

Factors Affecting D-Lactic Acid Production by Flocculant Saccharomyces cerevisiae Under Non-Neutralizing Conditions


 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Strains and Medium
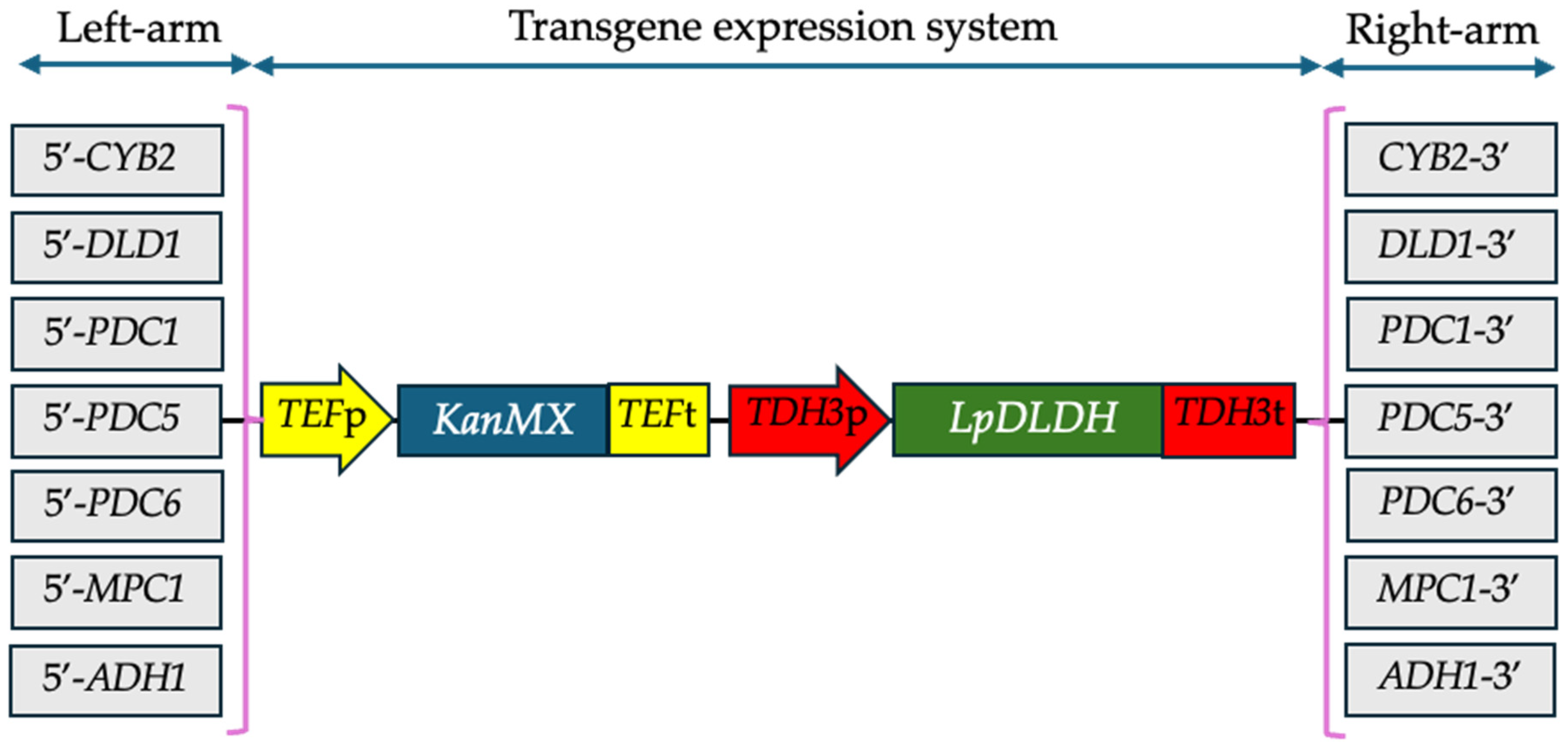
2.2. Construction of Transgene Expression System for D-Lactate Synthesis
2.3. Fermentation
2.4. Fermentation Product Measurements
2.5. Cell Growth Measurement
2.6. Cell Morphological Observation
2.7. Cell Wall Hydrophobicity Test
2.8. Quantitative PCR for Gene Expression Analysis
2.9. Statistical Analysis
2.10. Differential Expression Analysis
3. Results
3.1. D-LA Production Profiles by Engineered Flocculant S. cerevisiae Strains
3.2. Ideal Integration Spot of D-LDH in Flocculant S. cerevisiae F188 Strain for D-LA Production
3.3. Flocculation Behavior Across the Engineered Strains
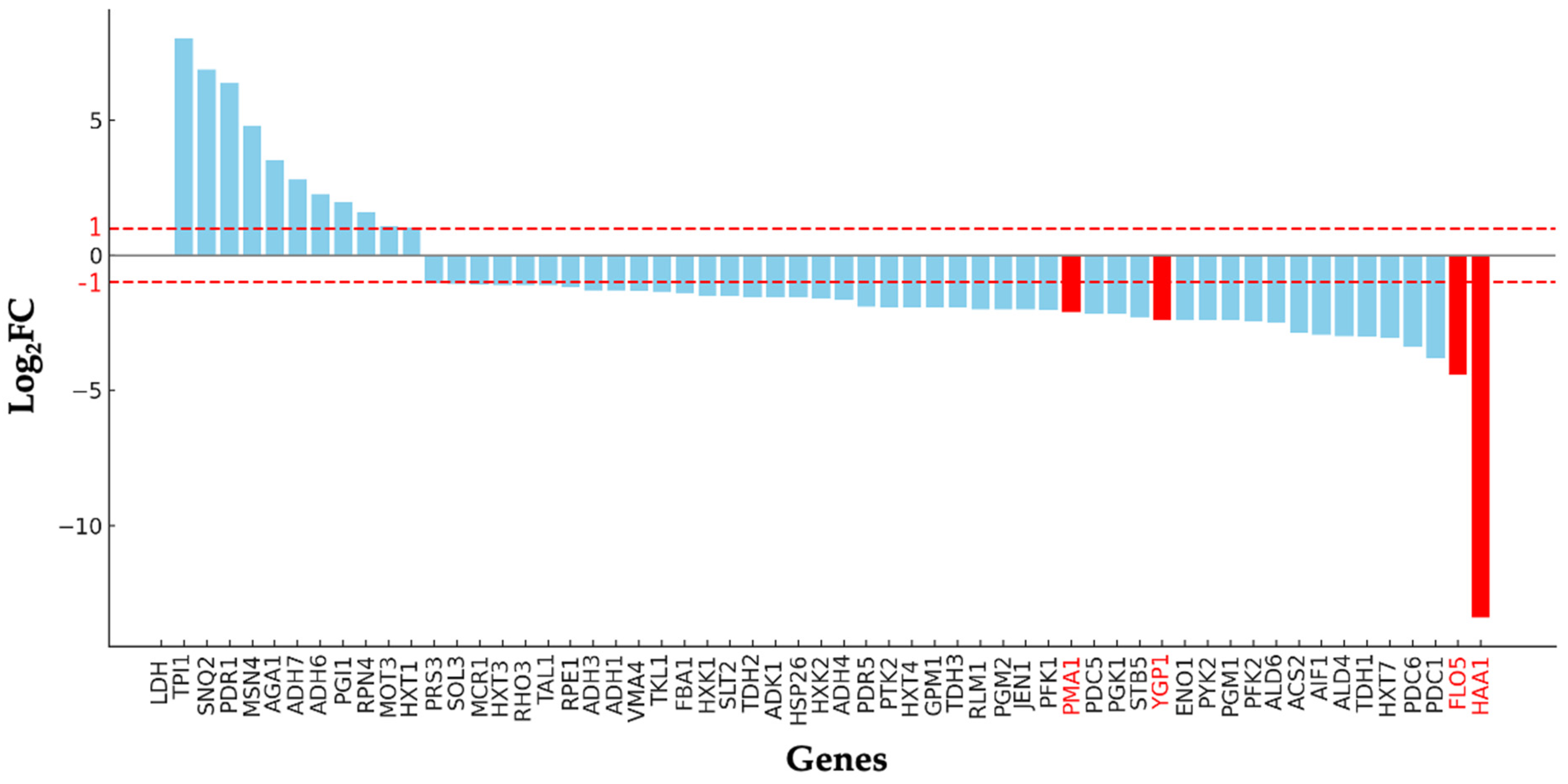
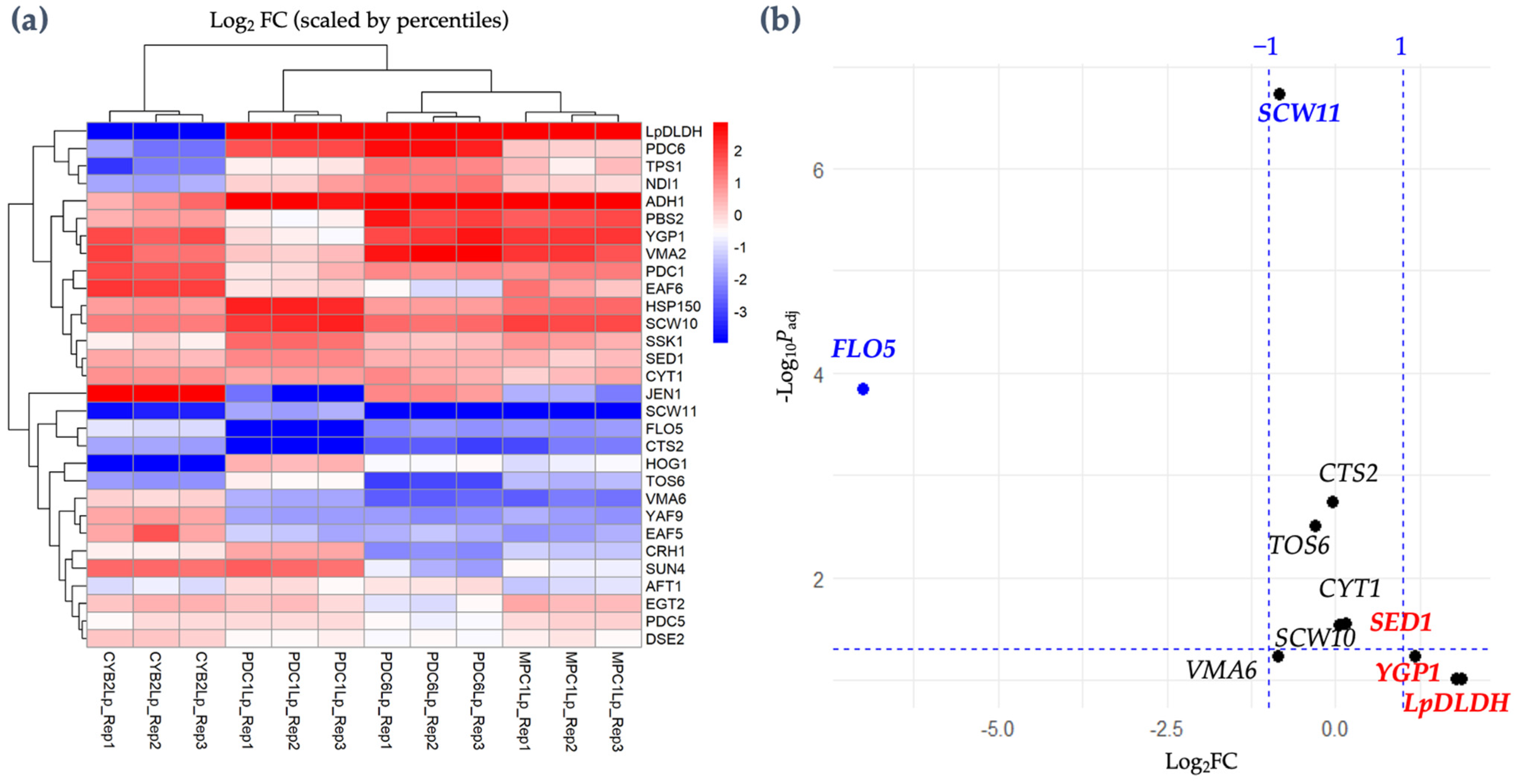
3.4. Gene Expression Analysis of D-LA-Producing Strains
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| LA | Lactic acid |
| D-LDH | D-lactate dehydrogenase |
| CYB2 | Cytochrome b2 |
| PDC1/5/6 | Pyruvate decarboxylase |
| DLD1 | D-lactate dehydrogenase |
| MPC1 | Mitochondrial pyruvate carrier |
| ADH1 | Alcohol dehydrogenase |
| GAP | Glyceraldehyde 3-phosphate |
| DHAP | Dihydroxyacetone phosphate |
| G3P | Glycerol 3-phosphate |
| Ct | Threshold cycle |
| Log2FC | Log fold change |
References
- Tan, J.; Abdel-Rahman, M.A.; Sonomoto, K. Biorefinery-Based Lactic Acid Fermentation: Microbial Production of Pure Monomer Product. In Synthesis, Structure and Properties of Poly (Lactic Acid); Di Lorenzo, M.L., Androsch, R., Eds.; Advances in Polymer Science; Springer International Publishing: Cham, Switzerland, 2017; Volume 279, pp. 27–66. ISBN 978-3-319-64229-1. [Google Scholar]
- Gallezot, P. Conversion of Biomass to Selected Chemical Products. Chem. Soc. Rev. 2012, 41, 1538–1558. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Rahman, M.A.; Tashiro, Y.; Sonomoto, K. Lactic Acid Production from Lignocellulose-Derived Sugars Using Lactic Acid Bacteria: Overview and Limits. J. Biotechnol. 2011, 156, 286–301. [Google Scholar] [CrossRef]
- Porro, D.; Brambilla, L.; Ranzi, B.M.; Martegani, E.; Alberghina, L. Development of Metabolically Engineered Saccharomyces Cerevisiae Cells for the Production of Lactic Acid. Biotechnol. Prog. 1995, 11, 294–298. [Google Scholar] [CrossRef]
- Park, H.J.; Bae, J.; Ko, H.; Lee, S.; Sung, B.H.; Han, J.; Sohn, J. Low-pH Production of D-lactic Acid Using Newly Isolated Acid Tolerant Yeast Pichia kudriavzevii NG7. Biotechnol. Bioeng. 2018, 115, 2232–2242. [Google Scholar] [CrossRef]
- Abdel-Rahman, M.A.; Sonomoto, K. Opportunities to Overcome the Current Limitations and Challenges for Efficient Microbial Production of Optically Pure Lactic Acid. J. Biotechnol. 2016, 236, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Lahtinen, S.; Ouwehand, A.C.; Salminen, S.; Von Wright, A. (Eds.) Lactic Acid Bacteria: Microbiological and Functional Aspects, 4th ed.; CRC Press: Boca Raton, FL, USA, 2011; ISBN 978-0-429-15145-3. [Google Scholar]
- Bai, H.; Deng, S.; Bai, D.; Zhang, Q.; Fu, Q. Recent Advances in Processing of Stereocomplex-Type Polylactide. Macromol. Rapid Commun. 2017, 38, 1700454. [Google Scholar] [CrossRef] [PubMed]
- Coelho, L.F.; De Lima, C.J.B.; Bernardo, M.P.; Contiero, J. D(−)-Lactic Acid Production by Leuconostoc mesenteroides B512 Using Different Carbon and Nitrogen Sources. Appl. Biochem. Biotechnol. 2011, 164, 1160–1171. [Google Scholar] [CrossRef]
- Baek, S.-H.; Kwon, E.Y.; Kim, Y.H.; Hahn, J.-S. Metabolic Engineering and Adaptive Evolution for Efficient Production of D-Lactic Acid in Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 2016, 100, 2737–2748. [Google Scholar] [CrossRef]
- Sauer, M.; Porro, D.; Mattanovich, D.; Branduardi, P. 16 Years Research on Lactic Acid Production with Yeast–Ready for the Market? Biotechnol. Genet. Eng. Rev. 2010, 27, 229–256. [Google Scholar] [CrossRef]
- He, L.-Y.; Zhao, X.-Q.; Bai, F.-W. Engineering Industrial Saccharomyces Cerevisiae Strain with the FLO1-Derivative Gene Isolated from the Flocculating Yeast SPSC01 for Constitutive Flocculation and Fuel Ethanol Production. Appl. Energy 2012, 100, 33–40. [Google Scholar] [CrossRef]
- Kahar, P.; Itomi, A.; Tsuboi, H.; Ishizaki, M.; Yasuda, M.; Kihira, C.; Otsuka, H.; Azmi, N.B.; Matsumoto, H.; Ogino, C.; et al. The Flocculant Saccharomyces cerevisiae Strain Gains Robustness via Alteration of the Cell Wall Hydrophobicity. Metab. Eng. 2022, 72, 82–96. [Google Scholar] [CrossRef] [PubMed]
- Daniel Gietz, R.; Woods, R.A. Transformation of Yeast by Lithium Acetate/Single-Stranded Carrier DNA/Polyethylene Glycol Method. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2002; Volume 350, pp. 87–96. ISBN 978-0-12-182253-8. [Google Scholar]
- Van Mulders, S.E.; Christianen, E.; Saerens, S.M.G.; Daenen, L.; Verbelen, P.J.; Willaert, R.; Verstrepen, K.J.; Delvaux, F.R. Phenotypic Diversity of Flo Protein Family-Mediated Adhesion in Saccharomyces cerevisiae. FEMS Yeast Res. 2009, 9, 178–190. [Google Scholar] [CrossRef] [PubMed]
- Ramil, E.; Agrimonti, C.; Shechter, E.; Gervais, M.; Guiard, B. Regulation of the CYB2 Gene Expression: Transcriptional Co-ordination by the Hap1p, Hap2/3/4/5p and Adr1p Transcription Factors. Mol. Microbiol. 2000, 37, 1116–1132. [Google Scholar] [CrossRef] [PubMed]
- Ookubo, A.; Hirasawa, T.; Yoshikawa, K.; Nagahisa, K.; Furusawa, C.; Shimizu, H. Improvement of L-Lactate Production by CYB2 Gene Disruption in a Recombinant Saccharomyces cerevisiae Strain under Low pH Condition. Biosci. Biotechnol. Biochem. 2008, 72, 3063–3066. [Google Scholar] [CrossRef]
- Pangestu, R.; Kahar, P.; Ogino, C.; Kondo, A. Comparative Responses of Flocculating and Nonflocculating Yeasts to Cell Density and Chemical Stress in Lactic Acid Fermentation. Yeast 2024, 41, 192–206. [Google Scholar] [CrossRef]
- Carmelo, V.; Santos, H.; Sá-Correia, I. Effect of Extracellular Acidification on the Activity of Plasma Membrane ATPase and on the Cytosolic and Vacuolar pH of Saccharomyces cerevisiae. Biochim. Biophys. Acta BBA-Biomembr. 1997, 1325, 63–70. [Google Scholar] [CrossRef]
- Ambesi, A.; Miranda, M.; Petrov, V.V.; Slayman, C.W. Biogenesis and Function of the Yeast Plasma-Membrane H+-ATPase. J. Exp. Biol. 2000, 203, 155–160. [Google Scholar] [CrossRef]
- Morsomme, P.; Slayman, C.W.; Goffeau, A. Mutagenic Study of the Structure, Function and Biogenesis of the Yeast Plasma Membrane H+-ATPase. Biochim. Biophys. Acta BBA-Rev. Biomembr. 2000, 1469, 133–157. [Google Scholar] [CrossRef]
- Fernandes, A.R.; Mira, N.P.; Vargas, R.C.; Canelhas, I.; Sá-Correia, I. Saccharomyces Cerevisiae Adaptation to Weak Acids Involves the Transcription Factor Haa1p and Haa1p-Regulated Genes. Biochem. Biophys. Res. Commun. 2005, 337, 95–103. [Google Scholar] [CrossRef]
- Mira, N.P.; Becker, J.D.; Sá-Correia, I. Genomic Expression Program Involving the Haa1p-Regulon in Saccharomyces cerevisiae Response to Acetic Acid. OMICS J. Integr. Biol. 2010, 14, 587–601. [Google Scholar] [CrossRef]
- Branduardi, P.; Sauer, M.; De Gioia, L.; Zampella, G.; Valli, M.; Mattanovich, D.; Porro, D. Lactate production yield from engineered yeasts is dependent from the host background, the lactate dehydrogenase source and the lactate export. Microb. Cell Factories 2006, 5, 4. [Google Scholar] [CrossRef]
- Zhong, W.; Yang, M.; Mu, T.; Wu, F.; Hao, X.; Chen, R.; Sharshar, M.M.; Thygesen, A.; Wang, Q.; Xing, J. Systematically Redesigning and Optimizing the Expression of D-Lactate Dehydrogenase Efficiently Produces High-Optical-Purity D-Lactic Acid in Saccharomyces cerevisiae. Biochem. Eng. J. 2019, 144, 217–226. [Google Scholar] [CrossRef]
- Tokuhiro, K.; Ishida, N.; Nagamori, E.; Saitoh, S.; Onishi, T.; Kondo, A.; Takahashi, H. Double Mutation of the PDC1 and ADH1 Genes Improves Lactate Production in the Yeast Saccharomyces cerevisiae Expressing the Bovine Lactate Dehydrogenase Gene. Appl. Microbiol. Biotechnol. 2009, 82, 883–890. [Google Scholar] [CrossRef]
- Van Maris, A.J.A.; Geertman, J.-M.A.; Vermeulen, A.; Groothuizen, M.K.; Winkler, A.A.; Piper, M.D.W.; Van Dijken, J.P.; Pronk, J.T. Directed Evolution of Pyruvate Decarboxylase-Negative Saccharomyces cerevisiae, Yielding a C2-Independent, Glucose-Tolerant, and Pyruvate-Hyperproducing Yeast. Appl. Environ. Microbiol. 2004, 70, 159–166. [Google Scholar] [CrossRef]
- Bender, T.; Pena, G.; Martinou, J. Regulation of Mitochondrial Pyruvate Uptake by Alternative Pyruvate Carrier Complexes. EMBO J. 2015, 34, 911–924. [Google Scholar] [CrossRef] [PubMed]
- Kong, S.; Yu, W.; Gao, N.; Zhai, X.; Zhou, Y.J. Expanding the Neutral Sites for Integrated Gene Expression in Saccharomyces cerevisiae. FEMS Microbiol. Lett. 2022, 369, fnac081. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-L.; Li, B.-Z.; Zhang, W.-Z.; Song, K.; Qi, H.; Dai, J.; Yuan, Y.-J. Genome-Wide Landscape of Position Effects on Heterogeneous Gene Expression in Saccharomyces cerevisiae. Biotechnol. Biofuels 2017, 10, 189. [Google Scholar] [CrossRef]
- Watcharawipas, A.; Sae-tang, K.; Sansatchanon, K.; Sudying, P.; Boonchoo, K.; Tanapongpipat, S.; Kocharin, K.; Runguphan, W. Systematic Engineering of Saccharomyces cerevisiae for D-Lactic Acid Production with near Theoretical Yield. FEMS Yeast Res. 2021, 21, foab024. [Google Scholar] [CrossRef]
- Zhu, P.; Luo, R.; Li, Y.; Chen, X. Metabolic Engineering and Adaptive Evolution for Efficient Production of l-Lactic Acid in Saccharomyces cerevisiae. Microbiol. Spectr. 2022, 10, e02277-22. [Google Scholar] [CrossRef]
- Ishida, N.; Saitoh, S.; Ohnishi, T.; Tokuhiro, K.; Nagamori, E.; Kitamoto, K.; Takahashi, H. Metabolic Engineering of Saccharomyces cerevisiae for Efficient Production of Pure L-(+)-Lactic Acid. Appl. Biochem. Biotechnol. 2006, 131, 795–807. [Google Scholar] [CrossRef]
- Sae-Tang, K.; Bumrungtham, P.; Mhuantong, W.; Champreda, V.; Tanapongpipat, S.; Zhao, X.-Q.; Liu, C.-G.; Runguphan, W. Engineering Flocculation for Improved Tolerance and Production of D-Lactic Acid in Pichia Pastoris. J. Fungi 2023, 9, 409. [Google Scholar] [CrossRef] [PubMed]
- Goossens, K.V.Y.; Ielasi, F.S.; Nookaew, I.; Stals, I.; Alonso-Sarduy, L.; Daenen, L.; Van Mulders, S.E.; Stassen, C.; Van Eijsden, R.G.E.; Siewers, V.; et al. Molecular Mechanism of Flocculation Self-Recognition in Yeast and Its Role in Mating and Survival. mBio 2015, 6, e00427-15. [Google Scholar] [CrossRef] [PubMed]
- Govender, P.; Domingo, J.L.; Bester, M.C.; Pretorius, I.S.; Bauer, F.F. Controlled Expression of the Dominant Flocculation Genes FLO1, FLO5, and FLO11 in Saccharomyces cerevisiae. Appl. Environ. Microbiol. 2008, 74, 6041–6052. [Google Scholar] [CrossRef] [PubMed]
- Soares, E.V. Flocculation in Saccharomyces cerevisiae: A Review: Yeast Flocculation: A Review. J. Appl. Microbiol. 2011, 110, 1–18. [Google Scholar] [CrossRef]
- Liu, T.; Sun, L.; Zhang, C.; Liu, Y.; Li, J.; Du, G.; Lv, X.; Liu, L. Combinatorial Metabolic Engineering and Process Optimization Enables Highly Efficient Production of L-Lactic Acid by Acid-Tolerant Saccharomyces cerevisiae. Bioresour. Technol. 2023, 379, 129023. [Google Scholar] [CrossRef]
- Lane, S.; Turner, T.L.; Jin, Y. Glucose Assimilation Rate Determines the Partition of Flux at Pyruvate between Lactic Acid and Ethanol in Saccharomyces cerevisiae. Biotechnol. J. 2023, 18, 2200535. [Google Scholar] [CrossRef]
- Drozdova, P.; Gurkov, A.; Saranchina, A.; Vlasevskaya, A.; Zolotovskaya, E.; Indosova, E.; Timofeyev, M.; Borvinskaya, E. Transcriptional Response of Saccharomyces cerevisiae to Lactic Acid Enantiomers. Appl. Microbiol. Biotechnol. 2024, 108, 121. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Time a (hour) | Final D-LA (g/L) | Final Ethanol (g/L) | Final Glycerol (g/L) | Flocculation/ Hydrophobicity (%) | Final Dry Cell Weight (g/L) |
|---|---|---|---|---|---|---|
| Wild type | 24 | 0.04 ± 0.03 | 40.63 ± 0.64 | 2.94 ± 0.26 | Flocculant/89.32 | 6.31 ± 0.73 |
| ΔCYB2::LpDLDH | 12 | 31.92 ± 0.98 *** | 31.64 ± 0.68 ** | 0 | Flocculant/97.16 | 4.77 ± 0.22 |
| ΔPDC1::LpDLDH | 48 | 51.36 ± 1.78 *** | 20.2 ± 1.69 ** | 0 | Less flocculant/98.38 | 2.97 ± 0.12 |
| ΔMPC1::LpDLDH | 24 | 10.32 ± 0.35 *** | 41.53 ± 0.22 *** | 0 | Flocculant/N.D. | 6.96 ± 0.15 |
| ΔPDC6::LpDLDH | 24 | 0.026 ± 0.01 *** | 45.11 ± 0.35 *** | 4.25 ± 0.42 ** | Flocculant/N.D. | 9.42 ± 0.44 |
| ΔDLD1::LpDLDH | 48 | 0 * | 33.84 ± 0.42 * | 1.39 ± 0.05 *** | Non-flocculant/1.57 | 5.05 ± 0.059 |
| ΔADH1::LpDLDH | 48 | 0 * | 38.29 ± 2.66 * | 1.31 ± 0.17 ** | Non-flocculant/N.D. | 6.9 ± 0.8 |
| ΔPDC5::LpDLDH | 48 | 0 * | 41.69 ± 1.33 *** | 1.39 ± 0.11 ** | Non-flocculant/N.D. | 6.63 ± 0.32 |
| ∆CYB2 | 24 | 0 | 45.96 ± 1.26 | 4.7 ± 1.15 | Flocculant/N.D. | 9.57 ± 1.5 |
| ∆PDC1 | 72 | 0 | 43.06 ± 1.07 | 4.41 ± 0.51 | Flocculant/N.D. | 3.95 ± 0.95 |
| ∆MPC1 | 21 | 0 | 45.72 ± 0.81 | 5.19 ± 0.61 | Flocculant/N.D. | 8.98 ± 0.17 |
| ∆PDC6 | 48 | 0 | 1.45 ± 2.43 | 0.57 ± 0.01 | Less flocculant/N.D. | N.D. b |
| ∆DLD1 | 24 | 0 | 46.03 ± 1.19 | 3.95 ± 0.09 | Flocculant/N.D. | 9.26 ± 0.48 |
| ∆ADH1 | 144 | 0 | 39.07 ± 2.1 * | 4.78 ± 0.21 | Less flocculant/N.D. | 3.71 ± 0.33 |
| ∆PDC5 | 48 | 0 | 44.61 ± 1.27 | 3.98 ± 0.11 | Flocculant/N.D. | 3.46 ± 0.37 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rahmasari, D.; Kahar, P.; de Oliveira, A.V.; Putra, F.J.N.; Kondo, A.; Ogino, C. Factors Affecting D-Lactic Acid Production by Flocculant Saccharomyces cerevisiae Under Non-Neutralizing Conditions. Microorganisms 2025, 13, 618. https://doi.org/10.3390/microorganisms13030618
Rahmasari D, Kahar P, de Oliveira AV, Putra FJN, Kondo A, Ogino C. Factors Affecting D-Lactic Acid Production by Flocculant Saccharomyces cerevisiae Under Non-Neutralizing Conditions. Microorganisms. 2025; 13(3):618. https://doi.org/10.3390/microorganisms13030618
Chicago/Turabian StyleRahmasari, Dianti, Prihardi Kahar, Arthur Vinícius de Oliveira, Filemon Jalu Nusantara Putra, Akihiko Kondo, and Chiaki Ogino. 2025. "Factors Affecting D-Lactic Acid Production by Flocculant Saccharomyces cerevisiae Under Non-Neutralizing Conditions" Microorganisms 13, no. 3: 618. https://doi.org/10.3390/microorganisms13030618
APA StyleRahmasari, D., Kahar, P., de Oliveira, A. V., Putra, F. J. N., Kondo, A., & Ogino, C. (2025). Factors Affecting D-Lactic Acid Production by Flocculant Saccharomyces cerevisiae Under Non-Neutralizing Conditions. Microorganisms, 13(3), 618. https://doi.org/10.3390/microorganisms13030618