

Locust Pathogen Aspergillus oryzae XJ1 Is Different from Aspergillus oryzae and Aspergillus flavus Based on Genomics Comparisons



Abstract
1. Introduction
2. Materials and Methods
2.1. Strain Culture
2.2. Genome Sequencing of A. oryzae XJ1
2.2.1. Extraction of Genomic DNA of A. oryzae XJ1
2.2.2. Library Construction of A. oryzae XJ1
- (1)
- PacBio Sequel platform (Pacific Biosciences, Menlo Park, CA, USA)
- (2)
- Illumina NovaSeq platform (Illumina, Inc., San Diego, CA, USA)
2.2.3. Sequencing
2.2.4. Genome Assembly
Preliminary Assembly with SMRT Link 5.0.1 software suite (Pacific Biosciences, Menlo Park, CA, USA)
Correction of the Preliminary Assembly
2.2.5. Genome Component Prediction
2.2.6. Prediction of Gene Functions
2.3. Comparative Genomic Analysis
2.3.1. Gene Family Analysis
2.3.2. Phylogenomic Tree Reconstruction Based on Genome Alignment
2.3.3. Core/Pan-Genome Analysis
2.3.4. Genomic Synteny Analysis
3. Results
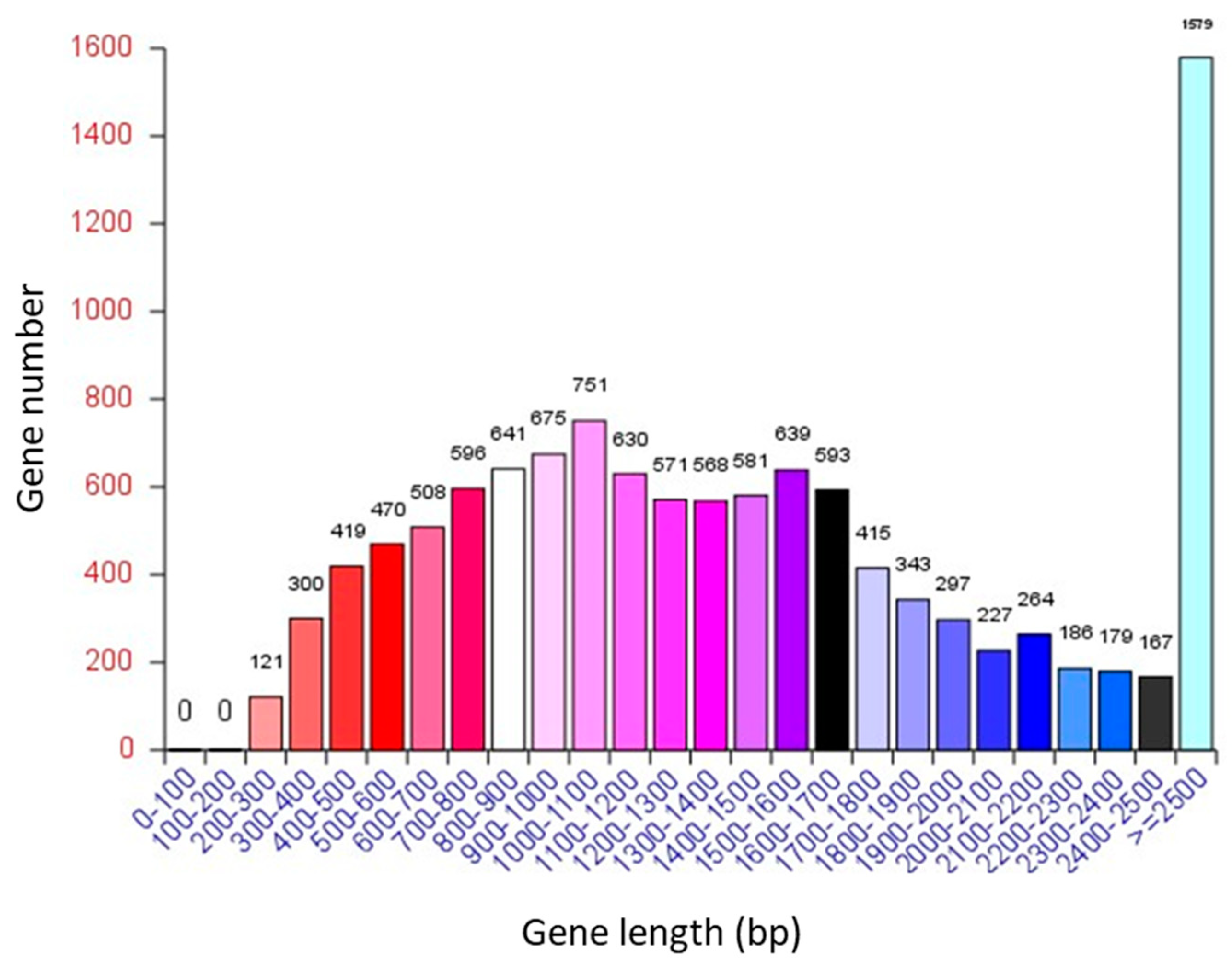
3.1. General Information on the A. oryzae XJ1 Genome
3.2. Analysis of Predicted Gene Functions in A. oryzae XJ1
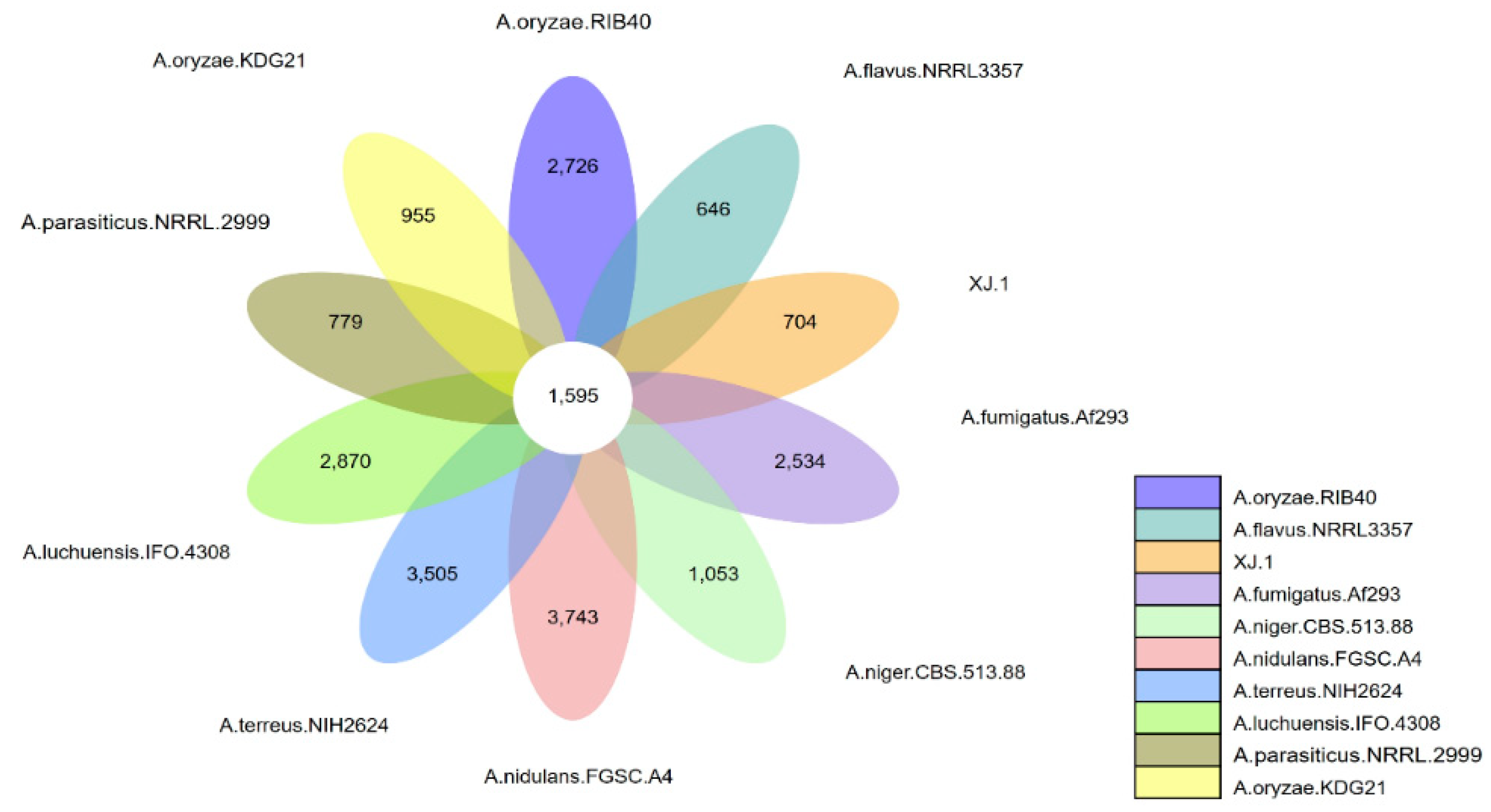
3.3. Evolutionary Analysis of the A. oryzae XJ1 Genome
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lomer, C.J.; Bateman, R.P.; Johnson, D.L.; Langewald, J.; Thomas, M. Biological control of locusts and grasshoppers. Annu. Rev. Entomol. 2001, 46, 667–702. [Google Scholar] [CrossRef] [PubMed]
- de Faria, M.R.; Wraight, S.P. Mycoinsecticides and Mycoacaricides: A comprehensive list with worldwide coverage and international classification of formulation types. Biol. Control 2007, 43, 237–256. [Google Scholar] [CrossRef]
- Zhang, P.; You, Y.; Song, Y.; Wang, Y.; Zhang, L. First record of Aspergillus oryzae (Eurotiales: Trichocomaceae) as an entomopathogenic fungus of the locust, Locusta migratoria (Orthoptera: Acrididae). Biocontrol Sci. Technol. 2015, 25, 1285–1298. [Google Scholar] [CrossRef]
- You, Y.; An, Z.; Zhang, X.; Liu, H.; Yang, W.; Yang, M.; Wang, T.; Xie, X.; Zhang, L. Virulence of the fungal pathogen, Aspergillus oryzae XJ-1 to adult locusts (Orthoptera: Acrididae) in both laboratory and field trials. Pest Manag. Sci. 2023, 79, 3767–3772. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Lu, X.; Xing, L.; Ho, S.W.A.; Kwan, H.S. Genomic and transcriptomic comparison of Aspergillus oryzae strains: A case study in soy sauce koji fermentation. J. Ind. Microbiol. Biotechnol. 2018, 45, 839–853. [Google Scholar] [CrossRef]
- Venkatesh, M.V.; Joshi, K.R.; Harjai, S.C.; Ramdeo, I.N. Aspergillosis in desert locust (Schistocerka gregaria Forsk). Mycopathologia 1975, 57, 135–138. [Google Scholar] [CrossRef]
- Kumar, S.; Slutana, R.; Wagan, M.S. Pathogenic application of Aspergillus species for the control of agricultural important grasshoppers. J. Biodivers. Environ. Sci. 2013, 3, 223–229. [Google Scholar]
- Sultana, R.; Wagan, Y.S.; Naeem, M.; Wagan, M.S.; Khatri, I. Susceptibility of three Hieroglyphus species (Hemiacridinae: Acrididae: Orthoptera) to some strains of the entomopathogenic fungi from Pakistan. Can. J. Pure Appl. Sci. 2013, 7, 2325–2332. [Google Scholar]
- He, B.; Tu, Y.; Jiang, C.; Zhang, Z.; Li, Y.; Zeng, B. Functional genomics of Aspergillus oryzae: Strategies and progress. Microorganisms 2019, 7, 103. [Google Scholar] [CrossRef]
- Watarai, N.; Yamamoto, N.; Sawada, K.; Yamada, T.; Watarai, N.; Yamamoto, N.; Sawada, K.; Yamada, T. Evolution of Aspergillus oryzae before and after domestication inferred by large-scale comparative genomic analysis. DNA Res. 2019, 26, 465–472. [Google Scholar] [CrossRef]
- Jeon, J.; Kim, J.A.; Park, S.Y.; Kim, G.W.; Park, C.S.; Kim, C.; Park, H.Y.; Yeo, J.H.; Lee, Y.H.; Kim, S. Draft genome sequence of Aspergillus oryzae BP2-1, isolated from traditional malted rice in South Korea. Microbiol. Resour. Announc. 2020, 9, e01405-19. [Google Scholar] [CrossRef] [PubMed]
- Chacón-Vargas, K.; McCarthy, C.O.; Choi, D.; Wang, L.; Yu, J.H.; Gibbons, J.G. Comparison of two Aspergillus oryzae genomes from different clades reveals independent evolution of alpha-amylase duplication, variation in secondary metabolism genes, and differences in primary metabolism. Front. Microbiol. 2021, 12, 691296. [Google Scholar] [CrossRef] [PubMed]
- Umemura, M.; Koike, H.; Yamane, N.; Koyama, Y.; Satou, Y.; Kikuzato, I.; Teruya, M.; Tsukahara, M.; Imada, Y.; Wachi, Y.; et al. Comparative genome analysis between Aspergillus oryzae strains reveals close relationship between sites of mutation localization and regions of highly divergent genes among Aspergillus species. DNA Res. 2012, 19, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Kjærbølling, I.; Vesth, T.; Frisvad, J.C.; Nybo, J.L.; Theobald, S.; Kildgaard, S.; Petersen, T.I.; Kuo, A.; Sato, A.; Lyhne, E.K.; et al. A comparative genomics study of 23 Aspergillus species from section Flavi. Nat. Commun. 2020, 11, 1106. [Google Scholar] [CrossRef] [PubMed]
- Han, D.M.; Baek, J.H.; Choi, D.G.; Jeon, M.S.; Eyun, S.I.; Jeon, C.O. Comparative pangenome analysis of Aspergillus flavus and Aspergillus oryzae reveals their phylogenetic, genomic, and metabolic homogeneity. Food Microbiol. 2024, 119, 104435. [Google Scholar] [CrossRef]
- Lim, H.J.; Lee, E.-H.; Yoon, Y.; Chua, B.; Son, A. Portable lysis apparatus for rapid single-step DNA extraction of Bacillus subtilis. J. Appl. Microbiol. 2016, 120, 379–387. [Google Scholar] [CrossRef]
- Machida, M.; Asai, K.; Sano, M.; Tanaka, T.; Kumagai, T.; Terai, G.; Kusumoto, K.; Arima, T.; Akita, O.; Kashiwagi, Y.; et al. Genome sequencing and analysis of Aspergillus oryzae. Nature 2005, 438, 1157–1161. [Google Scholar] [CrossRef]
- Nierman, W.C.; Yu, J.; Fedorova-Abrams, N.D.; Losada, L.; Cleveland, T.E.; Bhatnagar, D.; Bennett, J.W.; Dean, R.; Payne, G.A. Genome sequence of Aspergillus flavus NRRL 3357, a strain that causes aflatoxin contamination of food and feed. Genome Announc. 2015, 3, e00168-15. [Google Scholar] [CrossRef]
- Jørgensen, T.R. Identification and toxigenic potential of the industrially important fungi, Aspergillus oryzae and Aspergillus sojae. J. Food Prot. 2007, 70, 2916–2934. [Google Scholar] [CrossRef]
- Montiel, D.; Dickinson, M.J.; Lee, H.A.; Dyer, P.S.; Jeenes, D.J.; Roberts, I.N.; James, S.; Fuller, L.J.; Matsuchima, K.; Archer, D.B. Genetic differentiation of the Aspergillus section Flavi complex using AFLP fingerprints. Mycol. Res. 2003, 107 Pt 12, 1427–1434. [Google Scholar] [CrossRef]
- Abastabar, M.; Shabanzadeh, S.; Valadan, R.; Mayahi, S.; Haghani, I.; Khojasteh, S.; Nargesi, S.; Seyedmousavi, S.; Hedayati, M.T. Development of RFLP method for rapid differentiation of Aspergillus flavus and Aspergillus oryzae, two species with high importance in clinical and food microbiology. J. Mycol. Med. 2022, 32, 101274. [Google Scholar] [CrossRef] [PubMed]
- Kumeda, Y.; Asao, T. Heteroduplex panel analysis, a novel method for genetic identification of Aspergillus Section Flavi strains. Appl. Environ. Microbiol. 2001, 67, 4084–4090. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.K.; Ehrlich, K.C.; Hua, S.S. Cladal relatedness among Aspergillus oryzae isolates and Aspergillus flavus S and L morphotype isolates. Int. J. Food Microbiol. 2006, 108, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Nargesi, S.; Abastabar, M.; Valadan, R.; Mayahi, S.; Youn, J.H.; Hedayati, M.T.; Seyedmousavi, S. Differentiation of Aspergillus flavus from Aspergillus oryzae targeting the cyp51A gene. Pathogens 2021, 10, 1279. [Google Scholar] [CrossRef] [PubMed]
- Hedayati, M.T.; Taghizadeh-Armaki, M.; Zarrinfar, H.; Hoseinnejad, A.; Ansari, S.; Abastabar, M.; Er, H.; Özhak, B.; Öğünç, D.; Ilkit, M.; et al. Discrimination of Aspergillus flavus from Aspergillus oryzae by matrix-assisted laser desorption/ionisation time-of-flight (MALDI-TOF) mass spectrometry. Mycoses 2019, 62, 1182–1188. [Google Scholar] [CrossRef]
- Suleiman, W.B. A multi-aspect analysis of two analogous aspergillus spp. belonging to section Flavi: Aspergillus flavus and aspergillus oryzae. BMC Microbiol. 2023, 23, 71. [Google Scholar] [CrossRef]
- Barbesgaard, P.; Heldt-Hansen, H.P.; Diderichsen, B. On the safety of Aspergillus oryzae: A review. Appl. Microbiol. Biotechnol. 1992, 36, 569–572. [Google Scholar] [CrossRef]
- Geiser, D.M.; Pitt, J.I.; Taylor, J.W. Cryptic speciation and recombination in the aflatoxin–producing fungus Aspergillus flavus. Proc. Natl. Acad. Sci. USA 1998, 95, 388–393. [Google Scholar] [CrossRef]
- Tailor, M.J.; Richardson, T. Applications of microbial enzymes in food systems and in biotechnology. Adv. Appl. Microbiol. 1979, 25, 7–35. [Google Scholar] [CrossRef]
- FAO_WHO. Committee on Food Additives 31; World Health Organization Technical Report Series: Geneva, Switzerland, 1987. [Google Scholar]
- Abe, K.; Gomi, K.; Hasegawa, F.; Machida, M. Impact of Aspergillus oryzae genomics on industrial production of metabolites. Mycopathologia 2006, 162, 143–153. [Google Scholar] [CrossRef]
- Driver, F.; Milner, R.J.; Trueman, J.W.H. A taxonomic revision of Metarhizium based on a phylogenetic analysis of rDNA sequence data. Mycol. Res. 2000, 104, 134–150. [Google Scholar] [CrossRef]
- Bischoff, J.F.; Rehner, S.A.; Humber, R.A. A multilocus phylogeny of the Metarhizium anisopliae lineage. Mycologia 2009, 101, 512–530. [Google Scholar] [CrossRef] [PubMed]
- Blanford, S.; Thomas, M.B. Adult survival, maturation, and reproduction of the desert locust Schistocerca gregaria infected with the fungus Metarhizium anisopliae var acridum. J. Invertebr. Pathol. 2001, 78, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Nasseh, O.M.; Freres, T.; Wilps, J.; Kirkilionis, E.; Krall, S. Field cage trials on the effects of enriched neem oil, insect growth regulators and the pathogens Beauveria bassiana and Nosema locustae on desert locusts in the Republic of Niger. In Biological Control of Locusts and Grasshoppers; Moler, C.J., Prior, C., Eds.; CAB International: Wallingford, UK, 1992; pp. 311–320. [Google Scholar]
- Fu, X.; Liu, H.; Xu, X.; Guo, J.; Hu, S.; You, Y.; Zhang, L. Comparison of the virulence of space mutants of Aspergillus oryzae XJ-1 against adult Locusta migratoria. Agronomy 2024, 14, 116. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genome Information Summary | |
|---|---|
| Genome size (bp) | 37,874,054 |
| GC content of genome (%) | 47.31 |
| Gene number | 11,720 |
| Gene total length (bp) | 18,250,593 |
| GC content of gene (%) | 52.09 |
| Gene length/genome (%) | 48.19 |
| Gene average length (bp) | 1557 |
| Gene internal length (bp) | 19,623,461 |
| Gene internal GC content (%) | 42.88 |
| Gene internal length/genome (%) | 51.81 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
You, Y.; Xu, X.; Liu, H.; Zhang, L. Locust Pathogen Aspergillus oryzae XJ1 Is Different from Aspergillus oryzae and Aspergillus flavus Based on Genomics Comparisons. Microorganisms 2024, 12, 2501. https://doi.org/10.3390/microorganisms12122501
You Y, Xu X, Liu H, Zhang L. Locust Pathogen Aspergillus oryzae XJ1 Is Different from Aspergillus oryzae and Aspergillus flavus Based on Genomics Comparisons. Microorganisms. 2024; 12(12):2501. https://doi.org/10.3390/microorganisms12122501
Chicago/Turabian StyleYou, Yinwei, Xiao Xu, Hui Liu, and Long Zhang. 2024. "Locust Pathogen Aspergillus oryzae XJ1 Is Different from Aspergillus oryzae and Aspergillus flavus Based on Genomics Comparisons" Microorganisms 12, no. 12: 2501. https://doi.org/10.3390/microorganisms12122501
APA StyleYou, Y., Xu, X., Liu, H., & Zhang, L. (2024). Locust Pathogen Aspergillus oryzae XJ1 Is Different from Aspergillus oryzae and Aspergillus flavus Based on Genomics Comparisons. Microorganisms, 12(12), 2501. https://doi.org/10.3390/microorganisms12122501