
Wild-Mouse-Derived Gut Microbiome Transplantation in Laboratory Mice Partly Alleviates House-Dust-Mite-Induced Allergic Airway Inflammation

 , , , ,
, , , ,  ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethical Approval
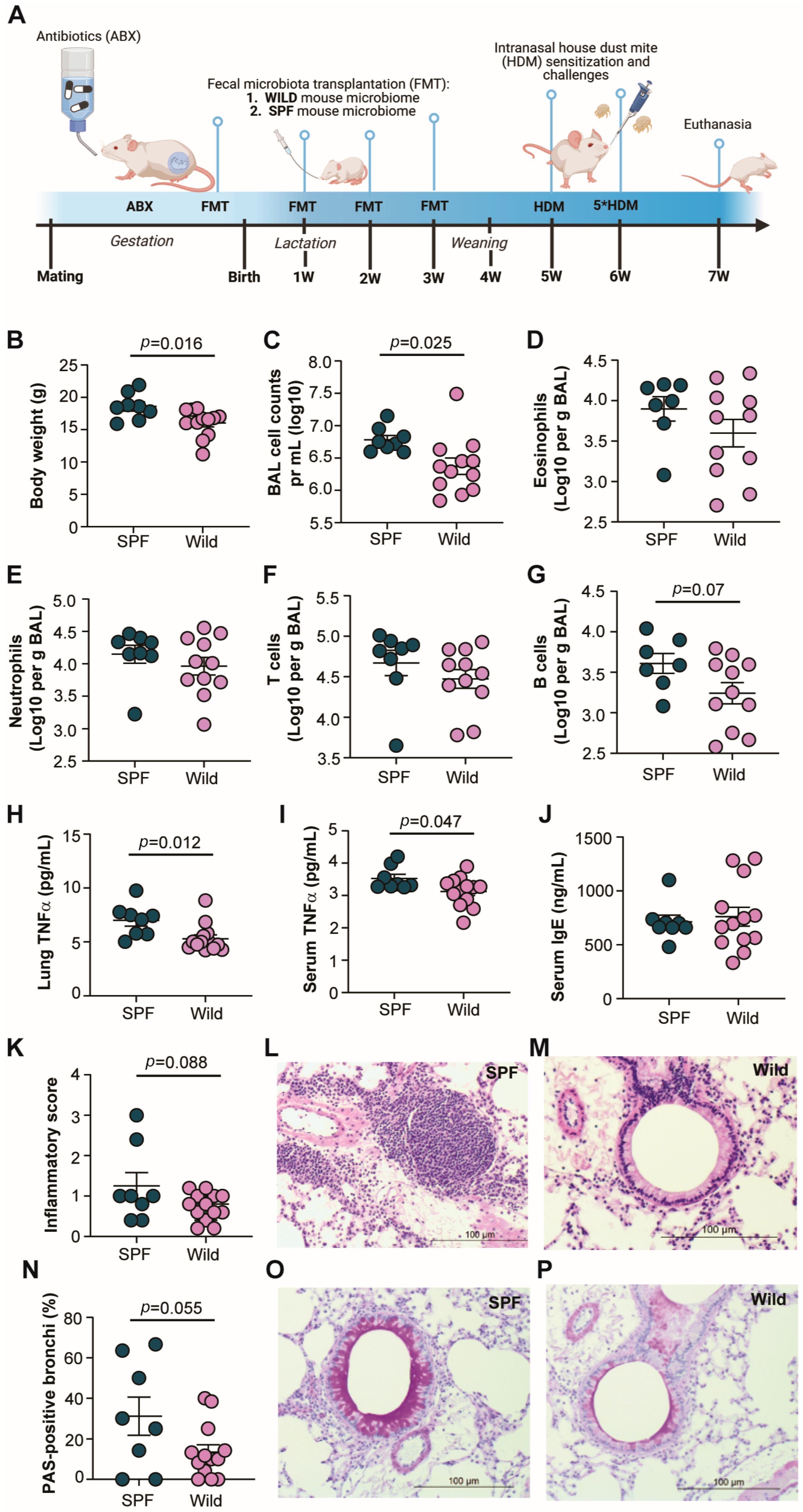
2.2. Experimental Design
2.3. Cecal Microbiome Transplant
2.4. House-Dust-Mite-Induced Allergic Airway Inflammation
2.5. Mouse Sacrifice and Specimen Collection
2.6. Flow Cytometry
2.7. Lung Histology
2.8. Cytokine Measurement
2.9. ELISA for IgG and IgE Antibodies
2.10. Gene Expression Analysis by qPCR
2.11. Serum Lipopolysaccharide (LPS)
2.12. High-Throughput Sequencing of the Gut Microbiota
2.13. Mycobiome Analysis
2.14. Metabolomics Analysis
2.15. Statistics
3. Results
3.1. Wild-Mouse-Derived Gut Microbiota Reduces Allergic Airway Inflammation in Transplanted Mice
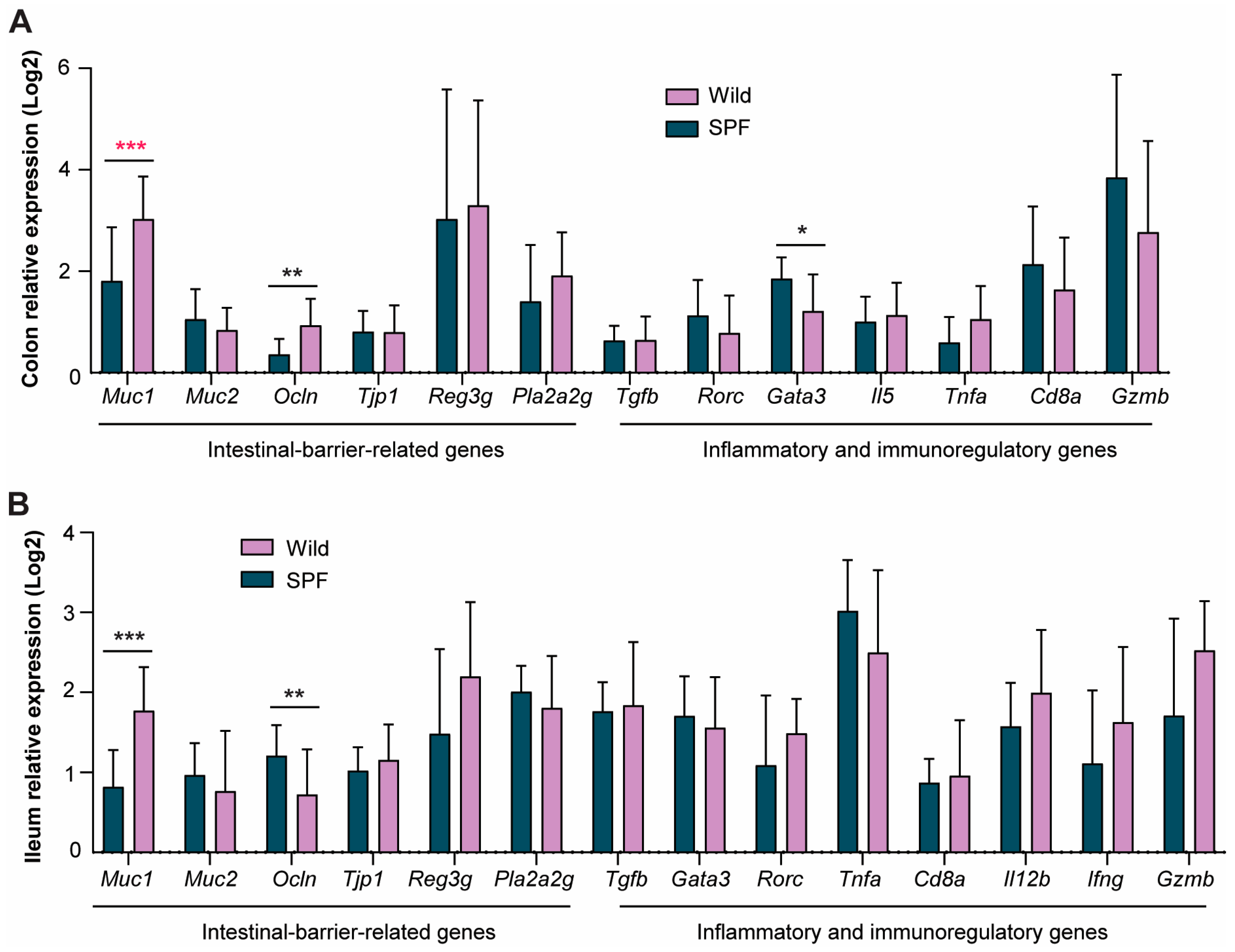
3.2. Wild-Derived Microbiome Transplantation Significantly Upregulated Intestinal Gene Expression Associated with Improved Gut Barrier Function
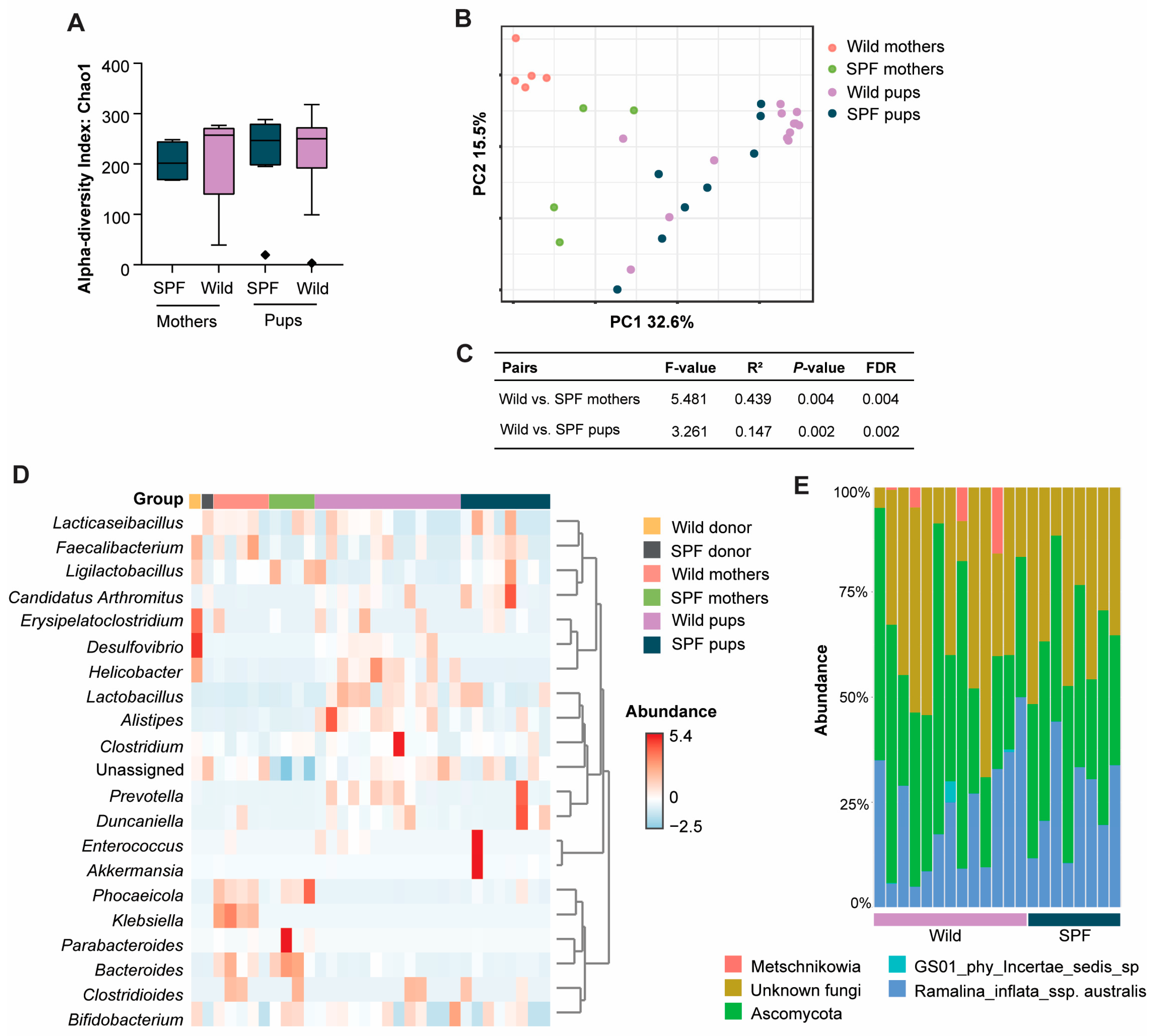
3.3. Distinct Microbiome Taxa Were Found Between Wild and SPF Recipient Mice
3.4. Presence of Intestinal Fungi, Protozoa, and Helminths Were Similar Between the Wild- and SPF-Microbiome-Associated Mice
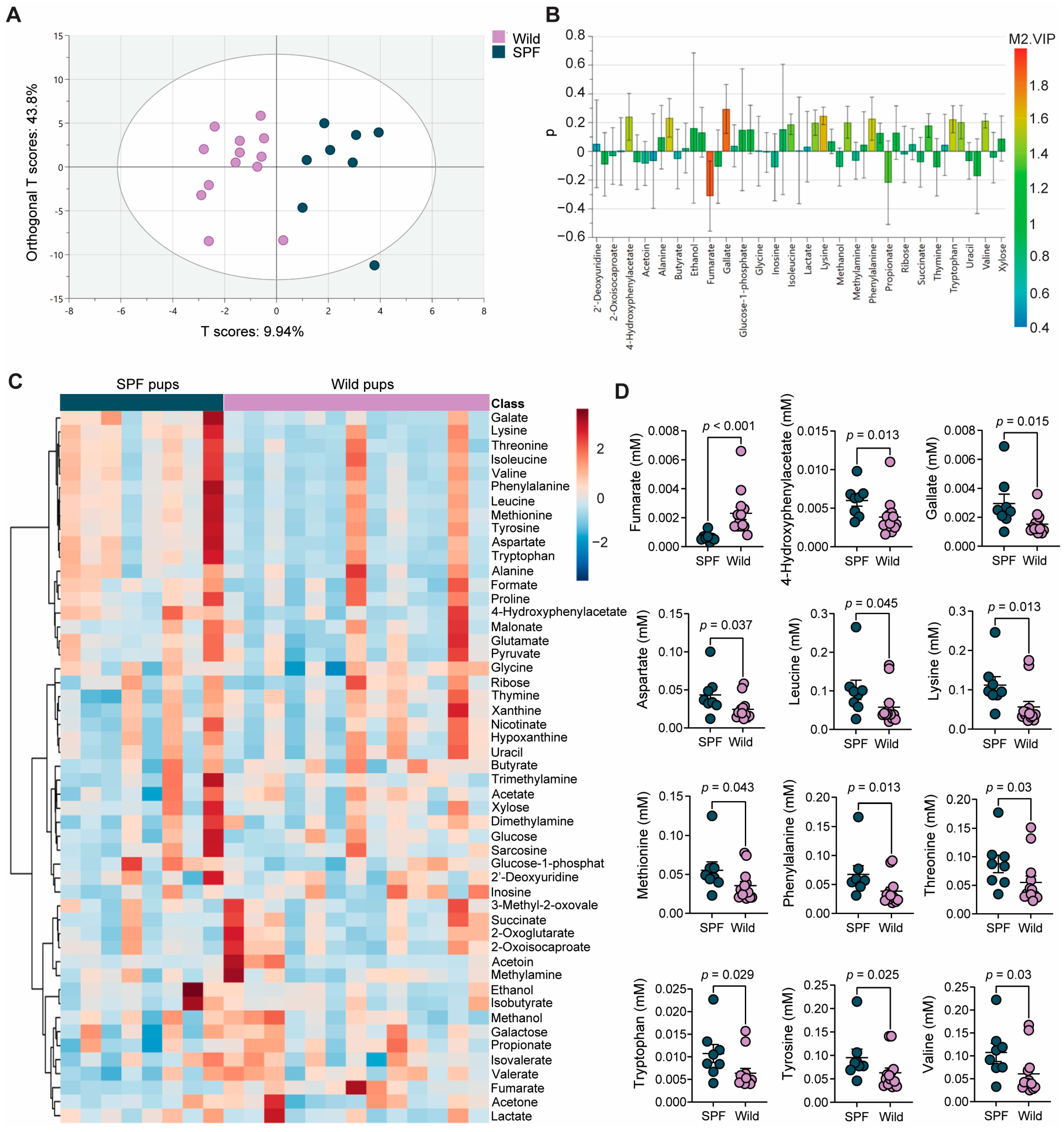
3.5. The Wild- and SPF-Mouse-Derived Microbiomes Induced Distinct Intestinal Metabolite Profiles in the Recipient Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rosshart, S.P.; Vassallo, B.G.; Angeletti, D.; Hutchinson, D.S.; Morgan, A.P.; Takeda, K.; Hickman, H.D.; McCulloch, J.A.; Badger, J.H.; Ajami, N.J.; et al. Wild Mouse Gut Microbiota Promotes Host Fitness and Improves Disease Resistance. Cell 2017, 171, 1015–1028.e1013. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.K.; Hansen, C.H.F. The microbiome and rodent models of immune mediated diseases. Mamm. Genome 2021, 32, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Bowerman, K.L.; Knowles, S.C.L.; Bradley, J.E.; Baltrūnaitė, L.; Lynch, M.D.J.; Jones, K.M.; Hugenholtz, P. Effects of laboratory domestication on the rodent gut microbiome. ISME Commun. 2021, 1, 49. [Google Scholar] [CrossRef] [PubMed]
- Beura, L.K.; Hamilton, S.E.; Bi, K.; Schenkel, J.M.; Odumade, O.A.; Casey, K.A.; Thompson, E.A.; Fraser, K.A.; Rosato, P.C.; Filali-Mouhim, A.; et al. Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature 2016, 532, 512–516. [Google Scholar] [CrossRef]
- Boysen, P.; Eide, D.M.; Storset, A.K. Natural killer cells in free-living Mus musculus have a primed phenotype. Mol. Ecol. 2011, 20, 5103–5110. [Google Scholar] [CrossRef]
- Rosshart, S.P.; Herz, J.; Vassallo, B.G.; Hunter, A.; Wall, M.K.; Badger, J.H.; McCulloch, J.A.; Anastasakis, D.G.; Sarshad, A.A.; Leonardi, I.; et al. Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science 2019, 365, eaaw4361. [Google Scholar] [CrossRef]
- Hild, B.; Dreier, M.S.; Oh, J.H.; McCulloch, J.A.; Badger, J.H.; Guo, J.; Thefaine, C.E.; Umarova, R.; Hall, K.D.; Gavrilova, O.; et al. Neonatal exposure to a wild-derived microbiome protects mice against diet-induced obesity. Nat. Metab. 2021, 3, 1042–1057. [Google Scholar] [CrossRef]
- Braman, S.S. The global burden of asthma. Chest 2006, 130, 4s–12s. [Google Scholar] [CrossRef] [PubMed]
- Debeuf, N.; Haspeslagh, E.; van Helden, M.; Hammad, H.; Lambrecht, B.N. Mouse Models of Asthma. Curr. Protoc. Mouse Biol. 2016, 6, 169–184. [Google Scholar] [CrossRef]
- Tan, H.T.; Hagner, S.; Ruchti, F.; Radzikowska, U.; Tan, G.; Altunbulakli, C.; Eljaszewicz, A.; Moniuszko, M.; Akdis, M.; Akdis, C.A.; et al. Tight junction, mucin, and inflammasome-related molecules are differentially expressed in eosinophilic, mixed, and neutrophilic experimental asthma in mice. Allergy 2019, 74, 294–307. [Google Scholar] [CrossRef]
- Milligan, K.L.; Matsui, E.; Sharma, H. Asthma in Urban Children: Epidemiology, Environmental Risk Factors, and the Public Health Domain. Curr. Allergy Asthma Rep. 2016, 16, 33. [Google Scholar] [CrossRef] [PubMed]
- Strachan, D.P. Hay fever, hygiene, and household size. BMJ 1989, 299, 1259–1260. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.; Brickley, E.; Rodrigues, L.; Normansell, R.A.; Barreto, M.; Cooper, P.J. Urbanisation and asthma in low-income and middle-income countries: A systematic review of the urban-rural differences in asthma prevalence. Thorax 2019, 74, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, E.G. Urbanization and childhood asthma: An African perspective. J. Allergy Clin. Immunol. 2000, 105, 224–231. [Google Scholar] [CrossRef]
- Herbst, T.; Sichelstiel, A.; Schär, C.; Yadava, K.; Bürki, K.; Cahenzli, J.; McCoy, K.; Marsland, B.J.; Harris, N.L. Dysregulation of allergic airway inflammation in the absence of microbial colonization. Am. J. Respir. Crit. Care Med. 2011, 184, 198–205. [Google Scholar] [CrossRef]
- Mähler Convenor, M.; Berard, M.; Feinstein, R.; Gallagher, A.; Illgen-Wilcke, B.; Pritchett-Corning, K.; Raspa, M. FELASA recommendations for the health monitoring of mouse, rat, hamster, guinea pig and rabbit colonies in breeding and experimental units. Lab. Anim. 2014, 48, 178–192. [Google Scholar] [CrossRef]
- Tournoy, K.G.; Kips, J.C.; Schou, C.; Pauwels, R.A. Airway eosinophilia is not a requirement for allergen-induced airway hyperresponsiveness. Clin. Exp. Allergy 2000, 30, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Arildsen, A.W.; Zachariassen, L.F.; Krych, L.; Hansen, A.K.; Hansen, C.H.F. Delayed Gut Colonization Shapes Future Allergic Responses in a Murine Model of Atopic Dermatitis. Front. Immunol. 2021, 12, 650621. [Google Scholar] [CrossRef]
- Zachariassen, L.F.; Ebert, M.B.B.; Mentzel, C.M.J.; Deng, L.; Krych, L.; Nielsen, D.S.; Stokholm, J.; Hansen, C.H.F. Cesarean section induced dysbiosis promotes type 2 immunity but not oxazolone-induced dermatitis in mice. Gut Microbes 2023, 15, 2271151. [Google Scholar] [CrossRef]
- Dhariwal, A.; Chong, J.; Habib, S.; King, I.L.; Agellon, L.B.; Xia, J. MicrobiomeAnalyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017, 45, W180–w188. [Google Scholar] [CrossRef]
- Amadi, C.N.; Orish, C.N.; Frazzoli, C.; Orisakwe, O.E. Dietary interventions for autism spectrum disorder: An updated systematic review of human studies. Psychiatriki 2022, 33, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.B.; Johansen, L.J.; Powell, L.D.; Quig, D.; Rubin, R.A. Gastrointestinal flora and gastrointestinal status in children with autism--comparisons to typical children and correlation with autism severity. BMC Gastroenterol. 2011, 11, 22. [Google Scholar] [CrossRef] [PubMed]
- Abarenkov, K.; Nilsson, R.H.; Larsson, K.H.; Taylor, A.F.S.; May, T.W.; Frøslev, T.G.; Pawlowska, J.; Lindahl, B.; Põldmaa, K.; Truong, C.; et al. The UNITE database for molecular identification and taxonomic communication of fungi and other eukaryotes: Sequences, taxa and classifications reconsidered. Nucleic Acids Res. 2024, 52, D791–D797. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, L.M.A.; Sundekilde, U.K.; Andersen, H.J.; Kot, W.; Mejia, J.L.C.; Nielsen, D.S.; Hansen, A.K.; Bertram, H.C. Administration of Bovine Milk Oligosaccharide to Weaning Gnotobiotic Mice Inoculated with a Simplified Infant Type Microbiota. Microorganisms 2021, 9, 1003. [Google Scholar] [CrossRef]
- Benard, A.; Desreumeaux, P.; Huglo, D.; Hoorelbeke, A.; Tonnel, A.B.; Wallaert, B. Increased intestinal permeability in bronchial asthma. J. Allergy Clin. Immunol. 1996, 97, 1173–1178. [Google Scholar] [CrossRef]
- Jakobsson, H.E.; Rodríguez-Piñeiro, A.M.; Schütte, A.; Ermund, A.; Boysen, P.; Bemark, M.; Sommer, F.; Bäckhed, F.; Hansson, G.C.; Johansson, M.E. The composition of the gut microbiota shapes the colon mucus barrier. EMBO Rep. 2015, 16, 164–177. [Google Scholar] [CrossRef]
- Mallick, H.; Rahnavard, A.; McIver, L.J.; Ma, S.; Zhang, Y.; Nguyen, L.H.; Tickle, T.L.; Weingart, G.; Ren, B.; Schwager, E.H.; et al. Multivariable association discovery in population-scale meta-omics studies. PLoS Comput. Biol. 2021, 17, e1009442. [Google Scholar] [CrossRef]
- Richter, S.H.; Garner, J.P.; Würbel, H. Environmental standardization: Cure or cause of poor reproducibility in animal experiments? Nat. Methods 2009, 6, 257–261. [Google Scholar] [CrossRef]
- Cahenzli, J.; Köller, Y.; Wyss, M.; Geuking, M.B.; McCoy, K.D. Intestinal microbial diversity during early-life colonization shapes long-term IgE levels. Cell Host Microbe 2013, 14, 559–570. [Google Scholar] [CrossRef]
- Mazmanian, S.K.; Liu, C.H.; Tzianabos, A.O.; Kasper, D.L. An immunomodulatory molecule of symbiotic bacteria directs maturation of the host immune system. Cell 2005, 122, 107–118. [Google Scholar] [CrossRef]
- Sonnenburg, E.D.; Sonnenburg, J.L. The ancestral and industrialized gut microbiota and implications for human health. Nat. Rev. Microbiol. 2019, 17, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Martínez, I.; Stegen, J.C.; Maldonado-Gómez, M.X.; Eren, A.M.; Siba, P.M.; Greenhill, A.R.; Walter, J. The gut microbiota of rural papua new guineans: Composition, diversity patterns, and ecological processes. Cell Rep. 2015, 11, 527–538. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef]
- Clemente, J.C.; Pehrsson, E.C.; Blaser, M.J.; Sandhu, K.; Gao, Z.; Wang, B.; Magris, M.; Hidalgo, G.; Contreras, M.; Noya-Alarcón, Ó.; et al. The microbiome of uncontacted Amerindians. Sci. Adv. 2015, 1, e1500183. [Google Scholar] [CrossRef]
- Blaser, M.J. The theory of disappearing microbiota and the epidemics of chronic diseases. Nat. Rev. Immunol. 2017, 17, 461–463. [Google Scholar] [CrossRef] [PubMed]
- Schuijs, M.J.; Willart, M.A.; Vergote, K.; Gras, D.; Deswarte, K.; Ege, M.J.; Madeira, F.B.; Beyaert, R.; van Loo, G.; Bracher, F.; et al. Farm dust and endotoxin protect against allergy through A20 induction in lung epithelial cells. Science 2015, 349, 1106–1110. [Google Scholar] [CrossRef] [PubMed]
- Alashkar Alhamwe, B.; Gao, Z.; Alhamdan, F.; Harb, H.; Pichene, M.; Garnier, A.; El Andari, J.; Kaufmann, A.; Graumann, P.L.; Kesper, D.; et al. Intranasal administration of Acinetobacter lwoffii in a murine model of asthma induces IL-6-mediated protection associated with cecal microbiota changes. Allergy 2023, 78, 1245–1257. [Google Scholar] [CrossRef]
- Weiss, K.; Wanner, N.; Queisser, K.; Frimel, M.; Nunn, T.; Myshrall, T.; Sangwan, N.; Erzurum, S.; Asosingh, K. Barrier Housing and Gender Effects on Allergic Airway Disease in a Murine House Dust Mite Model. Immunohorizons 2021, 5, 33–47. [Google Scholar] [CrossRef]
- Block, K.E.; Iijima, K.; Pierson, M.J.; Walsh, D.A.; Tei, R.; Kucaba, T.A.; Xu, J.; Khan, M.H.; Staley, C.; Griffith, T.S.; et al. Physiological microbial exposure transiently inhibits mouse lung ILC2 responses to allergens. Nat. Immunol. 2022, 23, 1703–1713. [Google Scholar] [CrossRef]
- Russell, S.L.; Gold, M.J.; Hartmann, M.; Willing, B.P.; Thorson, L.; Wlodarska, M.; Gill, N.; Blanchet, M.R.; Mohn, W.W.; McNagny, K.M.; et al. Early life antibiotic-driven changes in microbiota enhance susceptibility to allergic asthma. EMBO Rep. 2012, 13, 440–447. [Google Scholar] [CrossRef]
- Zaiss, M.M.; Rapin, A.; Lebon, L.; Dubey, L.K.; Mosconi, I.; Sarter, K.; Piersigilli, A.; Menin, L.; Walker, A.W.; Rougemont, J.; et al. The Intestinal Microbiota Contributes to the Ability of Helminths to Modulate Allergic Inflammation. Immunity 2015, 43, 998–1010. [Google Scholar] [CrossRef] [PubMed]
- Wilson, N.G.; Hernandez-Leyva, A.; Rosen, A.L.; Jaeger, N.; McDonough, R.T.; Santiago-Borges, J.; Lint, M.A.; Rosen, T.R.; Tomera, C.P.; Bacharier, L.B.; et al. The gut microbiota of people with asthma influences lung inflammation in gnotobiotic mice. iScience 2023, 26, 105991. [Google Scholar] [CrossRef]
- Ma, J.; Urgard, E.; Runge, S.; Classon, C.H.; Mathä, L.; Stark, J.M.; Cheng, L.; Álvarez, J.A.; von Zedtwitz, S.; Baleviciute, A.; et al. Laboratory mice with a wild microbiota generate strong allergic immune responses. Sci. Immunol. 2023, 8, eadf7702. [Google Scholar] [CrossRef]
- Devalapalli, A.P.; Lesher, A.; Shieh, K.; Solow, J.S.; Everett, M.L.; Edala, A.S.; Whitt, P.; Long, R.R.; Newton, N.; Parker, W. Increased levels of IgE and autoreactive, polyreactive IgG in wild rodents: Implications for the hygiene hypothesis. Scand. J. Immunol. 2006, 64, 125–136. [Google Scholar] [CrossRef]
- Abolins, S.R.; Pocock, M.J.; Hafalla, J.C.; Riley, E.M.; Viney, M.E. Measures of immune function of wild mice, Mus musculus. Mol. Ecol. 2011, 20, 881–892. [Google Scholar] [CrossRef] [PubMed]
- Fiege, J.K.; Block, K.E.; Pierson, M.J.; Nanda, H.; Shepherd, F.K.; Mickelson, C.K.; Stolley, J.M.; Matchett, W.E.; Wijeyesinghe, S.; Meyerholz, D.K.; et al. Mice with diverse microbial exposure histories as a model for preclinical vaccine testing. Cell Host Microbe 2021, 29, 1815–1827.e1816. [Google Scholar] [CrossRef]
- Sjaastad, F.V.; Huggins, M.A.; Lucas, E.D.; Skon-Hegg, C.; Swanson, W.; Martin, M.D.; Salgado, O.C.; Xu, J.; Pierson, M.; Dileepan, T.; et al. Reduced T Cell Priming in Microbially Experienced “Dirty” Mice Results from Limited IL-27 Production by XCR1+ Dendritic Cells. J. Immunol. 2022, 209, 2149–2159. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.W.; Xiang, Z. Mouse hygiene status-A tale of two environments for mast cells and allergy. Allergol. Int. 2024, 73, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Lehours, P.; Ferrero, R.L. Review: Helicobacter: Inflammation, immunology, and vaccines. Helicobacter 2019, 24 (Suppl. S1), e12644. [Google Scholar] [CrossRef]
- Chen, Y.; Blaser, M.J. Helicobacter pylori colonization is inversely associated with childhood asthma. J. Infect. Dis. 2008, 198, 553–560. [Google Scholar] [CrossRef]
- Amberbir, A.; Medhin, G.; Abegaz, W.E.; Hanlon, C.; Robinson, K.; Fogarty, A.; Britton, J.; Venn, A.; Davey, G. Exposure to Helicobacter pylori infection in early childhood and the risk of allergic disease and atopic sensitization: A longitudinal birth cohort study. Clin. Exp. Allergy 2014, 44, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Arnold, I.C.; Dehzad, N.; Reuter, S.; Martin, H.; Becher, B.; Taube, C.; Müller, A. Helicobacter pylori infection prevents allergic asthma in mouse models through the induction of regulatory T cells. J. Clin. Investig. 2011, 121, 3088–3093. [Google Scholar] [CrossRef] [PubMed]
- Stokholm, J.; Blaser, M.J.; Thorsen, J.; Rasmussen, M.A.; Waage, J.; Vinding, R.K.; Schoos, A.M.; Kunøe, A.; Fink, N.R.; Chawes, B.L.; et al. Maturation of the gut microbiome and risk of asthma in childhood. Nat. Commun. 2018, 9, 141. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Carroll-Portillo, A.; Lin, H.C. Desulfovibrio in the Gut: The Enemy within? Microorganisms 2023, 11, 1772. [Google Scholar] [CrossRef]
- Arnesen, H.; Markussen, T.; Birchenough, G.; Birkeland, S.; Nyström, E.E.L.; Hansson, G.C.; Carlsen, H.; Boysen, P. Microbial experience through housing in a farmyard-type environment alters intestinal barrier properties in mouse colons. Sci. Rep. 2023, 13, 13701. [Google Scholar] [CrossRef]
- Lee-Sarwar, K.A.; Lasky-Su, J.; Kelly, R.S.; Litonjua, A.A.; Weiss, S.T. Gut Microbial-Derived Metabolomics of Asthma. Metabolites 2020, 10, 97. [Google Scholar] [CrossRef] [PubMed]
- Yahsi, B.; Gunaydin, G. Immunometabolism—The Role of Branched-Chain Amino Acids. Front. Immunol. 2022, 13, 886822. [Google Scholar] [CrossRef]
- Comhair, S.A.; McDunn, J.; Bennett, C.; Fettig, J.; Erzurum, S.C.; Kalhan, S.C. Metabolomic Endotype of Asthma. J. Immunol. 2015, 195, 643–650. [Google Scholar] [CrossRef]
- Tao, J.L.; Chen, Y.Z.; Dai, Q.G.; Tian, M.; Wang, S.C.; Shan, J.J.; Ji, J.J.; Lin, L.L.; Li, W.W.; Yuan, B. Urine metabolic profiles in paediatric asthma. Respirology 2019, 24, 572–581. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Islam, M.Z.; Jozipovic, D.; Lopez, P.A.; Krych, L.; Correia, B.S.B.; Bertram, H.C.; Hansen, A.K.; Hansen, C.H.F. Wild-Mouse-Derived Gut Microbiome Transplantation in Laboratory Mice Partly Alleviates House-Dust-Mite-Induced Allergic Airway Inflammation. Microorganisms 2024, 12, 2499. https://doi.org/10.3390/microorganisms12122499
Islam MZ, Jozipovic D, Lopez PA, Krych L, Correia BSB, Bertram HC, Hansen AK, Hansen CHF. Wild-Mouse-Derived Gut Microbiome Transplantation in Laboratory Mice Partly Alleviates House-Dust-Mite-Induced Allergic Airway Inflammation. Microorganisms. 2024; 12(12):2499. https://doi.org/10.3390/microorganisms12122499
Chicago/Turabian StyleIslam, Md Zohorul, Danica Jozipovic, Pablo Atienza Lopez, Lukasz Krych, Banny Silva Barbosa Correia, Hanne Christine Bertram, Axel Kornerup Hansen, and Camilla Hartmann Friis Hansen. 2024. "Wild-Mouse-Derived Gut Microbiome Transplantation in Laboratory Mice Partly Alleviates House-Dust-Mite-Induced Allergic Airway Inflammation" Microorganisms 12, no. 12: 2499. https://doi.org/10.3390/microorganisms12122499
APA StyleIslam, M. Z., Jozipovic, D., Lopez, P. A., Krych, L., Correia, B. S. B., Bertram, H. C., Hansen, A. K., & Hansen, C. H. F. (2024). Wild-Mouse-Derived Gut Microbiome Transplantation in Laboratory Mice Partly Alleviates House-Dust-Mite-Induced Allergic Airway Inflammation. Microorganisms, 12(12), 2499. https://doi.org/10.3390/microorganisms12122499