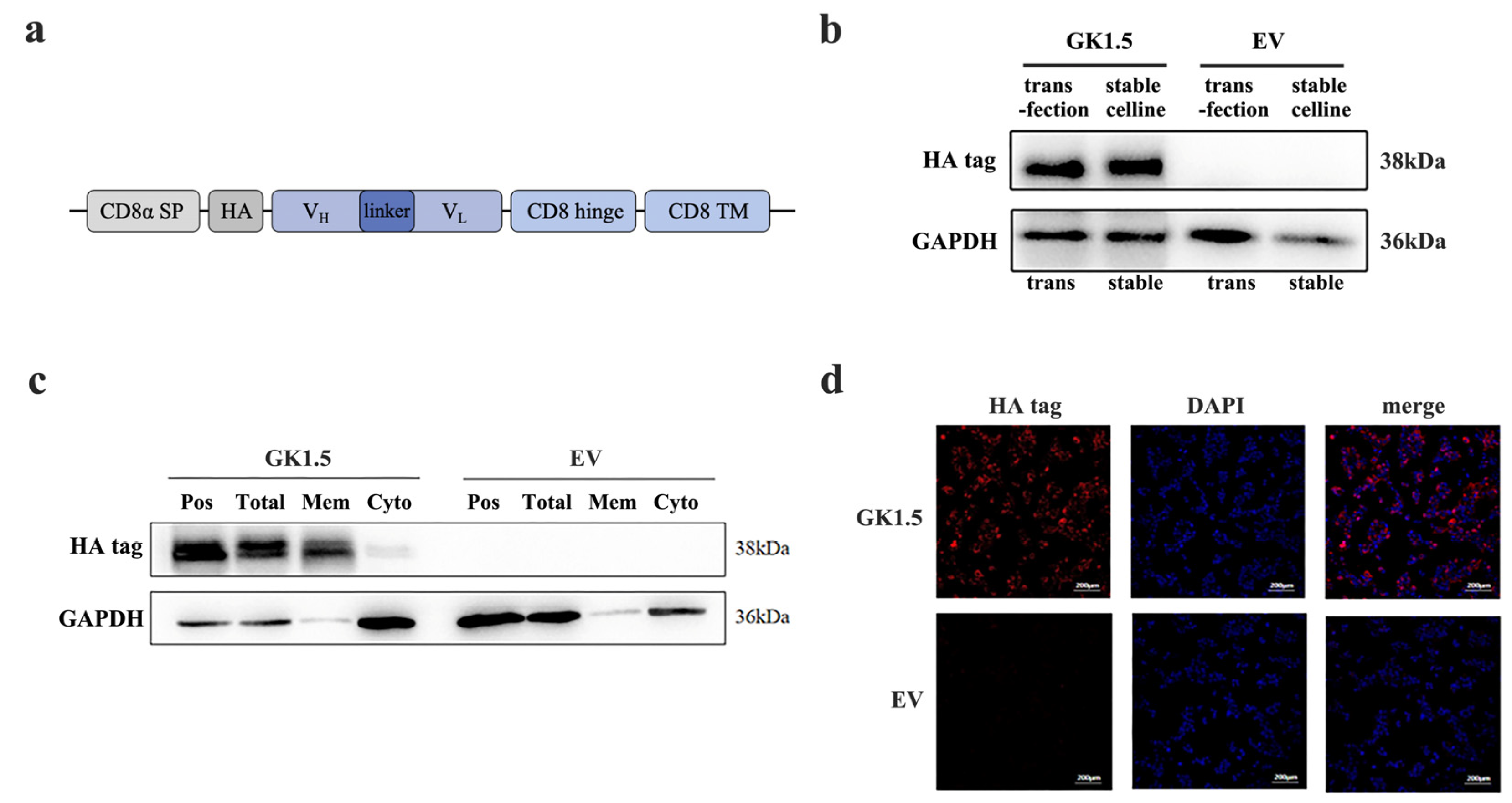
3.1. Establishment and Verification of Stable Transfected Cell Line Overexpressing CD4-Binding Protein
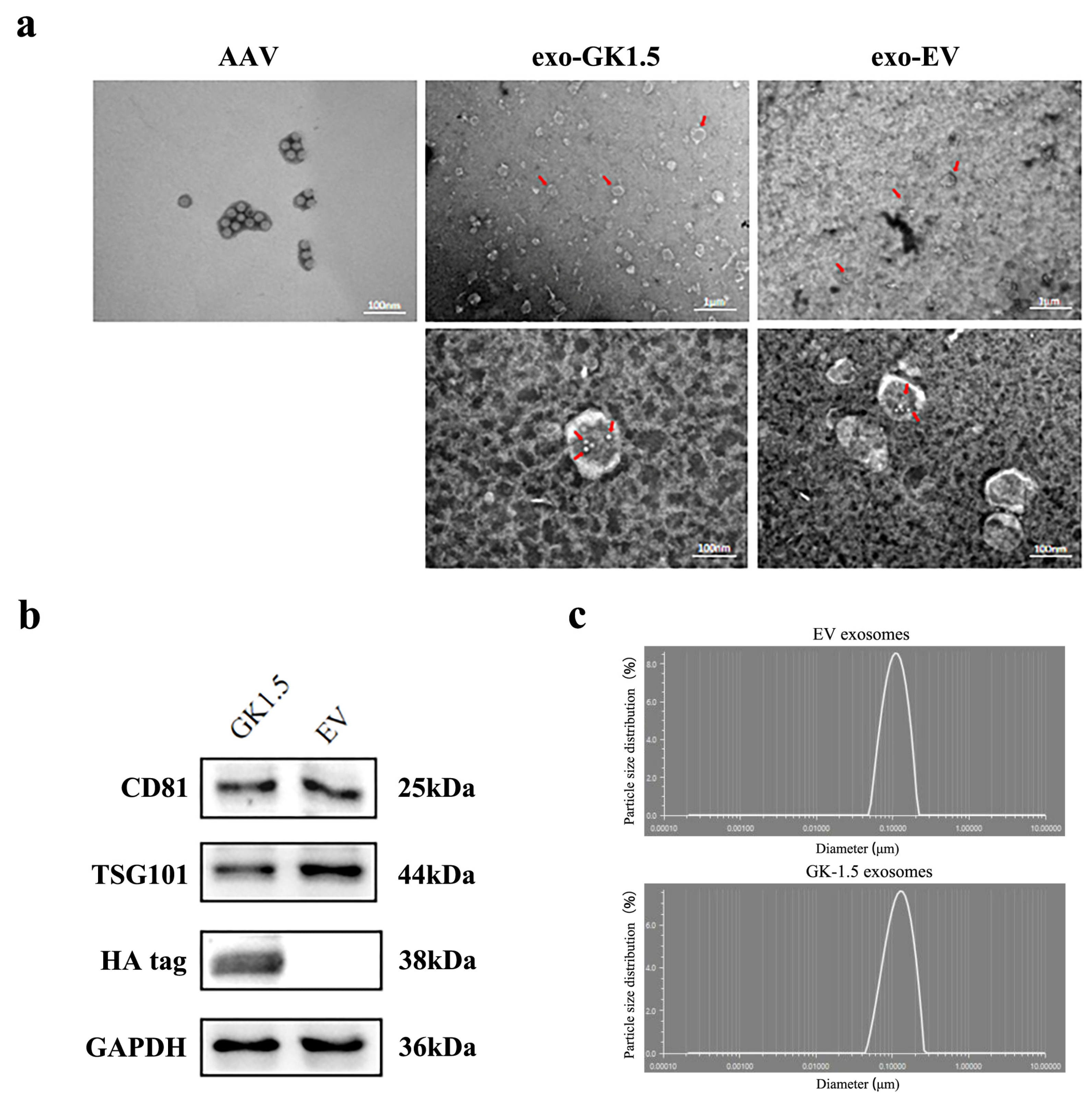
3.2. Exo-AAV Identification
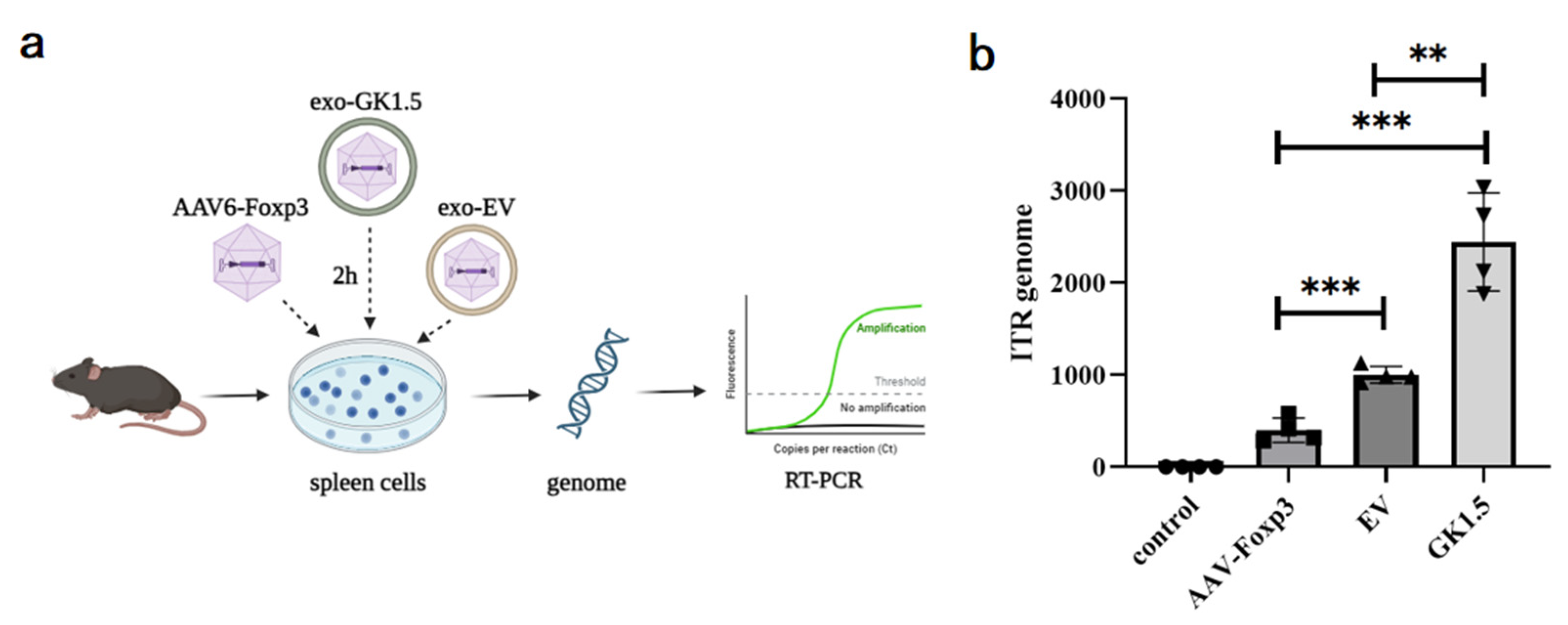
3.3. Detection of Exo-AAV Binding Ability In Vitro
3.4. Examination of Liver Damage in AIH Mice After Exo-AAV Treatment
3.5. Detection of Immune Cells and Inflammatory Cytokines in AIH Mice Liver After Exo-AAV Treatment
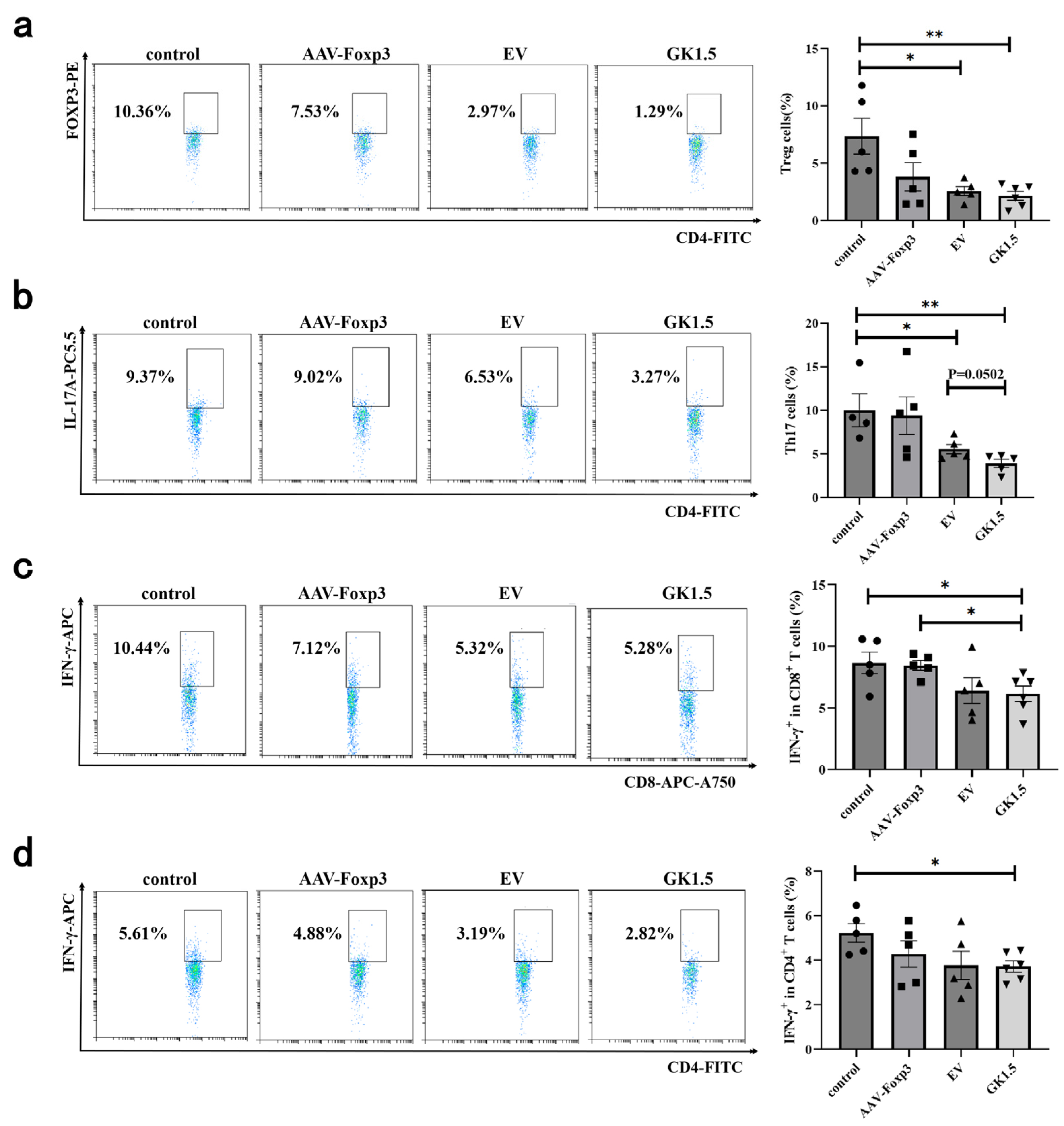
3.6. Identification of Spleen T Cells in AIH Mice After Exo-AAV Treatment
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Moy, L.; Levine, J. Autoimmune hepatitis: A classic autoimmune liver disease. Curr. Probl. Pediatr. Adolesc. Health Care 2014, 44, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Weiler-Normann, C.; Schramm, C.; Quaas, A.; Wiegard, C.; Glaubke, C.; Pannicke, N.; Moller, S.; Lohse, A.W. Infliximab as a rescue treatment in difficult-to-treat autoimmune hepatitis. J. Hepatol. 2013, 58, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Sierra, R.; Marenco-Flores, A.; Alsaqa, M.; Barba, R.; Cuellar-Lobo, M.; Barberan, C.; Sierra, L. Autoimmune Hepatitis Management: Recent Advances and Future Prospects. Livers 2024, 4, 240–252. [Google Scholar] [CrossRef]
- Shiffman, M.L. Autoimmune Hepatitis: Epidemiology, Subtypes, and Presentation. Clin. Liver Dis. 2024, 28, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Wang, J.; Liu, R.; Wang, Z.; Li, Y.; Zhang, Y.; Hao, X.; Huang, Y.; Xie, W.; Wei, H. Amelioration of concanavalin A-induced autoimmune hepatitis by magnesium isoglycyrrhizinate through inhibition of CD4+CD25−CD69+ subset proliferation. Drug Des. Devel. Ther. 2016, 10, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Feng, X.; Yan, W.; Tian, D. Regulatory T Cells in Autoimmune Hepatitis: Unveiling Their Roles in Mouse Models and Patients. Front. Immunol. 2020, 11, 575572. [Google Scholar] [CrossRef]
- Muratori, L.; Lohse, A.W.; Lenzi, M. Diagnosis and management of autoimmune hepatitis. BMJ 2023, 380, e070201. [Google Scholar] [CrossRef]
- Jimenez-Rivera, C.; Ling, S.C.; Ahmed, N.; Yap, J.; Aglipay, M.; Barrowman, N.; Graitson, S.; Critch, J.; Rashid, M.; Ng, V.L.; et al. Incidence and Characteristics of Autoimmune Hepatitis. Pediatrics 2015, 136, e1237–e1248. [Google Scholar] [CrossRef]
- Goel, A.; Kwo, P. Treatment of Autoimmune Hepatitis. Clin. Liver Dis. 2024, 28, 51–61. [Google Scholar] [CrossRef]
- van Gerven, N.M.; Verwer, B.J.; Witte, B.I.; van Hoek, B.; Coenraad, M.J.; van Erpecum, K.J.; Beuers, U.; van Buuren, H.R.; de Man, R.A.; Drenth, J.P.; et al. Relapse is almost universal after withdrawal of immunosuppressive medication in patients with autoimmune hepatitis in remission. J. Hepatol. 2013, 58, 141–147. [Google Scholar] [CrossRef]
- Terziroli Beretta-Piccoli, B.; Mieli-Vergani, G.; Vergani, D. Autoimmune hepatitis: Standard treatment and systematic review of alternative treatments. World J. Gastroenterol. 2017, 23, 6030–6048. [Google Scholar] [CrossRef] [PubMed]
- Krawitt, E.L. Autoimmune hepatitis. N. Engl. J. Med. 1996, 334, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Cassim, S.; Bilodeau, M.; Vincent, C.; Lapierre, P. Novel Immunotherapies for Autoimmune Hepatitis. Front. Pediatr. 2017, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Tiegs, G.; Hentschel, J.; Wendel, A. A T cell-dependent experimental liver injury in mice inducible by concanavalin A. J. Clin. Investig. 1992, 90, 196–203. [Google Scholar] [CrossRef]
- Heymann, F.; Hamesch, K.; Weiskirchen, R.; Tacke, F. The concanavalin A model of acute hepatitis in mice. Lab. Anim. 2015, 49, 12–20. [Google Scholar] [CrossRef]
- Varrin-Doyer, M.; Shetty, A.; Spencer, C.M.; Schulze-Topphoff, U.; Weber, M.S.; Bernard, C.C.; Forsthuber, T.; Cree, B.A.; Slavin, A.J.; Zamvil, S.S. MOG transmembrane and cytoplasmic domains contain highly stimulatory T-cell epitopes in MS. Neurol. Neuroimmunol. Neuroinflamm. 2014, 1, e20. [Google Scholar] [CrossRef] [PubMed]
- Keeler, G.D.; Kumar, S.; Palaschak, B.; Silverberg, E.L.; Markusic, D.M.; Jones, N.T.; Hoffman, B.E. Gene Therapy-Induced Antigen-Specific Tregs Inhibit Neuro-inflammation and Reverse Disease in a Mouse Model of Multiple Sclerosis. Mol. Ther. 2018, 26, 173–183. [Google Scholar] [CrossRef]
- Tian, L.; Yang, P.; Lei, B.; Shao, J.; Wang, C.; Xiang, Q.; Wei, L.; Peng, Z.; Kijlstra, A. AAV2-mediated subretinal gene transfer of hIFN-alpha attenuates experimental autoimmune uveoretinitis in mice. PLoS ONE 2011, 6, e19542. [Google Scholar] [CrossRef]
- Li, F.; Zhu, X.; Yang, Y.; Huang, L.; Xu, J. TIPE2 Alleviates Systemic Lupus Erythematosus Through Regulating Macrophage Polarization. Cell Physiol. Biochem. 2016, 38, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Bowles, D.E.; McPhee, S.W.; Li, C.; Gray, S.J.; Samulski, J.J.; Camp, A.S.; Li, J.; Wang, B.; Monahan, P.E.; Rabinowitz, J.E.; et al. Phase 1 gene therapy for Duchenne muscular dystrophy using a translational optimized AAV vector. Mol. Ther. 2012, 20, 443–455. [Google Scholar] [CrossRef]
- Wu, P.; Xiao, W.; Conlon, T.; Hughes, J.; Agbandje-McKenna, M.; Ferkol, T.; Flotte, T.; Muzyczka, N. Mutational analysis of the adeno-associated virus type 2 (AAV2) capsid gene and construction of AAV2 vectors with altered tropism. J. Virol. 2000, 74, 8635–8647. [Google Scholar] [CrossRef] [PubMed]
- Asokan, A.; Conway, J.C.; Phillips, J.L.; Li, C.; Hegge, J.; Sinnott, R.; Yadav, S.; DiPrimio, N.; Nam, H.J.; Agbandje-McKenna, M.; et al. Reengineering a receptor footprint of adeno-associated virus enables selective and systemic gene transfer to muscle. Nat. Biotechnol. 2010, 28, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.; Horowitz, E.D.; Troupes, A.N.; Brown, S.M.; Pulicherla, N.; Samulski, R.J.; Agbandje-McKenna, M.; Asokan, A. Engraftment of a galactose receptor footprint onto adeno-associated viral capsids improves transduction efficiency. J. Biol. Chem. 2013, 288, 28814–28823. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, E.; Song, L.; Llanga, T.; Bower, J.J.; Cullen, M.; Salmon, J.H.; Hirsch, M.L.; Gilger, B.C. AAV-mediated expression of HLA-G1/5 reduces severity of experimental autoimmune uveitis. Sci. Rep. 2019, 9, 19864. [Google Scholar] [CrossRef]
- Contini, P.; Murdaca, G.; Puppo, F.; Negrini, S. HLA-G Expressing Immune Cells in Immune Mediated Diseases. Front. Immunol. 2020, 11, 1613. [Google Scholar] [CrossRef]
- Alegre, E.; Rizzo, R.; Bortolotti, D.; Fernandez-Landazuri, S.; Fainardi, E.; Gonzalez, A. Some basic aspects of HLA-G biology. J. Immunol. Res. 2014, 2014, 657625. [Google Scholar] [CrossRef]
- Bakkum, D.J.; Radivojevic, M.; Frey, U.; Franke, F.; Hierlemann, A.; Takahashi, H. Parameters for burst detection. Front. Comput. Neurosci. 2013, 7, 193. [Google Scholar] [CrossRef]
- Salas, J.R.; Chen, B.Y.; Wong, A.; Cheng, D.; Van Arnam, J.S.; Witte, O.N.; Clark, P.M. 18F-FAC PET Selectively Images Liver-Infiltrating CD4 and CD8 T Cells in a Mouse Model of Autoimmune Hepatitis. J. Nucl. Med. 2018, 59, 1616–1623. [Google Scholar] [CrossRef]
- Zheng, C.; Yin, S.; Yang, Y.; Yu, Y.; Xie, X. CD24 aggravates acute liver injury in autoimmune hepatitis by promoting IFN-gamma production by CD4+ T cells. Cell Mol. Immunol. 2018, 15, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, P.; Djilali-Saiah, I.; Vitozzi, S.; Alvarez, F. A murine model of type 2 autoimmune hepatitis: Xenoimmunization with human antigens. Hepatology 2004, 39, 1066–1074. [Google Scholar] [CrossRef] [PubMed]
- Hardtke-Wolenski, M.; Fischer, K.; Noyan, F.; Schlue, J.; Falk, C.S.; Stahlhut, M.; Woller, N.; Kuehnel, F.; Taubert, R.; Manns, M.P.; et al. Genetic predisposition and environmental danger signals initiate chronic autoimmune hepatitis driven by CD4+ T cells. Hepatology 2013, 58, 718–728. [Google Scholar] [CrossRef] [PubMed]
- Holdener, M.; Hintermann, E.; Bayer, M.; Rhode, A.; Rodrigo, E.; Hintereder, G.; Johnson, E.F.; Gonzalez, F.J.; Pfeilschifter, J.; Manns, M.P.; et al. Breaking tolerance to the natural human liver autoantigen cytochrome P450 2D6 by virus infection. J. Exp. Med. 2008, 205, 1409–1422. [Google Scholar] [CrossRef] [PubMed]
- Longhi, M.S.; Meda, F.; Wang, P.; Samyn, M.; Mieli-Vergani, G.; Vergani, D.; Ma, Y. Expansion and de novo generation of potentially therapeutic regulatory T cells in patients with autoimmune hepatitis. Hepatology 2008, 47, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Longhi, M.S.; Liberal, R.; Holder, B.; Robson, S.C.; Ma, Y.; Mieli-Vergani, G.; Vergani, D. Inhibition of interleukin-17 promotes differentiation of CD25(-) cells into stable T regulatory cells in patients with autoimmune hepatitis. Gastroenterology 2012, 142, 1526–1535.e6. [Google Scholar] [CrossRef] [PubMed]
- Longhi, M.S.; Mieli-Vergani, G.; Vergani, D. Regulatory T cells in autoimmune hepatitis: An updated overview. J. Autoimmun. 2021, 119, 102619. [Google Scholar] [CrossRef]
- Grant, C.R.; Liberal, R.; Holder, B.S.; Cardone, J.; Ma, Y.; Robson, S.C.; Mieli-Vergani, G.; Vergani, D.; Longhi, M.S. Dysfunctional CD39(POS) regulatory T cells and aberrant control of T-helper type 17 cells in autoimmune hepatitis. Hepatology 2014, 59, 1007–1015. [Google Scholar] [CrossRef]
- Maguire, C.A.; Balaj, L.; Sivaraman, S.; Crommentuijn, M.H.; Ericsson, M.; Mincheva-Nilsson, L.; Baranov, V.; Gianni, D.; Tannous, B.A.; Sena-Esteves, M.; et al. Microvesicle-associated AAV vector as a novel gene delivery system. Mol. Ther. 2012, 20, 960–971. [Google Scholar] [CrossRef]
- Hull, J.A.; Mietzsch, M.; Chipman, P.; Strugatsky, D.; McKenna, R. Structural characterization of an envelope-associated adeno-associated virus type 2 capsid. Virology 2022, 565, 22–28. [Google Scholar] [CrossRef]
- Liu, B.; Li, Z.; Huang, S.; Yan, B.; He, S.; Chen, F.; Liang, Y. AAV-Containing Exosomes as a Novel Vector for Improved Gene Delivery to Lung Cancer Cells. Front. Cell Dev. Biol. 2021, 9, 707607. [Google Scholar] [CrossRef]
- Wassmer, S.J.; Carvalho, L.S.; Gyorgy, B.; Vandenberghe, L.H.; Maguire, C.A. Exosome-associated AAV2 vector mediates robust gene delivery into the murine retina upon intravitreal injection. Sci. Rep. 2017, 7, 45329. [Google Scholar] [CrossRef]
- Breuer, C.B.; Hanlon, K.S.; Natasan, J.S.; Volak, A.; Meliani, A.; Mingozzi, F.; Kleinstiver, B.P.; Moon, J.J.; Maguire, C.A. In vivo engineering of lymphocytes after systemic exosome-associated AAV delivery. Sci. Rep. 2020, 10, 4544. [Google Scholar] [CrossRef] [PubMed]
- You, Z.; Li, Y.; Wang, Q.; Zhao, Z.; Li, Y.; Qian, Q.; Li, B.; Zhang, J.; Huang, B.; Liang, J.; et al. The Clinical Significance of Hepatic CD69+ CD103+ CD8+ Resident-Memory T Cells in Autoimmune Hepatitis. Hepatology 2021, 74, 847–863. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Xu, J.; Bromberg, J.S. Regulatory T cell migration during an immune response. Trends Immunol. 2012, 33, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Tu, H.; Yin, X.; Peng, C.; Dou, C.; Yang, W.; Wu, W.; Guan, X.; Li, J.; Yan, H.; et al. Targeting Glutamine Metabolism Ameliorates Autoimmune Hepatitis via Inhibiting T Cell Activation and Differentiation. Front. Immunol. 2022, 13, 880262. [Google Scholar] [CrossRef]
- Wang, L.; Yan, F.; Zhang, J.; Xiao, Y.; Wang, C.; Zhu, Y.; Li, C.; Liu, Z.; Li, W.; Wang, C.; et al. Cornuside improves murine autoimmune hepatitis through inhibition of inflammatory responses. Phytomedicine 2023, 120, 155077. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shao, W.; Huang, W.; Wang, Y.; Sima, H.; Ma, K.; Chen, R.; Han, H.; Yang, Y.; Bao, Y.; Pei, X.; et al. Exosome-Modified AAV Gene Therapy Attenuates Autoimmune Hepatitis via Enhanced Regulatory T Cell Targeting and Immune Modulation. Microorganisms 2025, 13, 823. https://doi.org/10.3390/microorganisms13040823
Shao W, Huang W, Wang Y, Sima H, Ma K, Chen R, Han H, Yang Y, Bao Y, Pei X, et al. Exosome-Modified AAV Gene Therapy Attenuates Autoimmune Hepatitis via Enhanced Regulatory T Cell Targeting and Immune Modulation. Microorganisms. 2025; 13(4):823. https://doi.org/10.3390/microorganisms13040823
Chicago/Turabian StyleShao, Wenwei, Weilin Huang, Yixuan Wang, Helin Sima, Kai Ma, Rongtao Chen, Heqiao Han, Yixuan Yang, Yuchen Bao, Xiaolei Pei, and et al. 2025. "Exosome-Modified AAV Gene Therapy Attenuates Autoimmune Hepatitis via Enhanced Regulatory T Cell Targeting and Immune Modulation" Microorganisms 13, no. 4: 823. https://doi.org/10.3390/microorganisms13040823
APA StyleShao, W., Huang, W., Wang, Y., Sima, H., Ma, K., Chen, R., Han, H., Yang, Y., Bao, Y., Pei, X., & Zhang, L. (2025). Exosome-Modified AAV Gene Therapy Attenuates Autoimmune Hepatitis via Enhanced Regulatory T Cell Targeting and Immune Modulation. Microorganisms, 13(4), 823. https://doi.org/10.3390/microorganisms13040823