
Metagenome-Assembled Genomes of Pig Fecal Samples in Nine European Countries: Insights into Antibiotic Resistance Genes and Viruses

 , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Collection
2.2. MAG Collection Construction
2.3. ARG Analysis
2.4. Virus Analysis
3. Results
3.1. Metagenome-Assembled Genomes
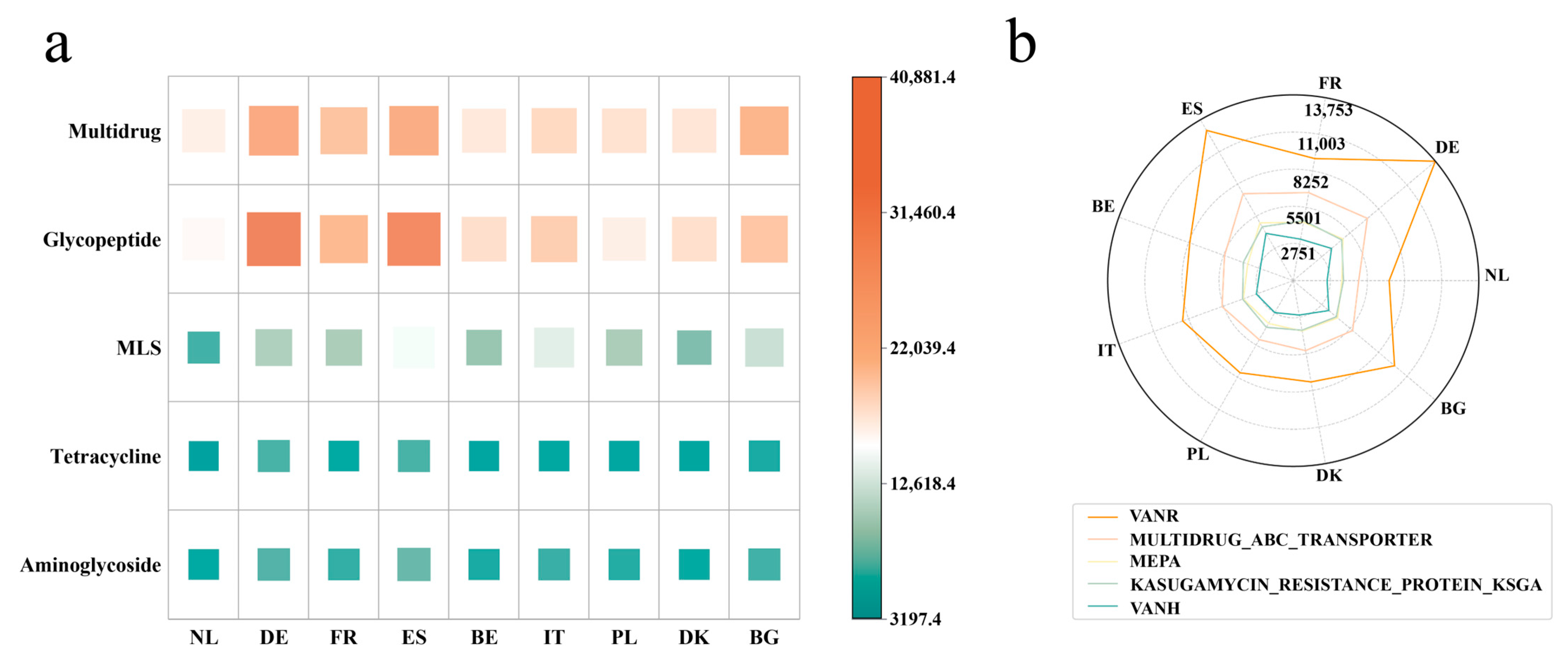
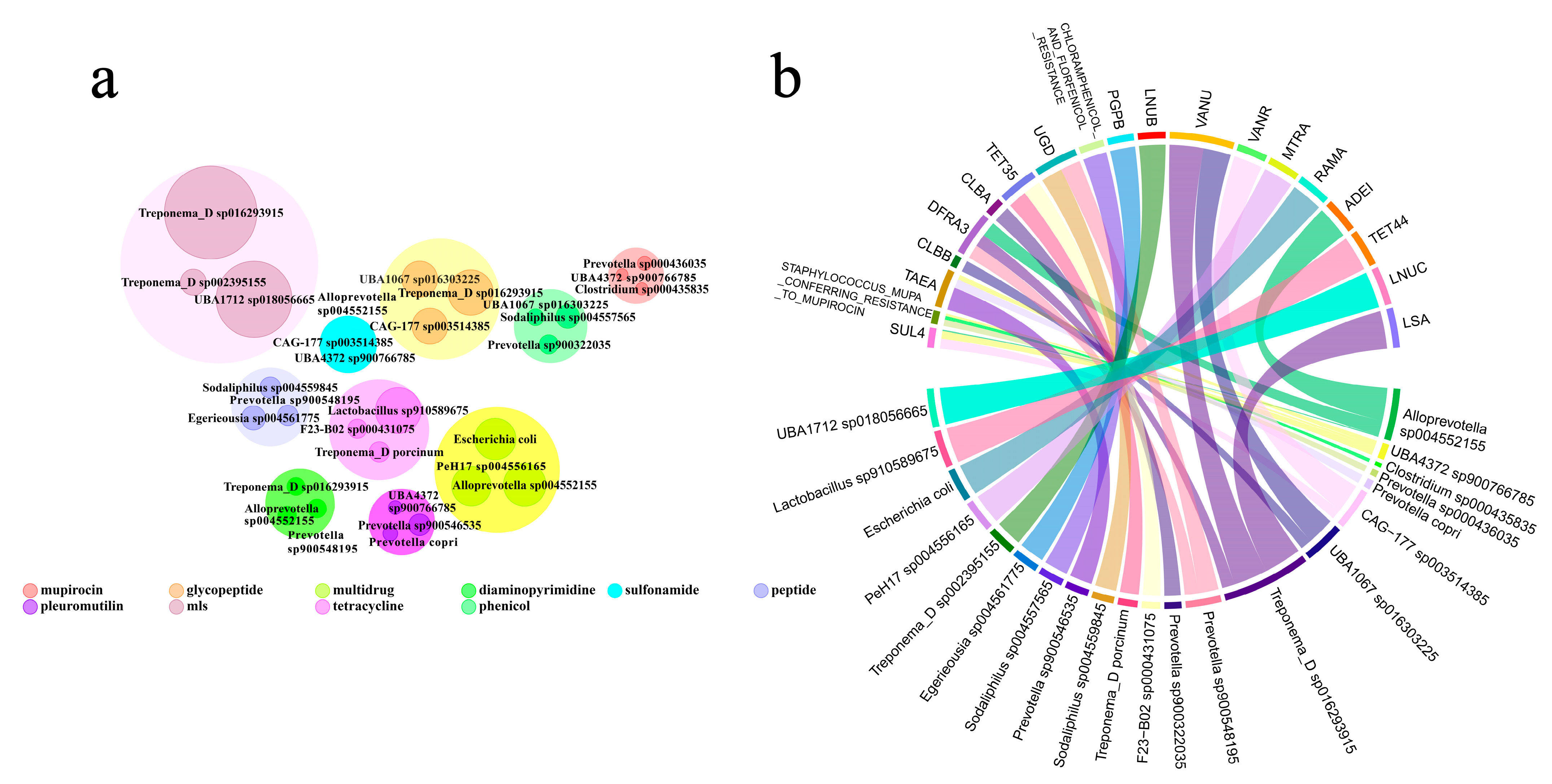
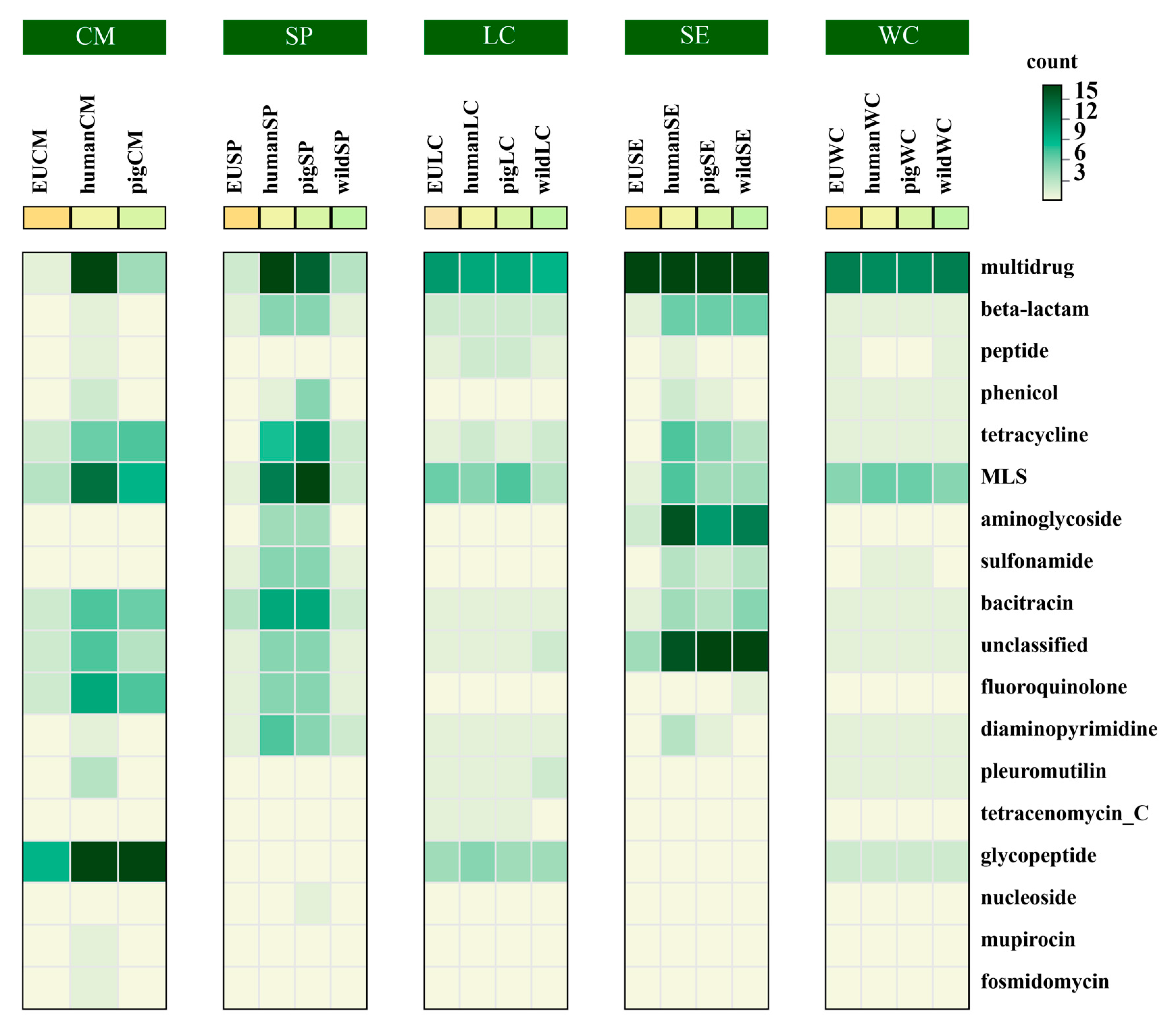
3.2. Antibiotic Resistance Gene Host Prediction
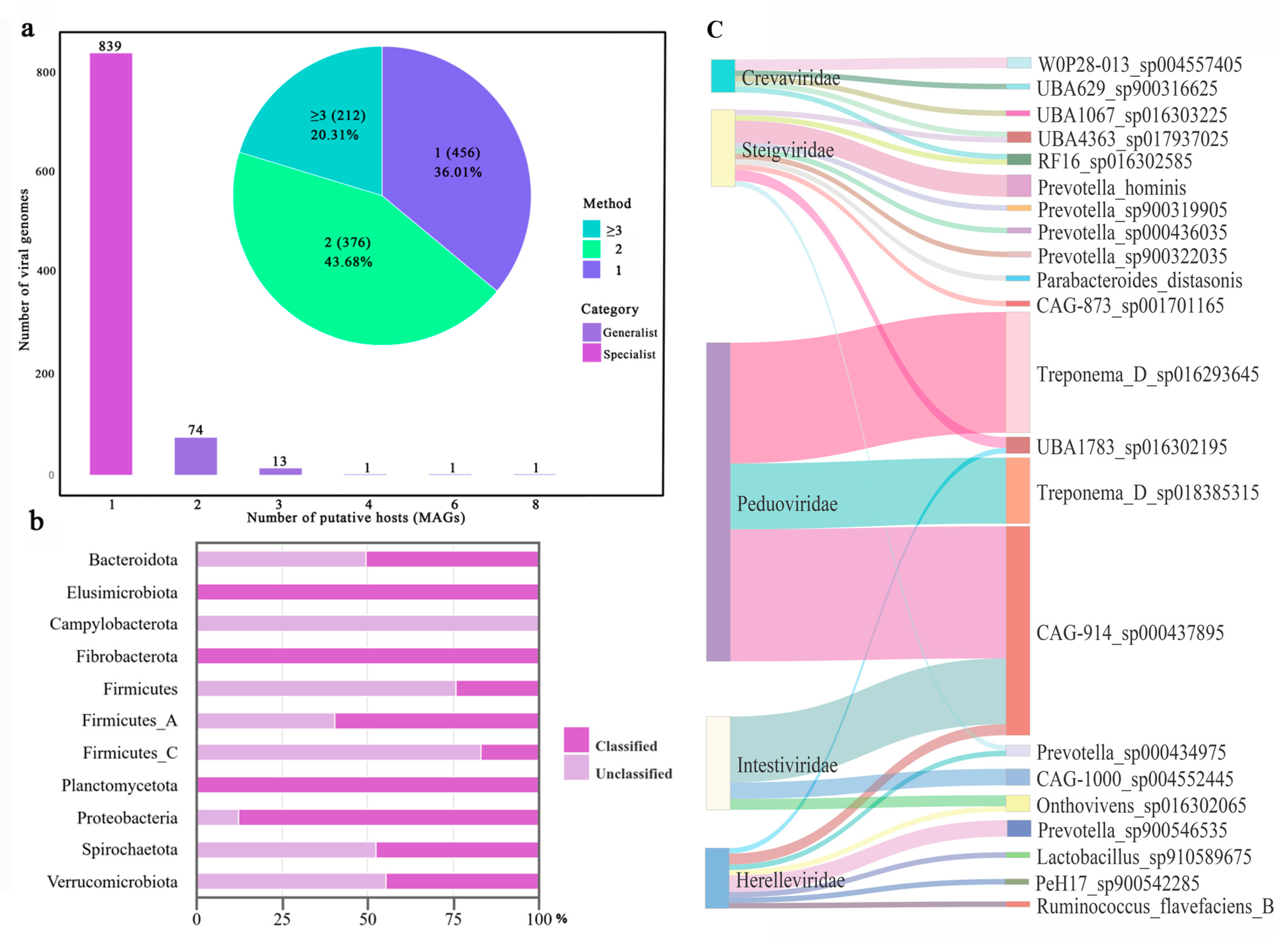
3.3. Viral Host Prediction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Li, Y.; Yang, Y.; Ma, L.; Liu, J.; An, Q.; Zhang, C.; Yin, G.; Cao, Z.; Pan, H. Comparative Analyses of Antibiotic Resistance Genes in Jejunum Microbiota of Pigs in Different Areas. Front. Cell. Infect. Microbiol. 2022, 12, 887428. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Song, Y.; Lyu, W.; Chen, Q.; Xiao, X.; Jin, Y.; Yang, H.; Wang, W.; Xiao, Y. Longitudinal metagenomic study reveals the dynamics of fecal antibiotic resistome in pigs throughout the lifetime. Anim. Microbiome 2023, 5, 55. [Google Scholar] [CrossRef] [PubMed]
- Ricker, N.; Trachsel, J.; Colgan, P.; Jones, J.; Choi, J.; Lee, J.; Coetzee, J.F.; Howe, A.; Brockmeier, S.L.; Loving, C.L. Toward antibiotic stewardship: Route of antibiotic administration impacts the microbiota and resistance gene diversity in swine feces. Front. Vet. Sci. 2020, 7, 255. [Google Scholar] [CrossRef] [PubMed]
- Authority, E.F.S. The European Union Summary Report on Antimicrobial Resistance in zoonotic and indicator bacteria from humans, animals and food in 2019–2020. EFSA J. 2020, 20, e07209. [Google Scholar]
- European Centre for Disease Prevention and Control. Antimicrobial Resistance Surveillance in Europe 2015; Annual Report of the European Antimicrobial Resistance Surveillance Network (EARS-Net); ECDC: Stockholm, Sweden, 2015.
- Zhang, N.; Liu, E.; Tang, A.; Ye, M.C.; Wang, K.; Jia, Q.; Huang, Z. Data-driven analysis of antimicrobial resistance in foodborne pathogens from six states within the US. Int. J. Environ. Res. Public Health 2019, 16, 1811. [Google Scholar] [CrossRef]
- Chai, J.; Zhuang, Y.; Cui, K.; Bi, Y.; Zhang, N. Metagenomics reveals the temporal dynamics of the rumen resistome and microbiome in goat kids. Microbiome 2024, 12, 14. [Google Scholar] [CrossRef]
- Yang, D.; Heederik, D.J.; Mevius, D.J.; Scherpenisse, P.; Luiken, R.E.; Van Gompel, L.; Skarżyńska, M.; Wadepohl, K.; Chauvin, C.; Van Heijnsbergen, E. Risk factors for the abundance of antimicrobial resistance genes aph (3′)-III, erm (B), sul2 and tet (W) in pig and broiler faeces in nine European countries. J. Antimicrob. Chemother. 2022, 77, 969–978. [Google Scholar] [CrossRef]
- Van Gompel, L.; Luiken, R.E.C.; Sarrazin, S.; Munk, P.; Knudsen, B.E.; Hansen, R.B.; Bossers, A.; Aarestrup, F.M.; Dewulf, J.; Wagenaar, J.A.; et al. The antimicrobial resistome in relation to antimicrobial use and biosecurity in pig farming, a metagenome-wide association study in nine European countries. J. Antimicrob. Chemother. 2019, 74, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Sui, L.; Kong, D.; Liu, D.; Gao, Y.; Jiang, Y.; Cui, W.; Li, J.; Li, Y.; Wang, L. Porcine epidemic diarrhea virus strain CH/HLJ/18 isolated in China: Characterization and phylogenetic analysis. Virol. J. 2024, 21, 28. [Google Scholar] [CrossRef]
- Li, C.; Wu, Q.; Song, H.; Lu, H.; Yang, K.; Liu, Z.; Liu, W.; Gao, T.; Yuan, F.; Zhu, J. Elucidating the biological characteristics and pathogenicity of the highly virulent G2a porcine epidemic diarrhea virus. J. Gen. Virol. 2024, 105, 001953. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Stockdale, S.R.; Lavelle, A.; Kondova, I.; Heuston, C.; Upadrasta, A.; Khokhlova, E.V.; Van Der Kamp, I.; Ouwerling, B.; Draper, L.A. Viral biogeography of the mammalian gut and parenchymal organs. Nat. Microbiol. 2022, 7, 1301–1311. [Google Scholar] [CrossRef] [PubMed]
- Munk, P.; Knudsen, B.E.; Lukjancenko, O.; Duarte, A.S.R.; Van Gompel, L.; Luiken, R.E.; Smit, L.A.; Schmitt, H.; Garcia, A.D.; Hansen, R.B. Abundance and diversity of the faecal resistome in slaughter pigs and broilers in nine European countries. Nat. Microbiol. 2018, 3, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Deng, F.; Han, Y.; Huang, Y.; Li, D.; Chai, J.; Deng, L.; Wei, M.; Wu, K.; Zhao, H.; Yang, G. A comprehensive analysis of antibiotic resistance genes in the giant panda gut. iMeta 2024, 3, e171. [Google Scholar] [CrossRef] [PubMed]
- Antipov, D.; Korobeynikov, A.; McLean, J.S.; Pevzner, P.A. hybridSPAdes: An algorithm for hybrid assembly of short and long reads. Bioinformatics 2016, 32, 1009–1015. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Olm, M.R.; Brown, C.T.; Brooks, B.; Banfield, J.F. dRep: A tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017, 11, 2864–2868. [Google Scholar] [CrossRef]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A Toolkit to Classify Genomes with the Genome Taxonomy Database; Oxford University Press: Oxford, UK, 2020. [Google Scholar]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed]
- Arango-Argoty, G.; Garner, E.; Pruden, A.; Heath, L.S.; Vikesland, P.; Zhang, L. DeepARG: A deep learning approach for predicting antibiotic resistance genes from metagenomic data. Microbiome 2018, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Mi, J.; Jing, X.; Ma, C.; Yang, Y.; Li, Y.; Zhang, Y.; Long, R.; Zheng, H. Massive expansion of the pig gut virome based on global metagenomic mining. Npj Biofilms Microbiomes 2024, 10, 76. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Feng, T.; Wu, Y.; Xu, Y.; Du, L.; Wang, T.; Luo, Y.; Wang, Y.; Li, Z.; Xuan, Z. The multi-kingdom microbiome of the goat gastrointestinal tract. Microbiome 2023, 11, 219. [Google Scholar] [CrossRef]
- Ryu, W.-S. Molecular Virology of Human Pathogenic Viruses; Academic Press: Cambridge, MA, USA, 2016. [Google Scholar]
- Nayfach, S.; Roux, S.; Seshadri, R.; Udwary, D.; Varghese, N.; Schulz, F.; Wu, D.; Paez-Espino, D.; Chen, I.-M.; Huntemann, M. A genomic catalog of Earth’s microbiomes. Nat. Biotechnol. 2021, 39, 499–509. [Google Scholar] [CrossRef]
- Deng, F.; Wang, C.; Li, D.; Peng, Y.; Deng, L.; Zhao, Y.; Zhang, Z.; Wei, M.; Wu, K.; Zhao, J. The unique gut microbiome of giant pandas involved in protein metabolism contributes to the host’s dietary adaption to bamboo. Microbiome 2023, 11, 180. [Google Scholar] [CrossRef]
- Jiminez, J.A.; Uwiera, T.C.; Abbott, D.W.; Uwiera, R.R.; Inglis, G.D. Impacts of resistant starch and wheat bran consumption on enteric inflammation in relation to colonic bacterial community structures and short-chain fatty acid concentrations in mice. Gut Pathog. 2016, 8, 67. [Google Scholar] [CrossRef]
- Wielgosz-Grochowska, J.P.; Domanski, N.; Drywień, M.E. Efficacy of an irritable bowel syndrome diet in the treatment of small intestinal bacterial overgrowth: A narrative review. Nutrients 2022, 14, 3382. [Google Scholar] [CrossRef]
- Bevins, C.L.; Salzman, N.H. The potter’s wheel: The host’s role in sculpting its microbiota. Cell. Mol. Life Sci. 2011, 68, 3675–3685. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Li, A.; Shi, C.; Chen, L.; Zhao, Z.; Yin, X.; Zhang, Q.; Huang, Y.; Pan, H. Mulberry branch fiber improved lipid metabolism and egg yolk fatty acid composition of laying hens via the enterohepatic axis. Microbiome 2024, 12, 73. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tsai, T.; Deng, F.; Wei, X.; Chai, J.; Knapp, J.; Apple, J.; Maxwell, C.V.; Lee, J.A.; Li, Y.; et al. Longitudinal investigation of the swine gut microbiome from birth to market reveals stage and growth performance associated bacteria. Microbiome 2019, 7, 109. [Google Scholar] [CrossRef] [PubMed]
- Holman, D.B.; Kommadath, A.; Tingley, J.P.; Abbott, D.W. Novel insights into the pig gut microbiome using metagenome-assembled genomes. Microbiol. Spectr. 2022, 10, e0238022. [Google Scholar] [CrossRef] [PubMed]
- Lapidus, A.L.; Korobeynikov, A.I. Metagenomic data assembly–the way of decoding unknown microorganisms. Front. Microbiol. 2021, 12, 613791. [Google Scholar] [CrossRef]
- Ma, L.; Lyu, W.; Zeng, T.; Wang, W.; Chen, Q.; Zhao, J.; Zhang, G.; Lu, L.; Yang, H.; Xiao, Y. Duck gut metagenome reveals the microbiome signatures linked to intestinal regional, temporal development, and rearing condition. iMeta 2024, 3, e198. [Google Scholar] [CrossRef]
- Sun, J.; Chen, C.; Cui, C.-Y.; Zhang, Y.; Liu, X.; Cui, Z.-H.; Ma, X.-Y.; Feng, Y.; Fang, L.-X.; Lian, X.-L. Plasmid-encoded tet (X) genes that confer high-level tigecycline resistance in Escherichia coli. Nat. Microbiol. 2019, 4, 1457–1464. [Google Scholar] [CrossRef]
- Deckert, A.; Gow, S.; Rosengren, L.; Leger, D.; Avery, B.; Daignault, D.; Dutil, L.; Reid-Smith, R.; Irwin, R. Canadian integrated program for antimicrobial resistance surveillance (CIPARS) farm program: Results from finisher pig surveillance. Zoonoses Public Health 2010, 57, 71–84. [Google Scholar] [CrossRef]
- Part IV, USDA. Health and Health Management on US Feedlots with a Capacity of 1000 or More Head; APHIS, Ed.; National Animal Health Monitoring System: Fort Collins, CO, USA, 2011.
- Zaheer, R.; Cook, S.R.; Barbieri, R.; Goji, N.; Cameron, A.; Petkau, A.; Polo, R.O.; Tymensen, L.; Stamm, C.; Song, J. Surveillance of Enterococcus spp. reveals distinct species and antimicrobial resistance diversity across a One-Health continuum. Sci. Rep. 2020, 10, 3937. [Google Scholar]
- Tang, K.L.; Caffrey, N.P.; Nóbrega, D.B.; Cork, S.C.; Ronksley, P.E.; Barkema, H.W.; Polachek, A.J.; Ganshorn, H.; Sharma, N.; Kellner, J.D. Restricting the use of antibiotics in food-producing animals and its associations with antibiotic resistance in food-producing animals and human beings: A systematic review and meta-analysis. Lancet Planet. Health 2017, 1, e316–e327. [Google Scholar] [CrossRef]
- Wang, Z.; Yun, H.; Li, S.; Ji, J.; Khan, A.; Fu, X.; Zhang, P.; Li, X. Factors influencing the transfer and abundance of antibiotic resistance genes in livestock environments in China. Int. J. Environ. Sci. Technol. 2023, 20, 2197–2208. [Google Scholar] [CrossRef]
- Andrade-Martínez, J.S.; Camelo Valera, L.C.; Chica Cardenas, L.A.; Forero-Junco, L.; López-Leal, G.; Moreno-Gallego, J.L.; Rangel-Pineros, G.; Reyes, A. Computational tools for the analysis of uncultivated phage genomes. Microbiol. Mol. Biol. Rev. 2022, 86, e0000421. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, B.; Yang, J.; Chen, R.; Chai, J.; Wei, X.; Zhao, J.; Zhao, Y.; Deng, F.; Li, Y. Metagenome-Assembled Genomes of Pig Fecal Samples in Nine European Countries: Insights into Antibiotic Resistance Genes and Viruses. Microorganisms 2024, 12, 2409. https://doi.org/10.3390/microorganisms12122409
Yang B, Yang J, Chen R, Chai J, Wei X, Zhao J, Zhao Y, Deng F, Li Y. Metagenome-Assembled Genomes of Pig Fecal Samples in Nine European Countries: Insights into Antibiotic Resistance Genes and Viruses. Microorganisms. 2024; 12(12):2409. https://doi.org/10.3390/microorganisms12122409
Chicago/Turabian StyleYang, Boxuan, Jianbo Yang, Routing Chen, Jianmin Chai, Xiaoyuan Wei, Jiangchao Zhao, Yunxiang Zhao, Feilong Deng, and Ying Li. 2024. "Metagenome-Assembled Genomes of Pig Fecal Samples in Nine European Countries: Insights into Antibiotic Resistance Genes and Viruses" Microorganisms 12, no. 12: 2409. https://doi.org/10.3390/microorganisms12122409
APA StyleYang, B., Yang, J., Chen, R., Chai, J., Wei, X., Zhao, J., Zhao, Y., Deng, F., & Li, Y. (2024). Metagenome-Assembled Genomes of Pig Fecal Samples in Nine European Countries: Insights into Antibiotic Resistance Genes and Viruses. Microorganisms, 12(12), 2409. https://doi.org/10.3390/microorganisms12122409