Evaluation of Cellular Responses of Heterotrophic Escherichia coli Cultured with Autotrophic Chlamydomonas reinhardtii as a Nutrient Source by Analyses Based on Microbiology and Transcriptome

 ,
, 
 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Obtainment of Green C. reinhardtii Cells
2.2. Preparation of Disrupted Cell Extracts from C. reinhardtii Cells
2.3. Cultivation of E. coli Cells
2.4. Microscope Observation of E. coli Treated with Gram Staining
2.5. Evaluation of Cell-Viability of C. reinhardtii
2.6. Quantification of Carbohydrate, Protein, Lipid and Organic Acid in Broth
2.7. Measurement of Levels of Gene Translation
3. Results
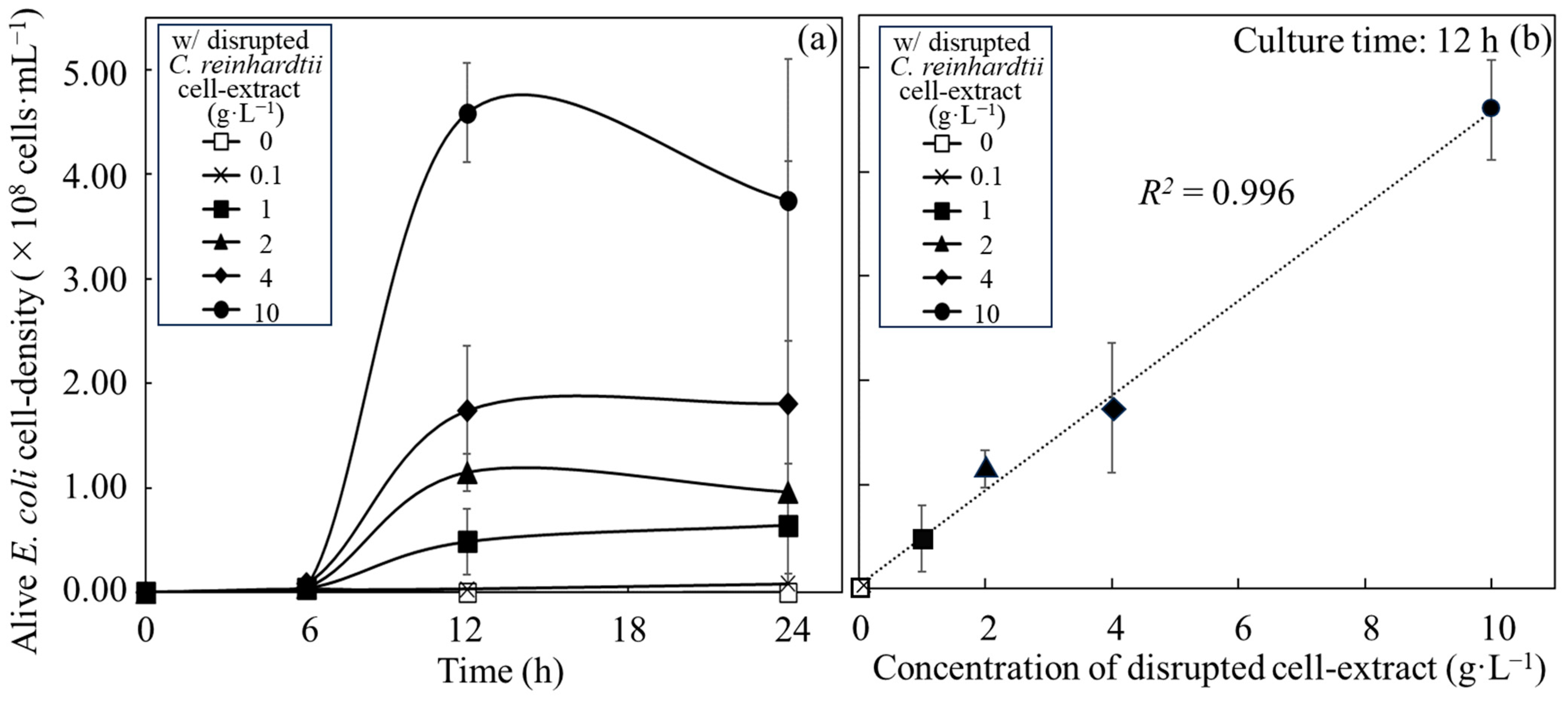
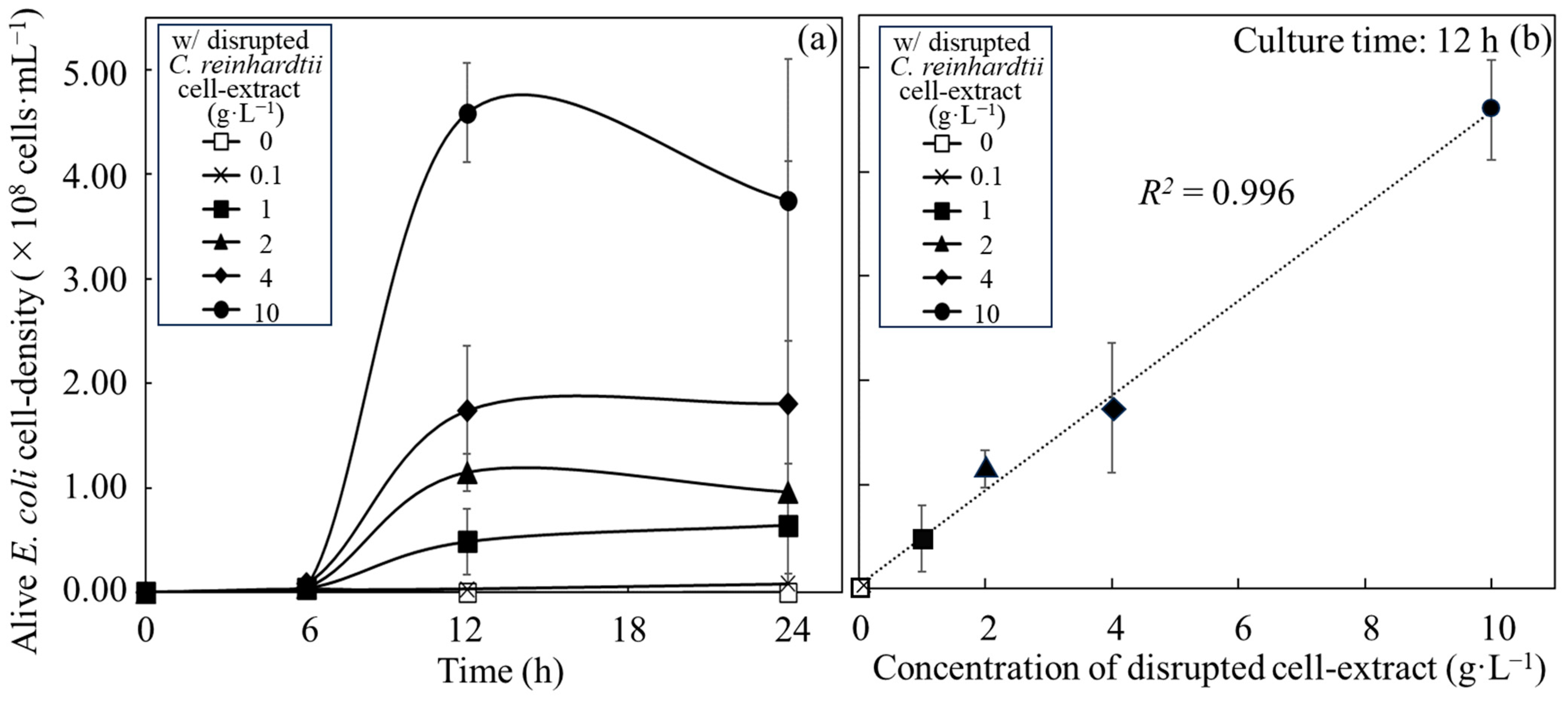
3.1. Growth of E. coli with Disrupted Cell Extracts from C. reinhardtii
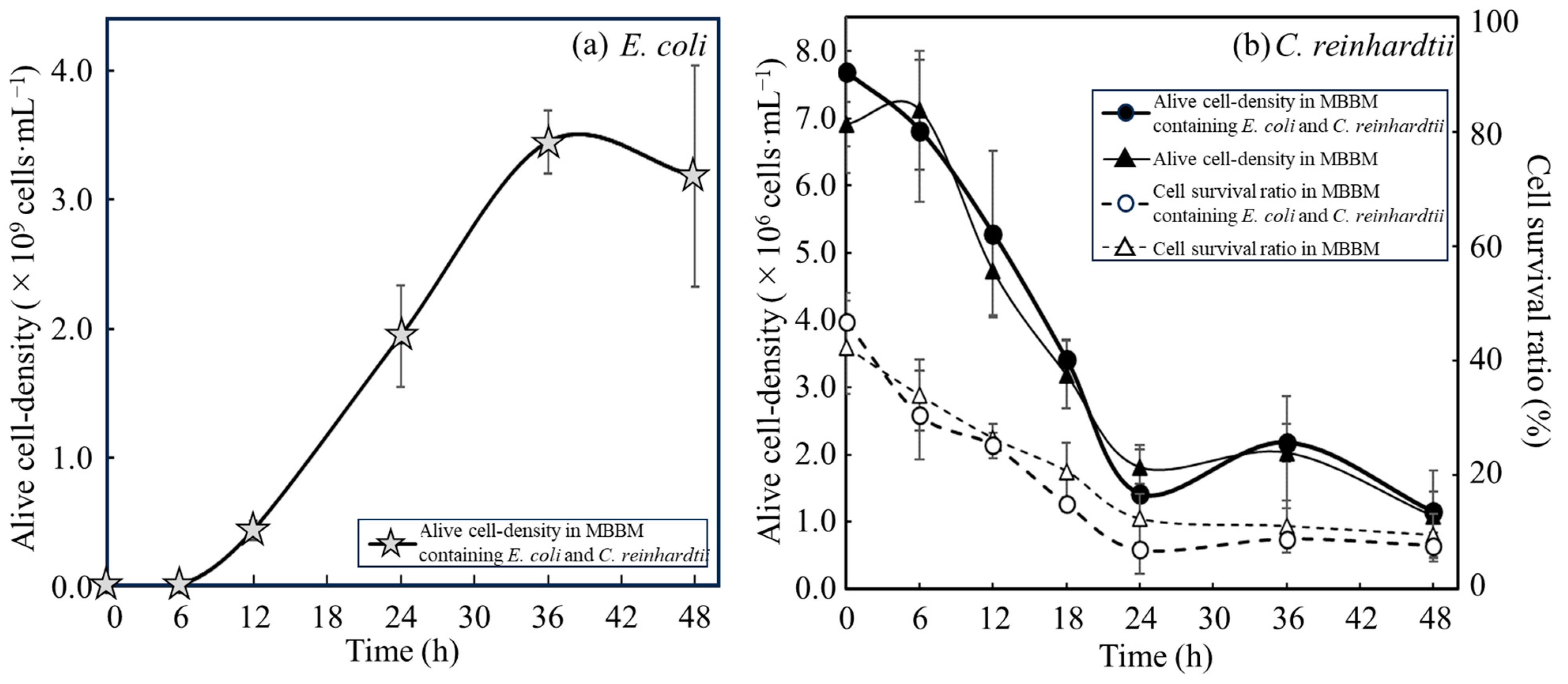
3.2. Comparison of Growth Activities of E. coli with C. reinhardtii Cells and with Disrupted Cell Extracts from C. reinhardtii
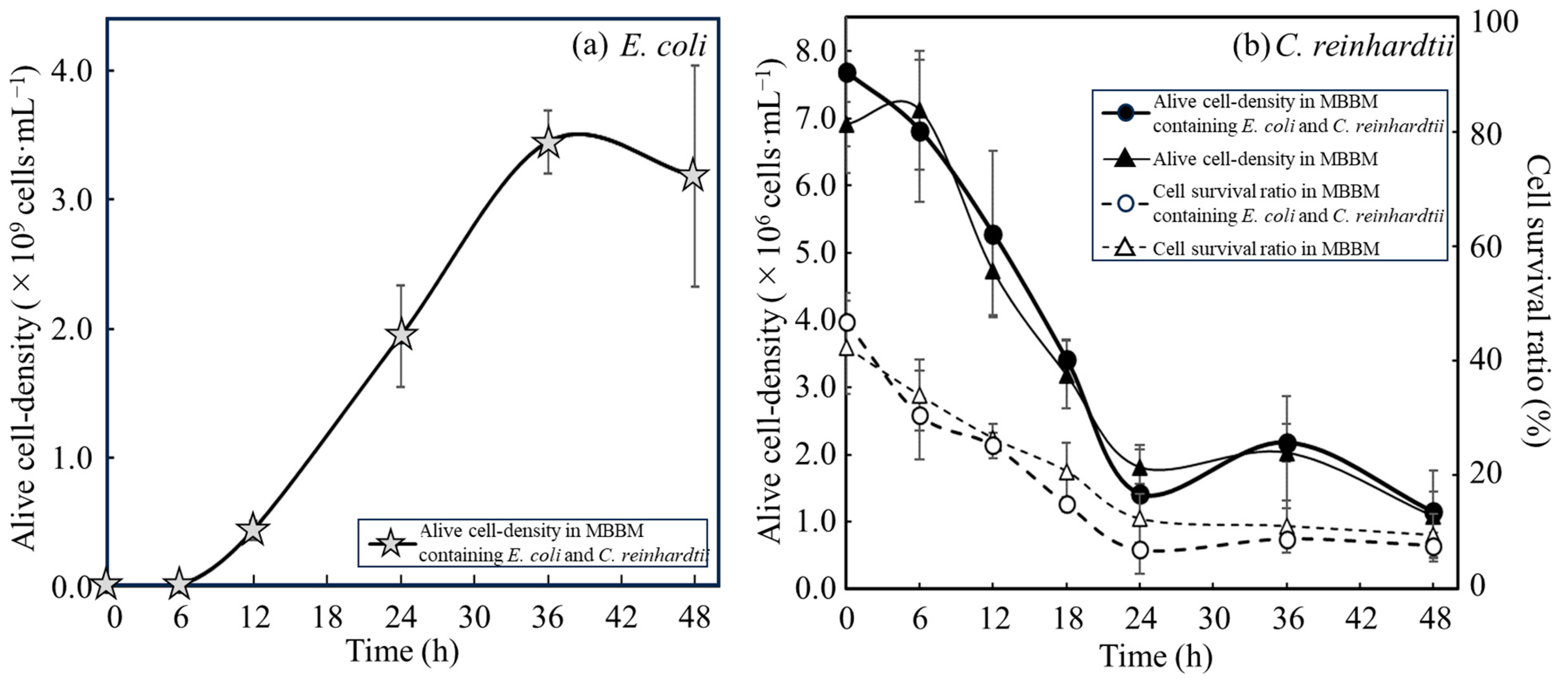
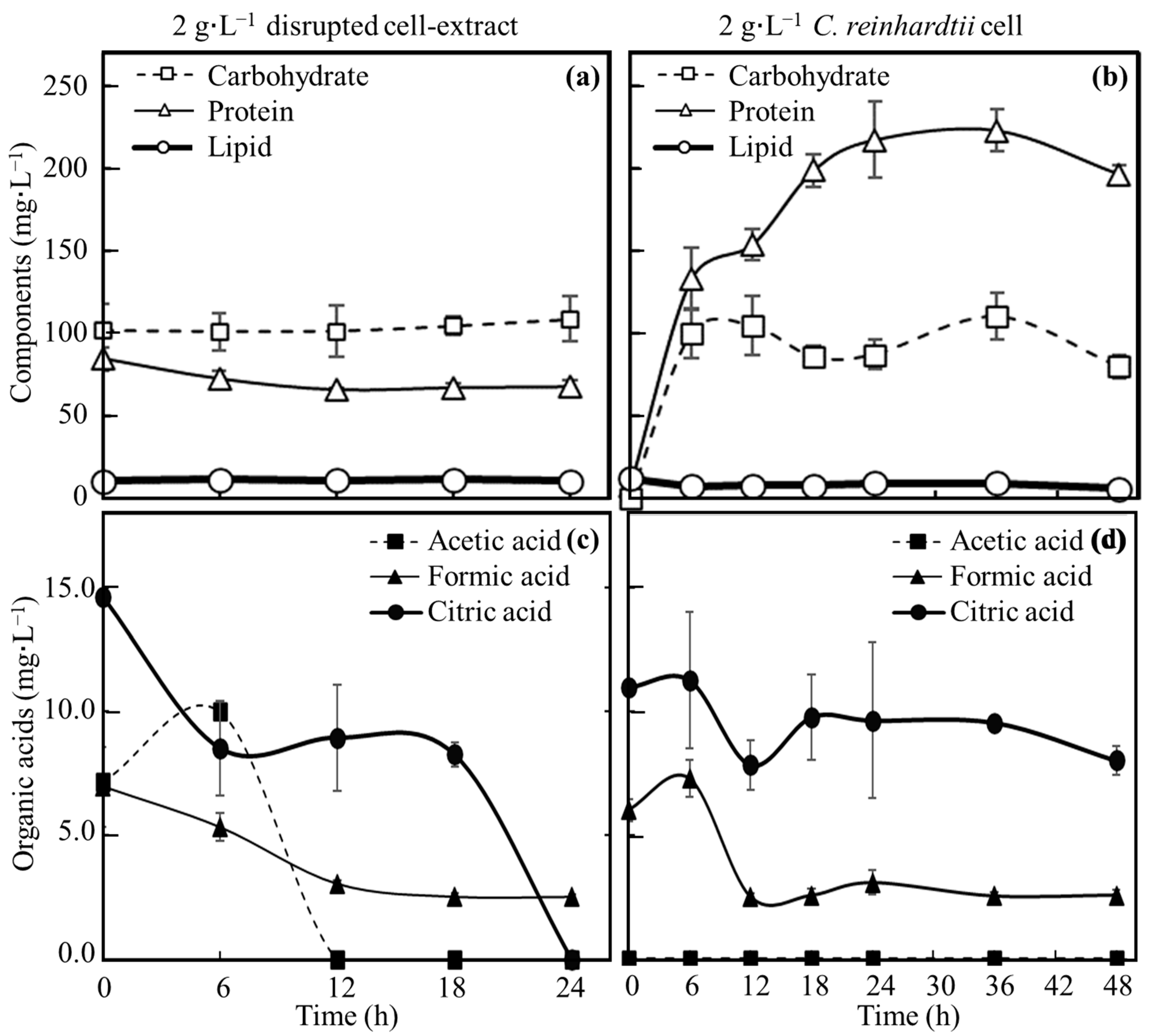
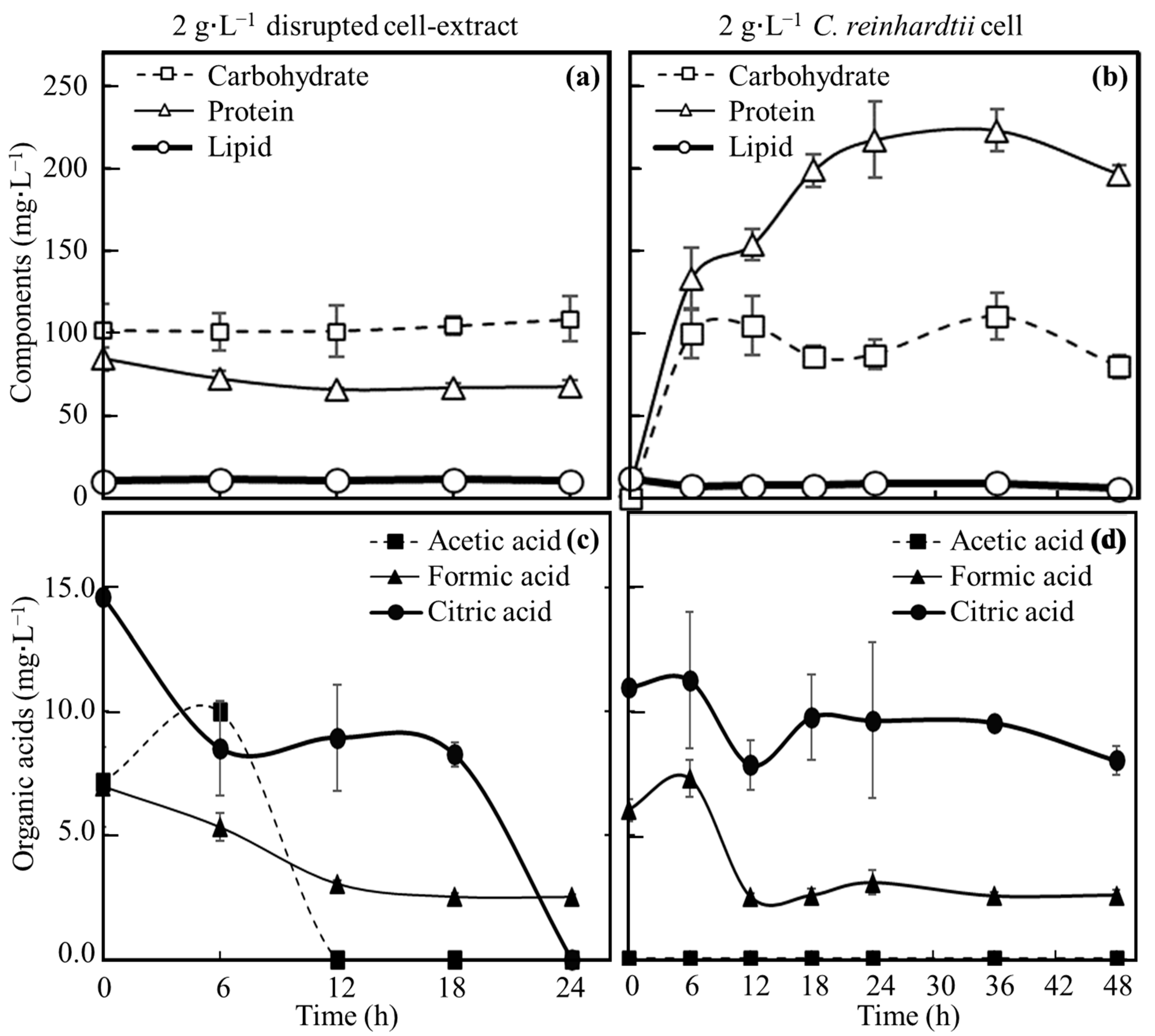
3.3. Evaluation of Nutrients Suplied from C. reinhardtii Consumed by E. coli
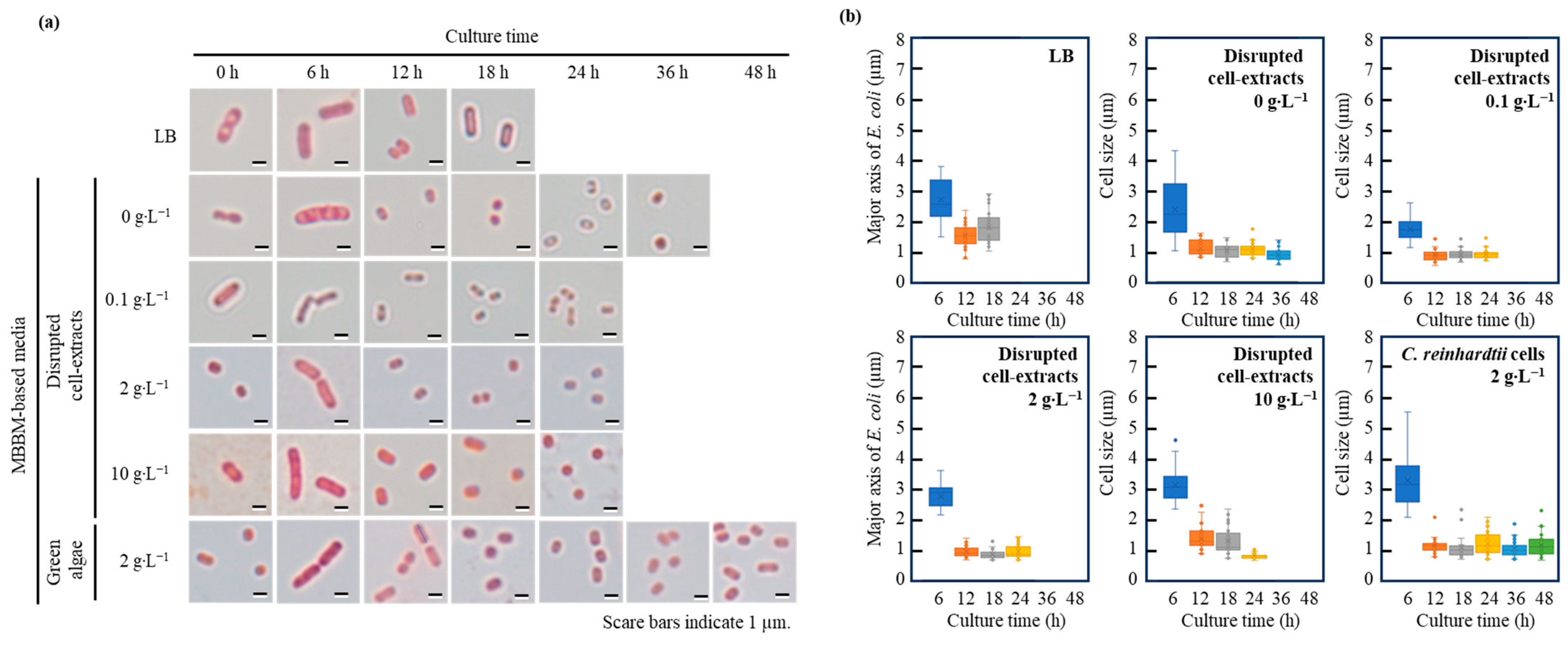
3.4. Aging Variation in Cell Size of E. coli in Broths Containing Disrupted Cell Extracts and C. reinhardtii Cells
3.5. Transcriptomic Analysis for E. coli in C. reinhardtii-Containing MBBM
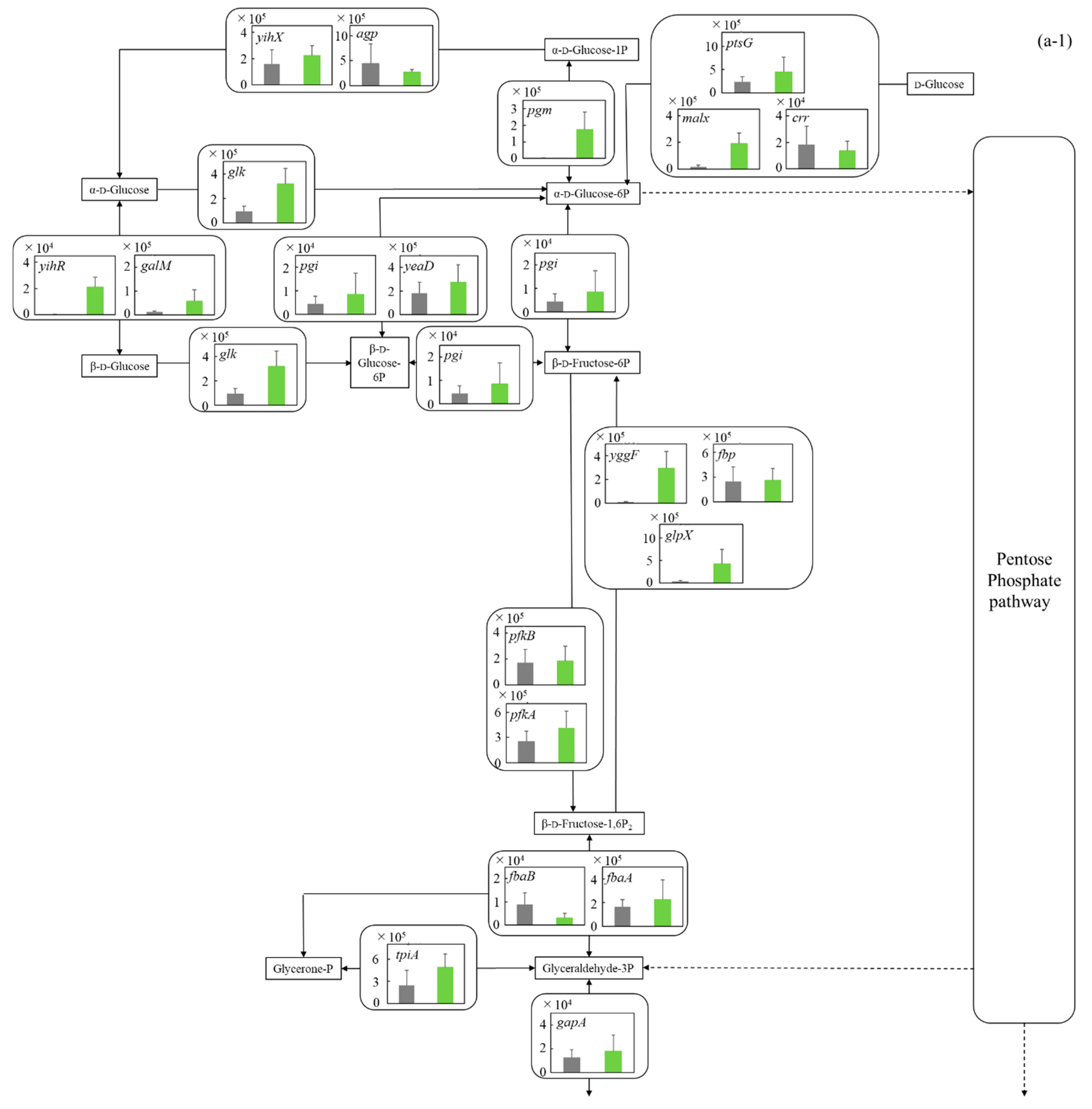
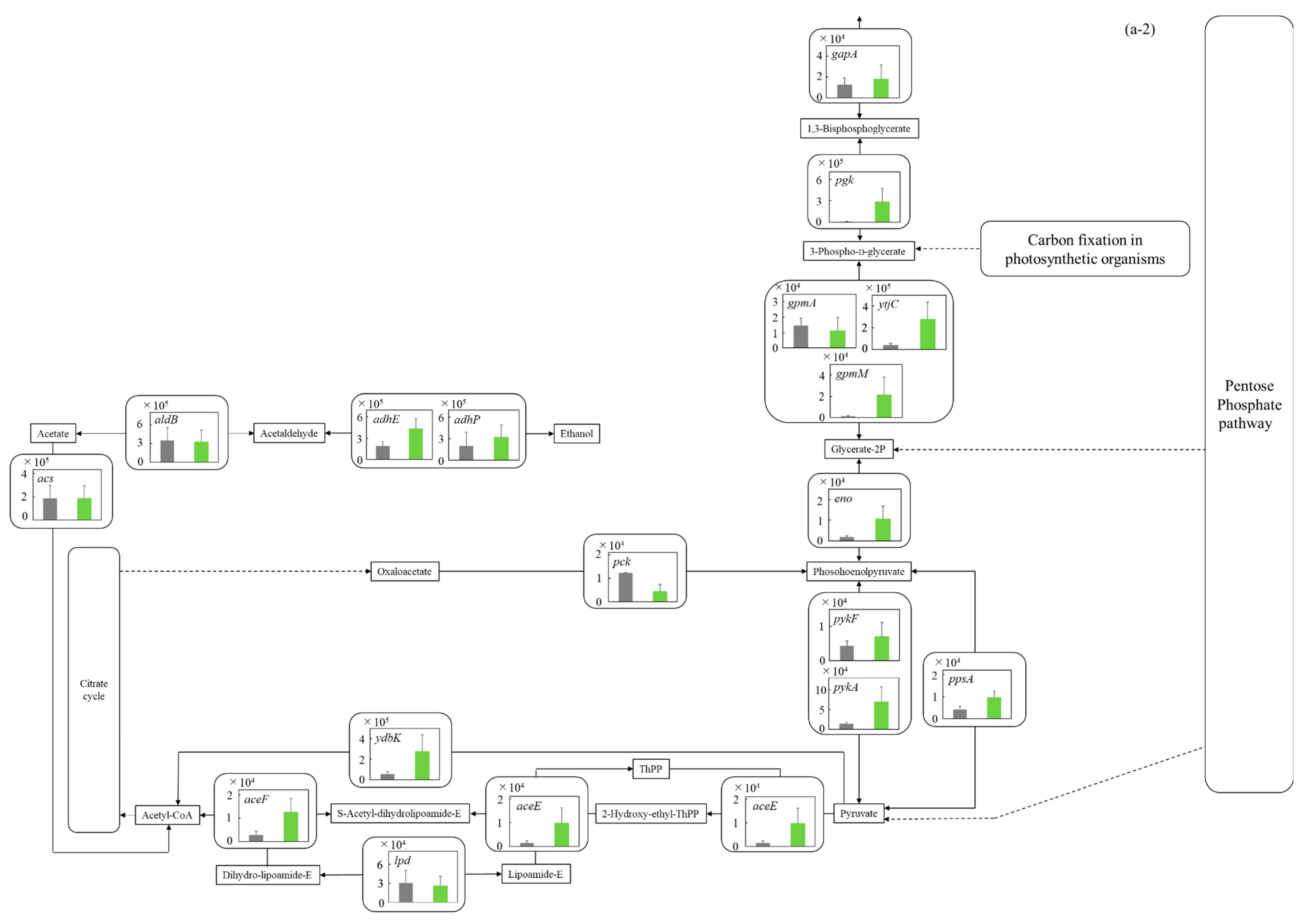
3.5.1. Glycolysis and Ethanol Assimilating Pathway
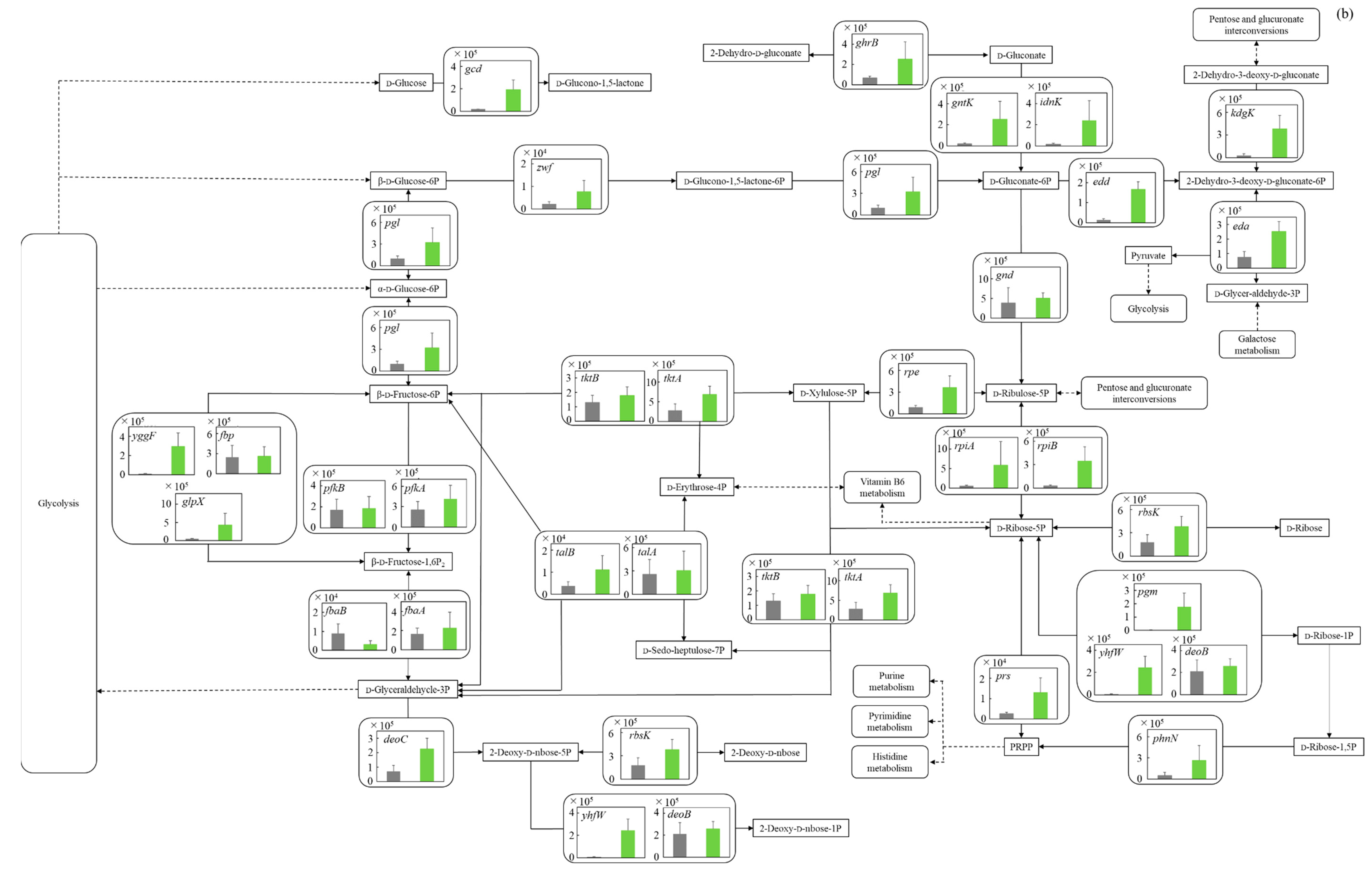
3.5.2. Pentose Phosphate Pathway
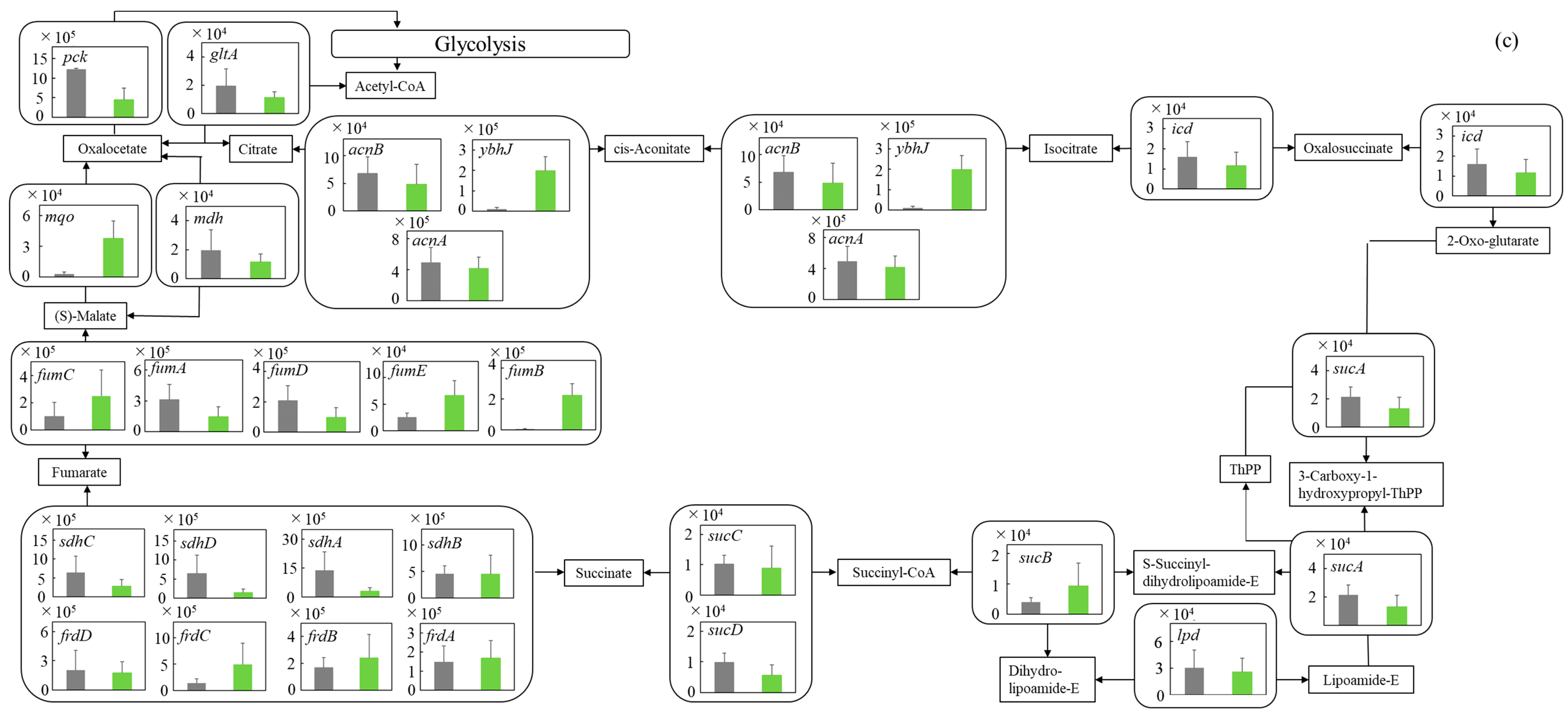
3.5.3. TCA Cycle
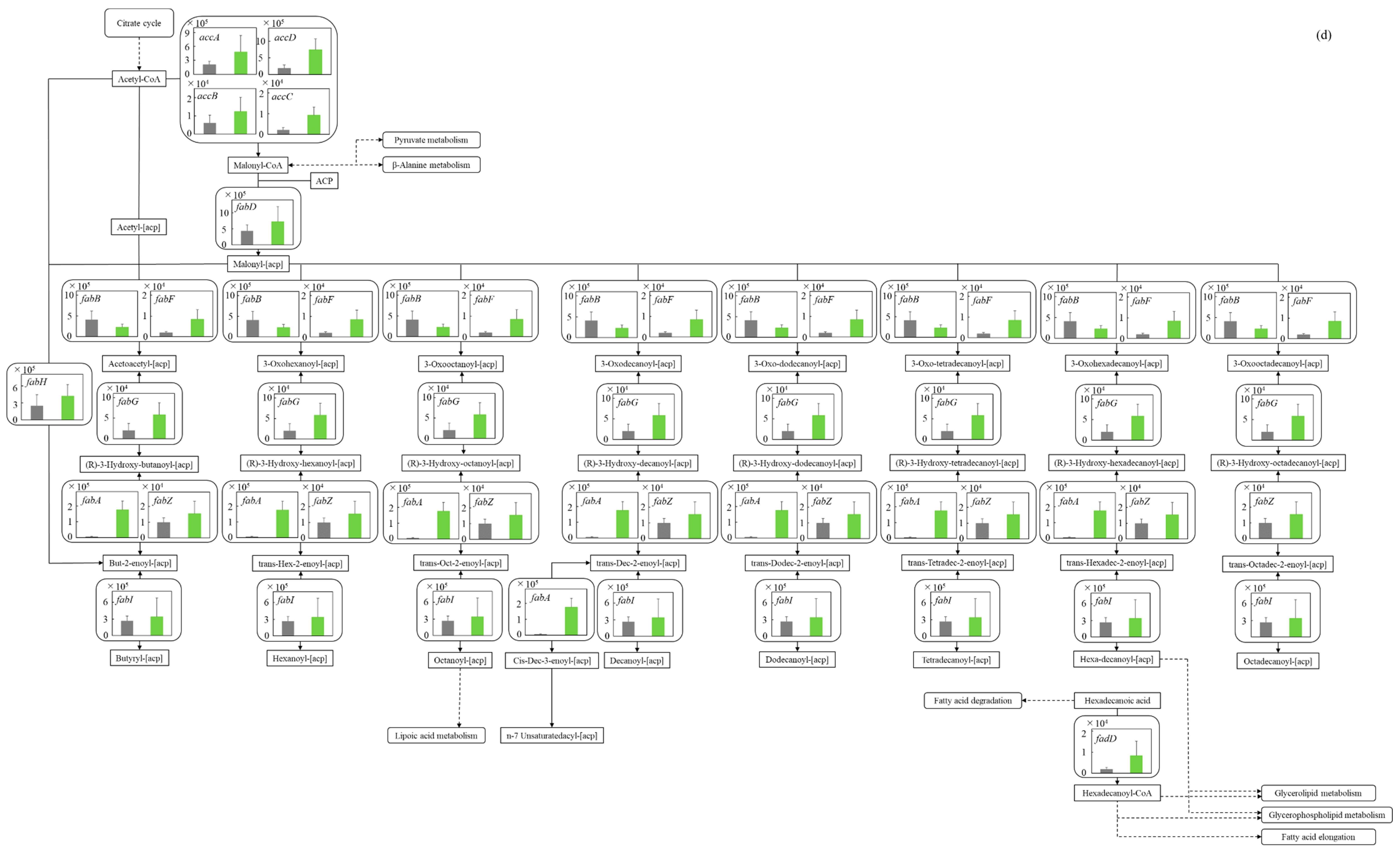
3.5.4. Fatty Acid Synthetic Pathway
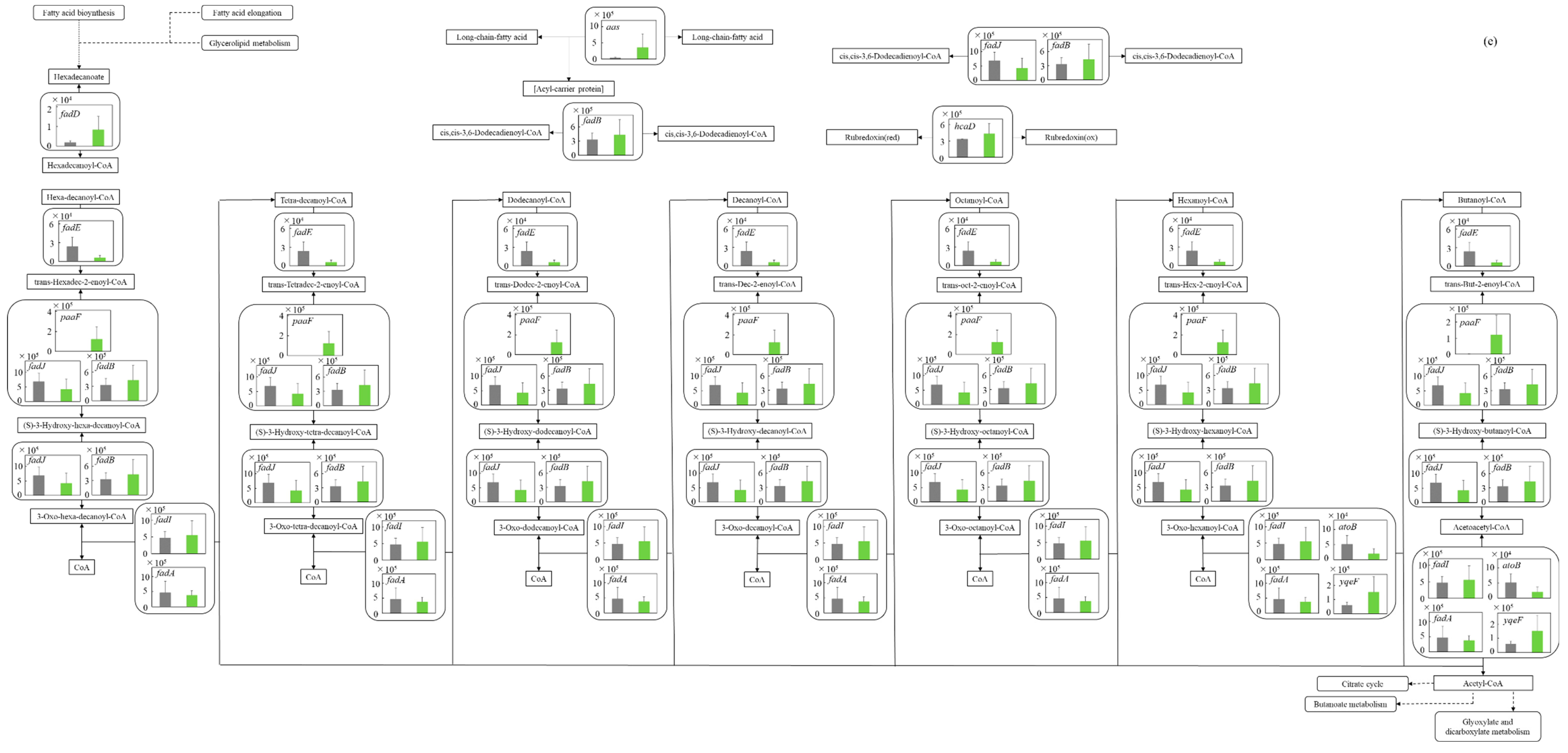
3.5.5. Fatty Acid Degradation Pathway
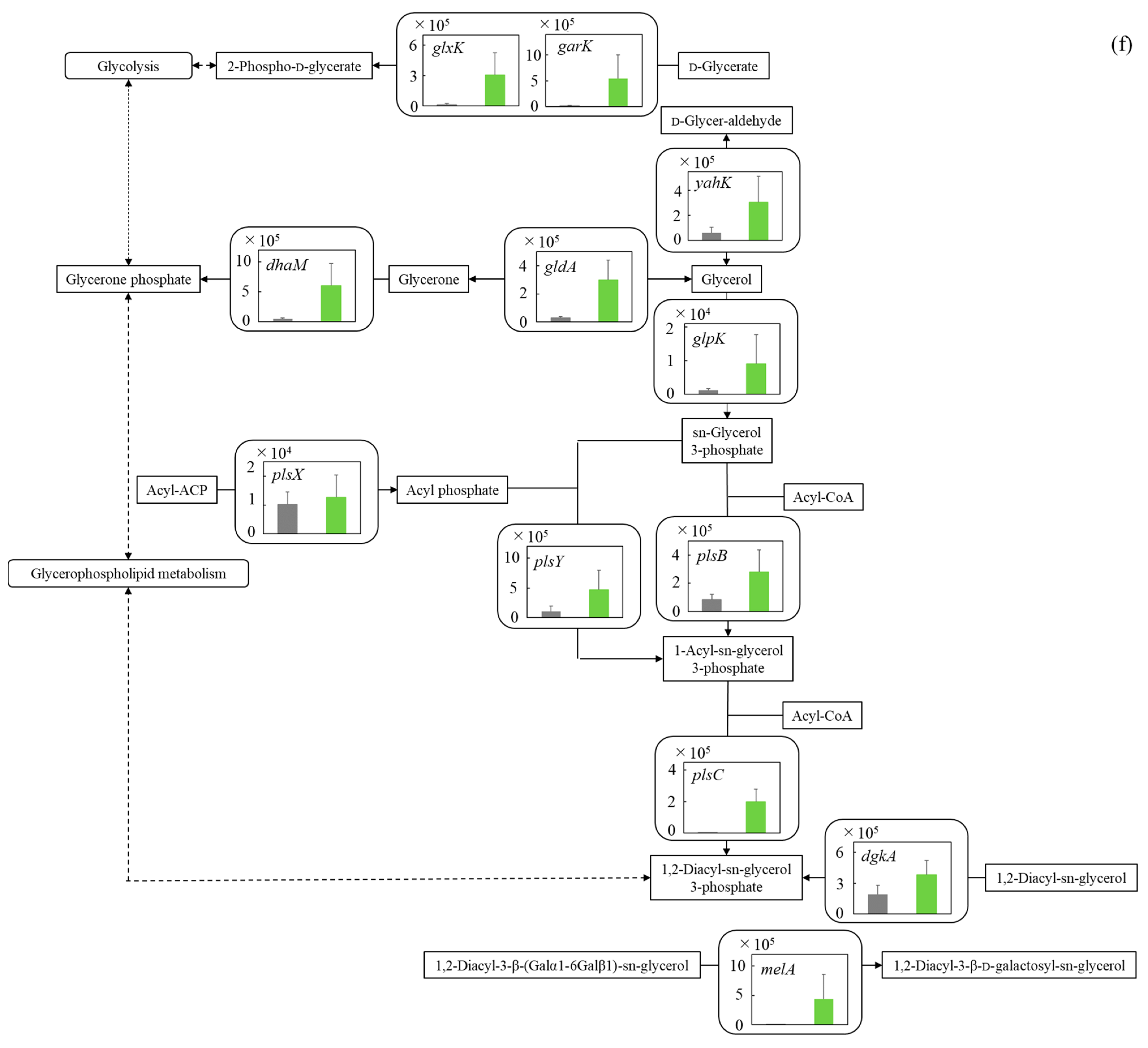
3.5.6. Glycerolipid Pathway
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parapouli, M.; Vasileiadis, A.; Afendra, A.; Hatziloukas, E. Saccharomyces cerevisiae and its industrial applications. AIMS Microbiol. 2020, 6, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Usui, Y.; Kimura, Y.; Satoh, T.; Takemura, N.; Ouchi, Y.; Ohmiya, H.; Kobayashi, K.; Suzuki, H.; Koyama, S.; Hagiwara, S.; et al. Effects of long-term intake of a yogurt fermented with Lactobacillus delbrueckii subsp. bulgaricus 2038 and Streptococcus thermophilus 1131 on mice. Int. Immunol. 2018, 30, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Song, M.W.; Chung, Y.; Kim, Y.T.; Hong, W.S.; Chang, H.J.; Paik, H.D. Probiotic characteristics of Lactobacillus brevis B13-2 isolated from kimchi and investigation of antioxidant and immune-modulating abilities of its heat-killed cells. LWT 2020, 128, 109452. [Google Scholar] [CrossRef]
- Lin, K.; Han, S.; Suiping, S. Application of Corynebacterium glutamicum engineering display system in three generations of biorefinery. Microb. Cell. Factories 2022, 21, 14. [Google Scholar] [CrossRef] [PubMed]
- De Lima Procópio, R.E.; Da Silva, I.R.; Martins, M.K.; De Azevedo, J.L.; De Araújo, J.M. Antibiotics produced by Streptomyces. Braz. J. Infect. Dis. 2012, 16, 466–471. [Google Scholar] [CrossRef]
- Lanigan, T.M.; Kopera, H.C.; Saunders, T.L. Principles of genetic engineering. Genes 2020, 11, 291. [Google Scholar] [CrossRef] [PubMed]
- Khalil, A.M. The genome editing revolution: Review. J. Genet. Eng. Biotechnol. 2020, 18, 68. [Google Scholar] [CrossRef] [PubMed]
- Huang, C., Jr.; Lin, H.; Yang, X. Industrial production of recombinant therapeutics in Escherichia coli and its recent advancements. J. Ind. Microbiol. Biotechnol. 2012, 39, 383–399. [Google Scholar] [CrossRef]
- Zhou, Y.; Lu, Z.; Wang, X.; Selvaraj, N.S.; Zhang, G. Genetic engineering modification and fermentation optimization for extracellular production of recombinant proteins using Escherichia coli. Appl. Microbiol. Biotechnol. 2012, 102, 1545–1556. [Google Scholar] [CrossRef]
- Clark, D.P. The fermentation pathways of Escherichia coli. FEMS Microbiol. Ecol. 1989, 5, 223–234. [Google Scholar] [CrossRef]
- Martínez-Gómez, K.; Flores, N.; Castañeda, H.M.; Martínez-Batallar, G.; Hernández-Chávez, G.; Ramírez, O.T.; Gosset, G.; Encarnación, S.; Bolivar, F. New insights into Escherichia coli metabolism: Carbon scavenging, acetate metabolism and carbon recycling responses during growth on glycerol. J. Clean. Prod. 2012, 11, 46. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Mu, H.; Guo, J.; Liu, H.; Zhang, R.; Liu, W.; Xian, M.; Liu, H. Metabolic engineering of Escherichia coli for the utilization of ethanol. J. Biol. Res. 2020, 27, 1. [Google Scholar] [CrossRef] [PubMed]
- Van Heeswijk, W.C.; Westerhoff, H.V.; Boogerd, F.C. Nitrogen assimilation in Escherichia coli: Putting molecular data into a systems perspective. Microbiol. Mol. Biol. Rev. 2013, 77, 4. [Google Scholar] [CrossRef] [PubMed]
- Baltz, H.R. Bacteriophage-resistant industrial fermentation strains: From the cradle to CRISPR/Cas9. J. Ind. Microbiol. Biotechnol. 2018, 45, 1003–1006. [Google Scholar] [CrossRef] [PubMed]
- Dayton, L. Atmospheric Carbon Dioxide Soars Past Crucial Milestone. Available online: https://www.science.org/content/article/atmospheric-carbon-dioxide-soars-past-crucial-milestone (accessed on 27 November 2023).
- Iglina, T.; Iglin, P.; Pashchenko, D. Industrial CO2 capture by algae: A review and recent advances. Sustainability 2022, 14, 3801. [Google Scholar] [CrossRef]
- Wang, B.; Li, Y.; Wu, N.; Lan, Q.C. CO2 bio-mitigation using microalgae. IOP AMB 2008, 79, 707–718. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, A.; Iritani, K.; Sakihama, Y.; Ozawa, N.; Mochizuki, A.; Watanabe, M. Construction of cell-plastics as neo-plastics consisted of cell-layer provided green alga Chlamydomonas reinhardtii covered by two-dimensional polymer. AMB Express 2020, 10, 112. [Google Scholar] [CrossRef]
- Jayshree, A.; Jayashree, S.; Thangaraju, N. Chlorella vulgaris and Chlamydomonas reinhardtii: Effective antioxidant, antibacterial and anticancer mediators. Indian J. Pharm. Sci. 2016, 78, 575–581. [Google Scholar] [CrossRef]
- Vadeboncoeur, Y.; Power, E.M. Attached algae: The cryptic base of inverted trophic pyramids in freshwaters. Annu. Rev. Ecol. Evol. Syst. 2017, 48, 255–279. [Google Scholar] [CrossRef]
- Berges, J.A.; Franklin, D.J.; Harrison, P.J. Evolution of an artificial seawater medium: Improvements in enriched seawater, artificial water over the last two decades. J. Phycol. 2001, 37, 1138–1145. [Google Scholar] [CrossRef]
- Spratt, S.K.; Ginsburgh, C.L.; Nunn, W.D. Isolation and genetic characterization of Escherichia coli mutants defective in propionate metabolism. J. Bacteriol. 1981, 146, 1166–1169. [Google Scholar] [CrossRef]
- Crippen, R.W.; Perrier, J.L. The use of neutral red and evans blue for live-dead determinations of marine plankton (with comments on the use of rotenone for inhibition of grazing). Stain Technol. 1974, 49, 97–104. [Google Scholar] [CrossRef]
- Zhou, K.; Zhou, L.; Lim, Q.E.; Zou, R.; Stephanopoulos, G.; Too, H.-P. Novel reference genes for quantifying transcriptional responses of Escherichia coli to protein overexpression by quantitative PCR. BMC Mol. Biol. 2011, 12, 18. [Google Scholar] [CrossRef]
- Fujiwara, R.; Noda, S.; Tanaka, T.; Kondo, A. Metabolic engineering of Escherichia coli for shikimate pathway derivative production from glucose-xylose co-substrate. Nat. Commun. 2020, 11, 279. [Google Scholar] [CrossRef] [PubMed]
- Lindner, N.S.; Aslan, S.; Muller, A.; Hoffart, E.; Behrens, P.; Edlich-Muth, C.; Blombach, B.; Bar-Even, A. A synthetic glycerol assimilation pathway demonstrates biochemical constraints of cellular metabolism. FEBS J. 2020, 287, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Baev, V.M.; Baev, D.; Radek, J.A.; Campbell, W.J. Growth of Escherichia coli MG1655 on LB medium: Monitoring utilization of sugars, alcohols, and organic acids with transcriptional microarrays. AMB 2006, 71, 310–316. [Google Scholar] [CrossRef]
- Zhou, Y.; Imlay, A.J. Escherichia coli K-12 lacks a high-affinity assimilatory cysteine importer. ASM 2019, 49, 120–128. [Google Scholar] [CrossRef]
- Peters, K.; Sargent, F. Formate hydrogenlyase, formic acid translocation and hydrogen production: Dynamic membrane biology during fermentation. Biochim. Biophys. Acta Bioenerg. 2023, 1864, 148919. [Google Scholar] [CrossRef]
- Xue, W.; Fan, D.; Shang, L.; Zhu, C.; Ma, X.; Zhu, X.; Yu, Y. Effects of acetic acid and its assimilation in fed-batch cultures of recombinant Escherichia coli containing human-like collagen cDNA. JBB 2010, 109, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Liu, D.; Chen, Z. Metabolic engineering of Escherichia coli to utilize methanol as a co-substrate for the production of (R)-1,3-butanediol. Biotechnol. Notes 2023, 103, 1441–1453. [Google Scholar] [CrossRef]
- Yao, Z.; Davis, R.M.; Kishony, R.; Kahne, D.; Ruiz, N. Regulation of cell size in response to nutrient availability by fatty acid biosynthesis in Escherichia coli. Proc. Natl. Acad. Sci. USA 2012, 109, E2561–E2568. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Ho, P.-Y.; Jiang, M.; Tang, B.; Liu, W.; Li, D.; Yu, X.; Kleckner, N.E.; Amir, A.; Liu, C. Interrogating the Escherichia coli cell cycle by cell dimension perturbations. Proc. Natl. Acad. Sci. USA 2016, 113, 15000–15005. [Google Scholar] [CrossRef]
- Nakanishi, A.; Mori, M.; Yamamoto, N.; Nemoto, S.; Kanamaru, N.; Yomogita, M.; Omino, N.; Matsumoto, R. Evaluation of cell responses of Saccharomyces cerevisiae under cultivation using wheat bran as a nutrient resource by analyses of growth activities and comprehensive gene transcription levels. Microorganisms 2023, 11, 2674. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, A.; Ho, S.H.; Kato, Y.; Yamasaki, H.; Chang, J.S.; Wang, Z.; Misawa, N.; Hirose, Y.; Minagawa, J.; Hasunuma, T.; et al. Dynamic metabolic profiling together with transcription analysis reveals salinity-induced starch-to-lipid biosynthesis in alga Chlamydomonas sp. JSC4. J. Sci. Rep. 2017, 7, 45471. [Google Scholar]
- Tome, D. Yeast extracts: Nutritional and flavoring food ingredients. ACS. Food. Sci. Technol. 2021, 1, 487–494. [Google Scholar] [CrossRef]
- Thomson, J.; Gerstenberger, P.D.; Goldberg, D.E.; Gociar, E.; De Silva, A.O.; Fraenkel, D.G. ColE1 hybrid plasmids for Escherichia coli genes of glycolysis and the hexose monophosphate shunt. J. Bacteriol. 1979, 137, 502–506. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.; Qiao, J.; Shanshan, C.; Dong, X.; Wu, Y.; Wang, Z.; Wang, X. Metabolic engineering for overproduction of colanic acid in Escherichia coli mutant with short lipopolysaccharide. J. Agric. Food Chem. 2022, 70, 8351–8364. [Google Scholar] [CrossRef]
- Singh, D.; Fairlamb, S.M.; Harrison, S.K.; Weeramange, C.; Meinhardt, S.; Tungtur, S.; Rau, F.B.; Hefty, P.S.; Fenton, W.A.; Swint-Kruse, L. Protein-protein interactions with fructose-I-kinase alter function of the central Escherichia coli transcription regulator, Cra. bioRxiv 2017. [CrossRef]
- Bren, A.; Park, J.O.; Towbin, B.D.; Dekel, E.; Rabinowitz, J.D.; Alona, U. Glucose becomes one of the worst carbon sources for E.coli on poor nitrogen sources due to suboptimal levels of cAMP. Sci. Rep. 2016, 6, 24834. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, X.; Sun, W.; Nie, T.; Quanquin, N.; Sun, Y. Escherichia coli increases its ATP concentration in weakly acidic environments principally through the glycolytic pathway. Genes 2020, 11, 991. [Google Scholar] [CrossRef]
- Liu, M.; Feng, X.; Ding, Y.; Zhao, G.; Liu, H.; Xian, M. Metabolic engineering of Escherichia coli to improve recombinant protein production. Appl. Microbiol. Biotechnol. 2015, 99, 10367–10377. [Google Scholar] [CrossRef]
- De Leon, R.P.; Creaser, E.H. The utilization of ethanol for biosynthesis in Escherichia coli. Can. J. Physiol. Pharmacol. 1958, 36, 839–845. [Google Scholar]
- Hove-Jensen, B.; Rosenkrantz, T.J.; Haldimann, A.; Wanner, B.L. Escherichia coli phnN, encoding Ribose 1,5-bisphosphokinase activity (phosphoribosyl diphosphate forming): Dual role in phosphonate degradation and NAD biosynthesis pathways. J. Bacteriol. 2003, 185, 2793–2801. [Google Scholar] [CrossRef]
- Lyngstadaas, A.; Sprenger, G.A.; Boye, E. Impaired growth of an Escherichia coli rpe mutant lacking ribulose-5-phosphate epimerase activity. Biochim. Et Biophys. Acta Gen. Subj. 1998, 1381, 319–330. [Google Scholar] [CrossRef]
- Sørensen, K.I.; Hove-Jensen, B. Ribose catabolism of Escherichia coli: Characterization of the rpiB gene encoding ribose phosphate isomerase B and of the rpiR gene, which is involved in regulation of rpiB expression. J. Bacteriol. 1996, 178, 1003–1011. [Google Scholar] [CrossRef]
- Van der Rest, M.E.; Frank, C.; Molenaar, D. Functions of the membrane-associated and cytoplasmic malate dehydrogenases in the citric acid cycle of Escherichia coli. J. Bacteriol. 2000, 182, 6892–6899. [Google Scholar] [CrossRef]
- Ford, T.J. Engineering Escherichia Coli Fatty Acid Metabolism for the Production of Biofuel Precursors. Available online: https://dash.harvard.edu/handle/1/17467357 (accessed on 16 June 2023).
- Zelcbuch, L.; Razo-Mejia, M.; Herz, E.; Yahav, S.; Antonovsky, N.; Kroytoro, H.; Milo, R.; Bar-Even, A. An in vivo metabolic approach for deciphering the product specificity of glycerate kinase proves that both E. coli’s glycerate kinases generate 2-phosphoglycerate. PLoS ONE 2015, 10, e0122957. [Google Scholar] [CrossRef] [PubMed]
- Gutknecht, R.; Beutler, R.; Garcia-Alles, L.F.; Baumann, U.; Erni, B. The dihydroxyacetone kinase of Escherichia coli utilizes a phosphoprotein instead of ATP as phosphoryl donor. EMBO J. 2001, 20, 2480–2486. [Google Scholar] [CrossRef] [PubMed]
- Noga, M.J.; Büke, F.; Van den Broek, N.J.F.; Imholz, N.C.E.; Scherer, N.; Yang, F.; Bokinsky, G. Posttranslational control of PlsB is sufficient to coordinate membrane synthesis with growth in Escherichia coli. mBio 2020, 11, e02703–e02719. [Google Scholar] [CrossRef] [PubMed]
- Kessels, J.M.M.; Ousen, H.; Van den Bosch, H. Facilitated utilization of endogenously synthesized lysophosphatidic acid by 1-acylglycerophosphate acyltransferase from Escherichia coli. Biochim. Biophys. Acta 1983, 753, 227–235. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Added Resource | Maximum Alive Cell Density | Growth Rate | |||||
|---|---|---|---|---|---|---|---|
| Time (h) | Alive Cell Density (×108 cells∙mL−1) | Time Range (h) | Specific Growth Rate (µmax, h−1) | Doubling Time (h) | Corresponding Figure | ||
| Disrupted cell extracts | 0.1 g∙L−1 | 12 | 0.03 ± 0.02 | 6~12 | 0.27 ± 0.10 | 2.75 ± 0.84 | Figure 1a |
| 12~24 | Growth not shown | Not calculated | |||||
| 1 g∙L−1 | 24 | 0.64 ± 0.59 | 6~12 | 0.45 ± 0.34 | 2.78 ± 2.71 | Figure 1a | |
| 12~24 | 0.03 ± 0.02 | 38.85 ± 32.51 | |||||
| 2 g∙L−1 | 12 | 1.15 ± 0.17 | 6~12 | 0.63 ± 0.29 | 1.26 ± 0.55 | Figure 1a | |
| 12~24 | Growth not shown | Not calculated | |||||
| 4 g∙L−1 | 24 | 1.80 ± 0.23 | 6~12 | 0.50 ± 0.11 | 1.44 ± 0.35 | Figure 1a | |
| 12~24 | 0.06 ± 0.05 | 11.76 ± 10.61 | |||||
| 10 g∙L−1 | 12 | 4.59 ± 0.48 | 6~12 | 0.82 ± 0.27 | 0.90 ± 0.25 | Figure 1a | |
| 12~24 | Growth not shown | Not calculated | |||||
| Green algae | 2 g∙L−1 | 36 | 34.48 ± 2.44 | 6~12 | 1.04 ± 0.27 | 0.71 ± 0.22 | Figure 2a |
| 12~24 | 0.12 ± 0.01 | 5.62 ± 0.61 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakanishi, A.; Omino, N.; Nakamura, T.; Goto, S.; Matsumoto, R.; Yomogita, M.; Narisawa, N.; Kimijima, M.; Iritani, K. Evaluation of Cellular Responses of Heterotrophic Escherichia coli Cultured with Autotrophic Chlamydomonas reinhardtii as a Nutrient Source by Analyses Based on Microbiology and Transcriptome. Microorganisms 2024, 12, 452. https://doi.org/10.3390/microorganisms12030452
Nakanishi A, Omino N, Nakamura T, Goto S, Matsumoto R, Yomogita M, Narisawa N, Kimijima M, Iritani K. Evaluation of Cellular Responses of Heterotrophic Escherichia coli Cultured with Autotrophic Chlamydomonas reinhardtii as a Nutrient Source by Analyses Based on Microbiology and Transcriptome. Microorganisms. 2024; 12(3):452. https://doi.org/10.3390/microorganisms12030452
Chicago/Turabian StyleNakanishi, Akihito, Natsumi Omino, Tomoyo Nakamura, Saki Goto, Riri Matsumoto, Misaki Yomogita, Naoki Narisawa, Manami Kimijima, and Kohei Iritani. 2024. "Evaluation of Cellular Responses of Heterotrophic Escherichia coli Cultured with Autotrophic Chlamydomonas reinhardtii as a Nutrient Source by Analyses Based on Microbiology and Transcriptome" Microorganisms 12, no. 3: 452. https://doi.org/10.3390/microorganisms12030452
APA StyleNakanishi, A., Omino, N., Nakamura, T., Goto, S., Matsumoto, R., Yomogita, M., Narisawa, N., Kimijima, M., & Iritani, K. (2024). Evaluation of Cellular Responses of Heterotrophic Escherichia coli Cultured with Autotrophic Chlamydomonas reinhardtii as a Nutrient Source by Analyses Based on Microbiology and Transcriptome. Microorganisms, 12(3), 452. https://doi.org/10.3390/microorganisms12030452