

Biocontrol Effect of Bacillus velezensis D7-8 on Potato Common Scab and Its Complete Genome Sequence Analysis

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Pathogens Resources and Culture Medium for the Experiment
2.2. Isolation of Endophytic Bacteria
2.3. Antagonistic Screening of S. acidiscabies Strains
2.4. Detection of Broad-Spectrum Fungistatic Ability of D7-8
2.5. Pot Trials
2.6. Preliminary Molecular Identification of D7-8 Strain
2.7. Genome Sequencing and Annotation of D7-8
2.8. Comparative Genomic Analysis of D7-8
2.9. Identification of Lipopeptide Compounds by UPLC-Q-Exactive HRMS
2.10. Statistical Analysis
3. Results
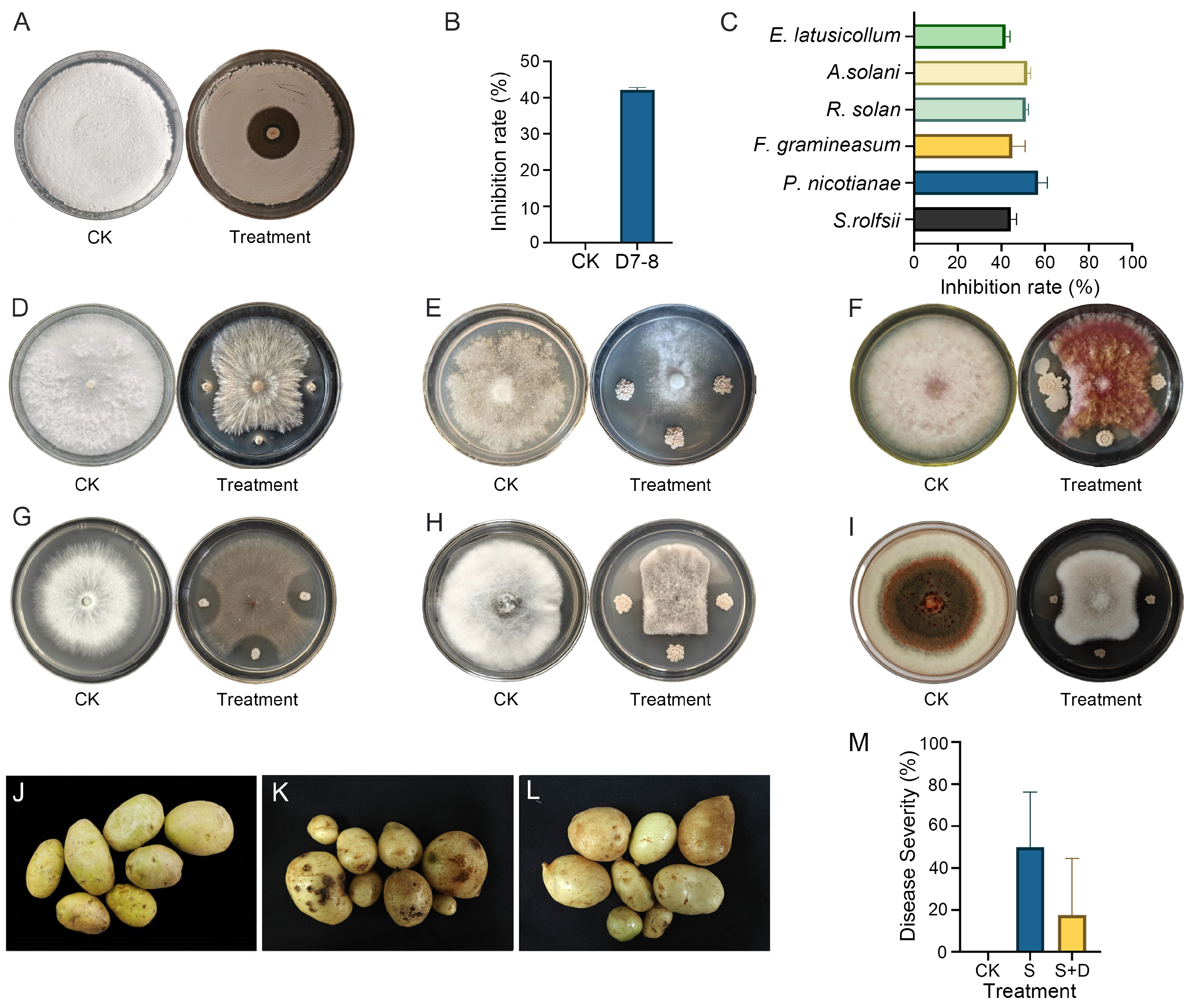
3.1. Inhibition and Control Effect of D7-8 on Potato Common Scab and Its Broad-Spectrum Fungal Inhibition Ability
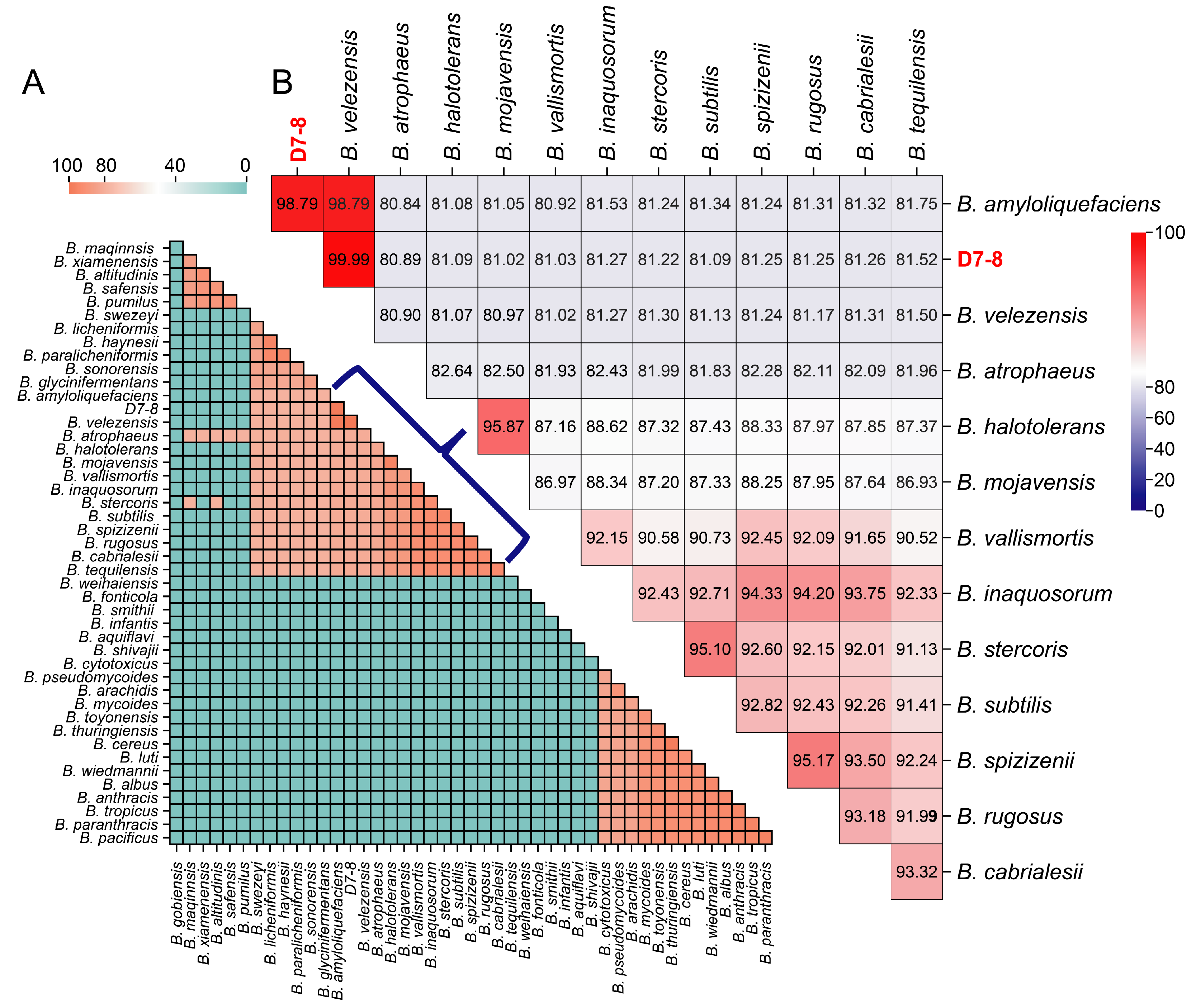
3.2. Phylogenomic Analysis of D7-8
3.3. Complete Genome Sequencing and Annotation of D7-8
3.4. Comparative Genomic Analysis of Bacillus Species
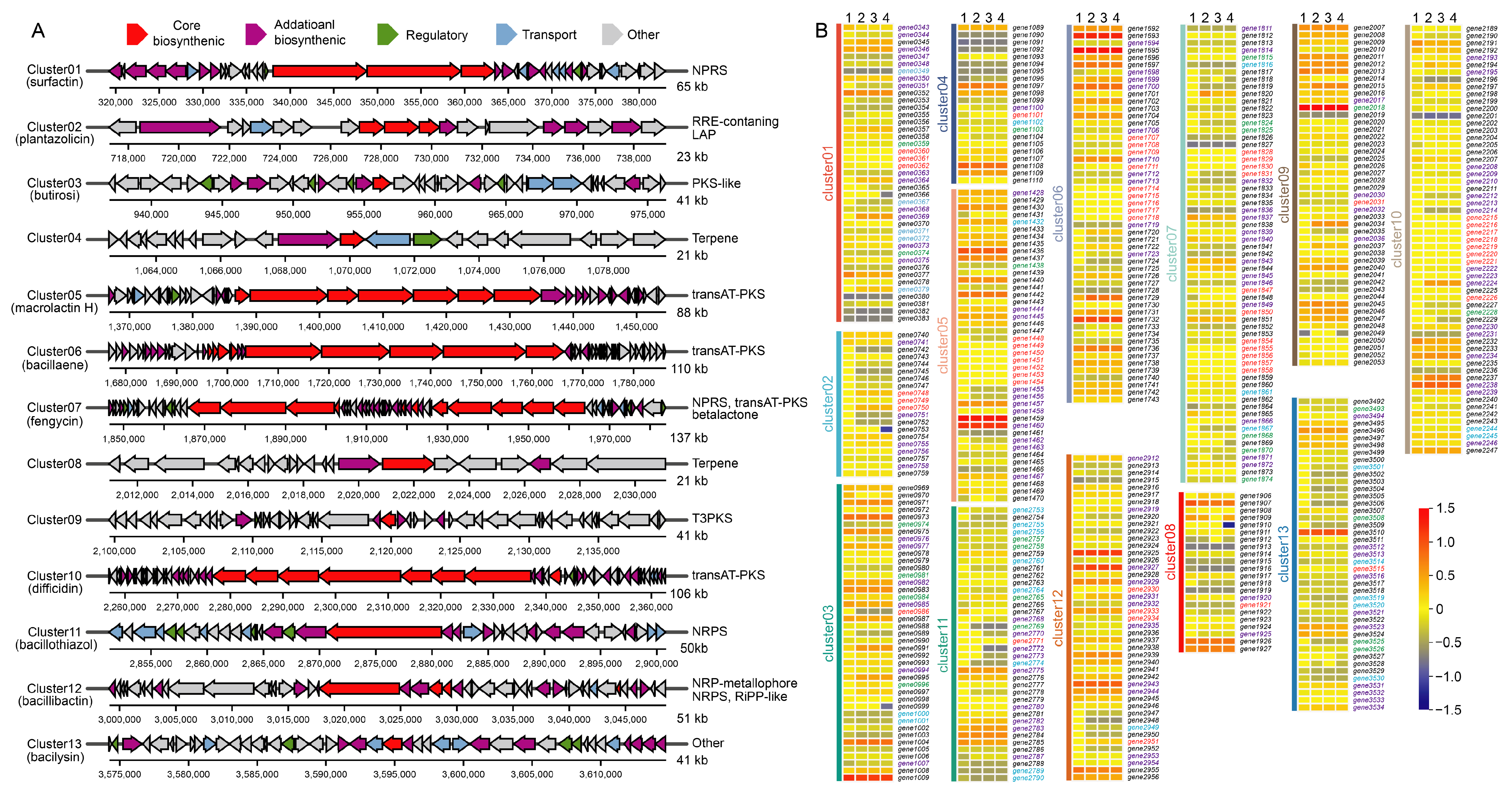
3.5. Carbohydrate Metabolism and Biosynthesis Gene Clusters
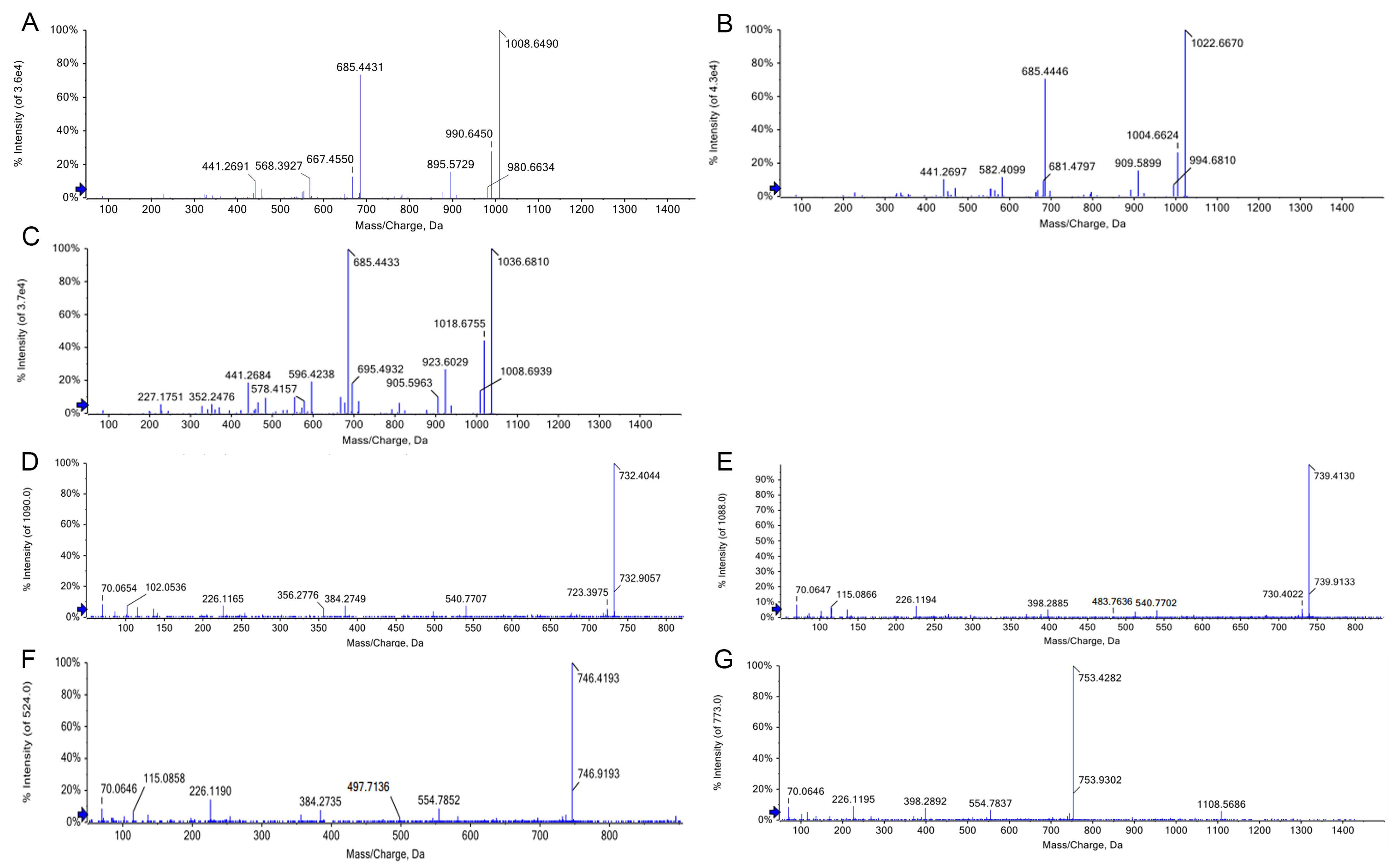
3.6. Identification of Crude Extracts of Lipopeptides
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dees, M.W.; Wanner, L.A. In search of better management of potato common scab. Potato Res. 2012, 55, 249–268. [Google Scholar] [CrossRef]
- Braun, S.; Gevens, A.; Charkowski, A.; Allen, C. Potato common scab: A review of the causal pathogens, management practices, varietal resistance screening methods, and host resistance. Am. J. Potato Res. 2017, 94, 58–72. [Google Scholar] [CrossRef]
- Wanner, L.A. A survey of genetic variation in Streptomyces isolates causing potato common scab in the United States. Phytopathology 2006, 96, 1363–1371. [Google Scholar] [CrossRef] [PubMed]
- Dees, M.W.; Sletten, A.; Hermansen, A. Isolation and characterization of Streptomyces species from potato common scab lesions in Norway. Plant Pathol. 2013, 62, 217–225. [Google Scholar] [CrossRef]
- Park, D.H.; Yu, Y.M.; Kim, J.S.; Cho, J.M.; Hur, J.H.; Lee, J.H. Multiple resistance of Streptomyces scabies isolates causing common scab of potato in Korea. Plant Dis. 2003, 87, 1290–1296. [Google Scholar] [CrossRef]
- Muzhinji, N.; van der Waals, J.E. Population biology and genetic variation of Spongospora subterranea f. sp. subterranea, the causal pathogen of powdery scab and root galls on potatoes in South Africa. Phytopathology 2019, 109, 1957–1965. [Google Scholar] [CrossRef] [PubMed]
- Bignell, D.R.D.; Fyans, J.K.; Cheng, Z. Phytotoxins produced by plant pathogenic Streptomyces species. J. Appl. Microbiol. 2014, 116, 223–235. [Google Scholar] [CrossRef]
- Marques, H.M.C.; Appy, M.P.; Destéfano, S.A.L. Effect of pH soil and irrigation regimes on management of potato scab. Arq. Inst. Biol. 2021, 88, e00552020. [Google Scholar] [CrossRef]
- Leiminger, J.; Frank, M.; Wenk, C.; Poschenrieder, G.; Kellermann, A.; Schwarzfischer, A. Distribution and characterization of Streptomyces species causing potato common scab in Germany. Plant Pathol. 2013, 62, 611–623. [Google Scholar] [CrossRef]
- Tomihama, T.; Nishi, Y.; Sakai, M.; Ikenaga, M.; Okubo, T.; Ikeda, S. Draft genome sequences of Streptomyces scabiei S58, Streptomyces turgidiscabies T45, and Streptomyces acidiscabies a10, the pathogens of potato common scab, isolated in Japan. Genome Announc. 2016, 4, e00217-16. [Google Scholar] [CrossRef]
- Hosny, M.; Abo-Elyousr, K.A.; Asran, M.R.; Saead, F.A. Chemical control of potato common scab disease under field conditions. Arch. Phytopathol. Plant Prot. 2014, 47, 2193–2199. [Google Scholar] [CrossRef]
- Tegg, R.S.; Corkrey, R.; Wilson, C.R. Relationship between the application of foliar chemicals to reduce common scab disease of potato and correlation with thaxtomin A toxicity. Plant Dis. 2012, 96, 97–103. [Google Scholar] [CrossRef]
- Singh, B.P.; Jeswani, M.D. Managing common scab of potato through chemical and cultural practices. Potato J. 1987, 14, 26–32. [Google Scholar]
- Abd El-Rahman, A.F.; El-Kafrawy, A.A.; Abd El-Hafez, O.A.; El-Ghany, A.B.D.; Rady, E. Evaluation of some fungicides effectiveness in control of blackleg and common scab of potato. Egypt. J. Agric. Res. 2018, 96, 1307–1323. [Google Scholar] [CrossRef]
- Kopecky, J.; Rapoport, D.; Sarikhani, E.; Stovicek, A.; Patrmanova, T.; Sagova-Mareckova, M. Micronutrients and soil microorganisms in the suppression of potato common scab. Agronomy 2021, 11, 383. [Google Scholar] [CrossRef]
- Lin, C.; Tsai, C.H.; Chen, P.Y.; Wu, C.Y.; Chang, Y.L.; Yang, Y.L.; Chen, Y.L. Biological control of potato common scab by Bacillus amyloliquefaciens Ba01. PLoS ONE 2018, 13, e0196520. [Google Scholar] [CrossRef]
- Porto, J.S.; Rebouças, T.N.H.; José, A.R.S.; José, A.R.S.; Tebaldi, N.D.; Luz, J.M.Q. Biocontrol of potato common scab cultivated on different soil mulch. Agronomy 2022, 12, 904. [Google Scholar] [CrossRef]
- Singhai, P.K.; Sarma, B.K.; Srivastava, J.S. Biological management of common scab of potato through Pseudomonas species and vermicompost. Biol. Control 2011, 57, 150–157. [Google Scholar] [CrossRef]
- Irmawatie, L.; Adviany, I.; Hamdani, D.; Nurdin, R.; Suswana, S. Effects of Gliocladium sp. on suppression of the intensity of scab disease (Streptomyces scabies Lambert and Loria) and the quality of potato seeds (Solanum tuberosum L.). Contrib. Cent. Res. Inst. Agric. 2025, 19, 9–14. [Google Scholar]
- Afzal, I.; Shinwari, Z.K.; Sikandar, S.; Shahzad, S. Plant beneficial endophytic bacteria: Mechanisms, diversity, host range and genetic determinants. Microbiol. Res. 2019, 221, 36–49. [Google Scholar] [CrossRef]
- Land, M.; Hauser, L.; Jun, S.R.; Nookaew, I.; Leuze, M.R.; Ahn, T.H.; Karpinets, T.; Lund, O.; Kora, G.; Wassenaar, T.; et al. Insights from 20 years of bacterial genome sequencing. Funct. Integr. Genom. 2015, 15, 141–161. [Google Scholar] [CrossRef]
- Cole, S.T.; Saint-Girons, I. Bacterial genomes—All shapes and sizes. In Organization of the Prokaryotic Genome; American Society of Microbiology: Washington, DC, USA, 1999; pp. 35–62. [Google Scholar]
- Delihas, N. Impact of small repeat sequences on bacterial genome evolution. Genome Biol. Evol. 2011, 3, 959–973. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Mei, W.; Lin, Q.; Wang, H.; Wang, J.; Peng, S.; Li, H.; Zhu, J.; Li, W.; Wang, P.; et al. Genome sequence of the agarwood tree Aquilaria sinensis (Lour.) Spreng: The first chromosome-level draft genome in the Thymelaeceae family. Gigascience 2020, 9, giaa013. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Islam, M.S.; Wang, J.; Zhao, Y.; Dong, W. Isolation of potato endophytes and screening of Chaetomium globosum antimicrobial genes. Int. J. Mol. Sci. 2022, 23, 4611. [Google Scholar] [CrossRef]
- Elbing, K.L.; Brent, R. Growth of E. coli on solid media. CP Mol. Biol. 2019, 125, e82. [Google Scholar] [CrossRef]
- Cui, L.; Yang, C.; Wei, L.; Li, T.; Chen, X. Isolation and identification of an endophytic bacteria Bacillus velezensis 8-4 Exhibiting Biocontrol Activity Against Potato Scab. Biol. Control 2020, 141, 104156. [Google Scholar] [CrossRef]
- Al-Mutar, D.M.K.; Noman, M.; Alzawar, N.S.A.; Qasim, H.H.; Li, D.; Song, F. The extracellular lipopeptides and volatile organic compounds of Bacillus subtilis DHA41 display broad-spectrum antifungal activity against soil-borne phytopathogenic fungi. J. Fungi 2023, 9, 797. [Google Scholar] [CrossRef]
- Hu, Y.; Li, Y.; Yang, X.; Li, C.; Wang, L.; Feng, J.; Chen, S.; Li, X.; Yang, Y. Effects of integrated biocontrol on bacterial wilt and rhizosphere bacterial community of tobacco. Sci. Rep. 2021, 11, 2653. [Google Scholar] [CrossRef]
- Kim, S.H.; Shin, J.H. Identification of nontuberculous mycobacteria using multilocus sequence analysis of 16S rRNA, hsp65, and rpoB. J. Clin. Lab. Anal. 2018, 32, e22184. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Chen, S. Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp. iMeta 2023, 2, e107. [Google Scholar] [CrossRef]
- Merda, D.; Vila-Nova, M.; Bonis, M.; Boutigny, A.L.; Brauge, T.; Cavaiuolo, M.; Cunty, A.; Regnier, A.; Sayeb, M.; Vingadassalon, N.; et al. Unraveling the impact of genome assembly on bacterial typing: A one health perspective. BMC Genom. 2024, 25, 1059. [Google Scholar] [CrossRef]
- Schmartz, G.P.; Hartung, A.; Hirsch, P.; Kern, F.; Fehlmann, T.; Müller, R.; Keller, A. PLSDB: Advancing a comprehensive database of bacterial plasmids. Nucleic Acids Res. 2022, 50, D273–D278. [Google Scholar] [CrossRef] [PubMed]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef] [PubMed]
- Tarailo-Graovac, M.; Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2009, 25, 4–10. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Ding, X.; Mei, W.; Huang, S.; Wang, H.; Zhu, J.; Hu, W.; Ding, Z.; Tie, W.; Peng, S.; Dai, H. Genome survey sequencing for the characterization of genetic background of Dracaena cambodiana and its defense response during dragon’s blood formation. PLoS ONE 2018, 13, e0209258. [Google Scholar] [CrossRef]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef]
- Yang, Z.; Mei, W.; Wang, H.; Zeng, J.; Dai, H.; Ding, X. Comprehensive analysis of NAC transcription factors reveals their evolution in malvales and functional characterization of AsNAC019 and AsNAC098 in Aquilaria sinensis. Int. J. Mol. Sci. 2023, 24, 17384. [Google Scholar] [CrossRef]
- Marçais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef]
- Goel, M.; Sun, H.; Jiao, W.B.; Schneeberger, K. SyRI: Finding genomic rearrangements and local sequence differences from whole-genome assemblies. Genome Biol. 2019, 20, 1–13. [Google Scholar] [CrossRef]
- Shao, B.; Yan, Y.; Fu, G. Predicting microbial transcriptome using genome sequence. bioRxiv 2024. [Google Scholar] [CrossRef]
- Ma, Y.; Kong, Q.; Qin, C.; Chen, Y.; Chen, Y.; Lv, R.; Zhou, G. Identification of lipopeptides in Bacillus megaterium by two-step ultrafiltration and LC-ESI-MS/MS. AMB Express 2016, 6, 79. [Google Scholar] [CrossRef]
- Zhan, Q.; Thakur, K.; Feng, J.Y.; Zhu, Y.Y.; Zhang, J.G.; Wei, Z.J. LC-MS based metabolomics analysis of okara fermented by Bacillus subtilis DC-15: Insights into nutritional and functional profile. Food Chem. 2023, 413, 135656. [Google Scholar] [CrossRef]
- Ismail, S.; Jiang, B.; Nasimi, Z.; Inam-ul-Haq, M.; Yamamoto, N.; Danso Ofori, A.; Khan, N.; Arshad, M.; Abbas, K.; Zheng, A. Investigation of Streptomyces scabies causing potato scab by various detection techniques, its pathogenicity and determination of host-disease resistance in potato germplasm. Pathogens 2020, 9, 760. [Google Scholar] [CrossRef]
- Zhao, P.; Liu, L.; Cao, J.; Wang, Z.; Zhao, Y.; Zhong, N. Transcriptome analysis of tryptophan-induced resistance against potato common scab. Int. J. Mol. Sci. 2022, 23, 8420. [Google Scholar] [CrossRef]
- Biessy, A.; Filion, M. Biological control of potato common scab by plant-beneficial bacteria. Biol. Control 2022, 165, 104808. [Google Scholar] [CrossRef]
- Tao, H.; Wang, S.; Li, X.; Li, X.; Cai, J.; Zhao, L.; Wang, J.; Zeng, J.; Qin, Y.; Xiong, X.; et al. Biological control of potato common scab and growth promotion of potato by Bacillus velezensis Y6. Front. Microbiol. 2023, 14, 1295107. [Google Scholar] [CrossRef]
- Cao, J.; Ma, Y.; Fu, J.; Wang, Z.; Zhao, Y.; Zhong, N.; Zhao, P. Bacillus atrophaeus DX-9 biocontrol against potato common scab involves significant changes in the soil microbiome and metabolome. aBIOTECH 2025, 6, 33–49. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, Y.; Zhuang, L.; Yu, Y.; Liu, J.; Zhang, L.; Gao, Z.; Wu, Y.; Gao, W.; Ding, G.C.; et al. A rhizosphere-derived consortium of Bacillus subtilis and Trichoderma harzianum suppresses common scab of potato and increases yield. Comput. Struct. Biotechnol. J. 2019, 17, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Hassan, M.M.; Elfarash, A.E.; Abo-Elyousr, K.A.; Hussein, M.A. Efficacy of some biocontrol agents against Streptomyces scabiei the causative of common scab disease in potatoes. Egypt. J. Phytopathol. 2021, 49, 168–178. [Google Scholar] [CrossRef]
- Etesami, H.; Jeong, B.R.; Glick, B.R. Biocontrol of plant diseases by Bacillus spp. Physiol. Mol. Plant Pathol. 2023, 126, 102048. [Google Scholar] [CrossRef]
- Ongena, M.; Jacques, P. Bacillus lipopeptides: Versatile weapons for plant disease biocontrol. Trends Microbiol. 2008, 16, 115–125. [Google Scholar] [CrossRef]
- Fira, D.; Dimkić, I.; Berić, T.; Lozo, J.; Stanković, S. Biological control of plant pathogens by Bacillus species. J. Biotechnol. 2018, 285, 44–55. [Google Scholar] [CrossRef] [PubMed]
- Cawoy, H.; Bettiol, W.; Fickers, P.; Ongena, M. Bacillus-based biological control of plant diseases. In Pesticides in the Modern World—Pesticides Use and Management; IntechOpen: London, UK, 2011; pp. 273–302. [Google Scholar]
- Dimkić, I.; Janakiev, T.; Petrović, M.; Degrassi, G.; Fira, D. Plant-associated Bacillus and Pseudomonas antimicrobial activities in plant disease suppression via biological control mechanisms—A review. Physiol. Mol. Plant Pathol. 2022, 117, 101754. [Google Scholar] [CrossRef]
- Wang, X.Q.; Zhao, D.L.; Shen, L.L.; Jing, C.L.; Zhang, C.S. Application and mechanisms of Bacillus subtilis in biological control of plant disease. In Role of Rhizospheric Microbes in Soil: Volume 1: Stress Management and Agricultural Sustainability; Springer: Singapore, 2018; pp. 225–250. [Google Scholar]
- Volynchikova, E.; Kim, K.D. Biological control of oomycete soilborne diseases caused by Phytophthora capsici, Phytophthora infestans, and Phytophthora nicotianae in solanaceous crops. Mycobiology 2022, 50, 269–293. [Google Scholar] [CrossRef]
- Ma, S.; Wang, Y.; Teng, W. Bacillus velezensis K-9 as a potential biocontrol agent for managing potato scab. Plant Dis. 2023, 107, 3943–3951. [Google Scholar] [CrossRef]
- Zheng, Z.; Zhang, Y.; Liu, Z.; Dong, Z.; Xie, C.; Bravo, A.; Soberón, M.; Mahillon, J.; Sun, M.; Peng, D. The CRISPR-Cas systems were selectively inactivated during evolution of Bacillus cereus group for adaptation to diverse environments. ISME J. 2020, 14, 1479–1493. [Google Scholar] [CrossRef]
- Carlton, B.C.; Gawron-Burke, C. Genetic improvement of Bacillus thuringiensis for bioinsecticide development. In Advanced Engineered Pesticides; CRC Press: Boca Raton, FL, USA, 2024; pp. 43–61. [Google Scholar]
- Janda, J.M.; Abbott, S.L. 16S rRNA gene sequencing for bacterial identification in the diagnostic laboratory: Pluses, perils, and pitfalls. J. Clin. Microbiol. 2007, 45, 2761–2764. [Google Scholar] [CrossRef] [PubMed]
- Zaid, D.S.; Cai, S.; Hu, C.; Li, Z.; Li, Y. Comparative genome analysis reveals phylogenetic identity of Bacillus velezensis HNA3 and genomic insights into its plant growth promotion and biocontrol effects. Microbiol. Spectr. 2022, 10, e02169-21. [Google Scholar] [CrossRef]
- Yang, W.; Zhang, L.; Zhang, H.; Li, H.; Yang, Y.; Yu, J.; Xiang, H. First report of root rot of tobacco caused by Fusarium concentricum in China. Plant Dis. 2024. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlić, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Na, S.I.; Kim, D.; Chun, J. UBCG2: Up-to-date bacterial core genes and pipeline for phylogenomic analysis. J. Microbiol. 2021, 59, 609–615. [Google Scholar] [CrossRef] [PubMed]
- Goodswen, S.J.; Barratt, J.L.; Kennedy, P.J.; Kaufer, A.; Calarco, L.; Ellis, J.T. Machine learning and applications in microbiology. FEMS Microbiol. Rev. 2021, 45, fuab015. [Google Scholar] [CrossRef]
- Jiang, Y.; Luo, J.; Huang, D.; Liu, Y.; Li, D.D. Machine learning advances in microbiology: A review of methods and applications. Front. Microbiol. 2022, 13, 925454. [Google Scholar] [CrossRef]
- Rani, P.; Kotwal, S.; Manhas, J.; Sharma, V.; Sharma, S. Machine learning and deep learning based computational approaches in automatic microorganisms image recognition: Methodologies, challenges, and developments. Arch. Comput. Methods Eng. 2022, 29, 1801–1837. [Google Scholar] [CrossRef]
- Liu, Y.; Gao, Y.; Niu, R.; Zhang, Z.; Lu, G.W.; Hu, H.; Liu, T.; Cheng, Z. Rapid and accurate bacteria identification through deep-learning-based two-dimensional Raman spectroscopy. Anal. Chim. Acta 2024, 1332, 343376. [Google Scholar] [CrossRef]
- Stewart, R.D.; Auffret, M.D.; Warr, A.; Wiser, A.H.; Press, M.O.; Langford, K.W.; Liachko, I.; Snelling, T.J.; Dewhurst, R.J.; Walker, A.W.; et al. Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen. Nat. Commun. 2018, 9, 870. [Google Scholar] [CrossRef]
- Cuperus, J.T.; Groves, B.; Kuchina, A.; Rosenberg, A.B.; Jojic, N.; Fields, S.; Seelig, G. Deep learning of the regulatory grammar of yeast 5′ untranslated regions from 500,000 random sequences. Genome Res. 2017, 27, 2015–2024. [Google Scholar] [CrossRef] [PubMed]
- Chu, Y.; Yu, D.; Li, Y.; Huang, K.; Shen, Y.; Cong, L.; Zhang, J.; Wang, M. A 5′ UTR language model for decoding untranslated regions of mRNA and function predictions. Nat. Mach. Intell. 2024, 6, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Rabbee, M.F.; Ali, M.S.; Choi, J.; Hwang, B.S.; Jeong, S.C.; Baek, K.-H. Bacillus velezensis: A valuable member of bioactive molecules within plant microbiomes. Molecules 2019, 24, 1046. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, Y.; Yang, Q.; Zhao, W.; Cui, L.; Wang, B.; Zhang, L.; Cheng, H.; Song, S.; Zhang, L. Genomics and LC-MS reveal diverse active secondary metabolites in Bacillus amyloliquefaciens WS-8. J. Microbiol. Biotechnol. 2020, 30, 417–426. [Google Scholar] [CrossRef]
- Sa, R.B.; An, X.; Sui, J.K.; Wang, X.H.; Ji, C.; Wang, C.Q.; Liu, X. Purification and structural characterization of fengycin homologues produced by Bacillus subtilis from poplar wood bark. Australas. Plant Pathol. 2018, 47, 259–268. [Google Scholar] [CrossRef]
- Feng, R.Y.; Chen, Y.H.; Lin, C.; Tsai, C.H.; Yang, Y.L.; Chen, Y.L. Surfactin secreted by Bacillus amyloliquefaciens Ba01 is required to combat Streptomyces scabies causing potato common scab. Front. Plant Sci. 2022, 13, 998707. [Google Scholar] [CrossRef]
- Yin, Y.; Wang, X.; Zhang, P.; Wang, P.; Wen, J. Strategies for improving fengycin production: A review. Microb. Cell Fact. 2024, 23, 144. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jiang, Y.; He, P.; Kong, H.; He, P.; Wu, Y.; Tang, G.; Tang, P.; Di, Y.; Li, X.; Liu, L.; et al. Biocontrol Effect of Bacillus velezensis D7-8 on Potato Common Scab and Its Complete Genome Sequence Analysis. Microorganisms 2025, 13, 770. https://doi.org/10.3390/microorganisms13040770
Jiang Y, He P, Kong H, He P, Wu Y, Tang G, Tang P, Di Y, Li X, Liu L, et al. Biocontrol Effect of Bacillus velezensis D7-8 on Potato Common Scab and Its Complete Genome Sequence Analysis. Microorganisms. 2025; 13(4):770. https://doi.org/10.3390/microorganisms13040770
Chicago/Turabian StyleJiang, Yu, Pengfei He, Huihui Kong, Pengbo He, Yixin Wu, Guowen Tang, Ping Tang, Yining Di, Xingyu Li, Lufeng Liu, and et al. 2025. "Biocontrol Effect of Bacillus velezensis D7-8 on Potato Common Scab and Its Complete Genome Sequence Analysis" Microorganisms 13, no. 4: 770. https://doi.org/10.3390/microorganisms13040770
APA StyleJiang, Y., He, P., Kong, H., He, P., Wu, Y., Tang, G., Tang, P., Di, Y., Li, X., Liu, L., Munir, S., & He, Y. (2025). Biocontrol Effect of Bacillus velezensis D7-8 on Potato Common Scab and Its Complete Genome Sequence Analysis. Microorganisms, 13(4), 770. https://doi.org/10.3390/microorganisms13040770