
Large-Scale Integration of Amplicon Data Reveals Massive Diversity within Saprospirales, Mostly Originating from Saline Environments


Abstract
1. Introduction
2. Materials and Methods
3. Results
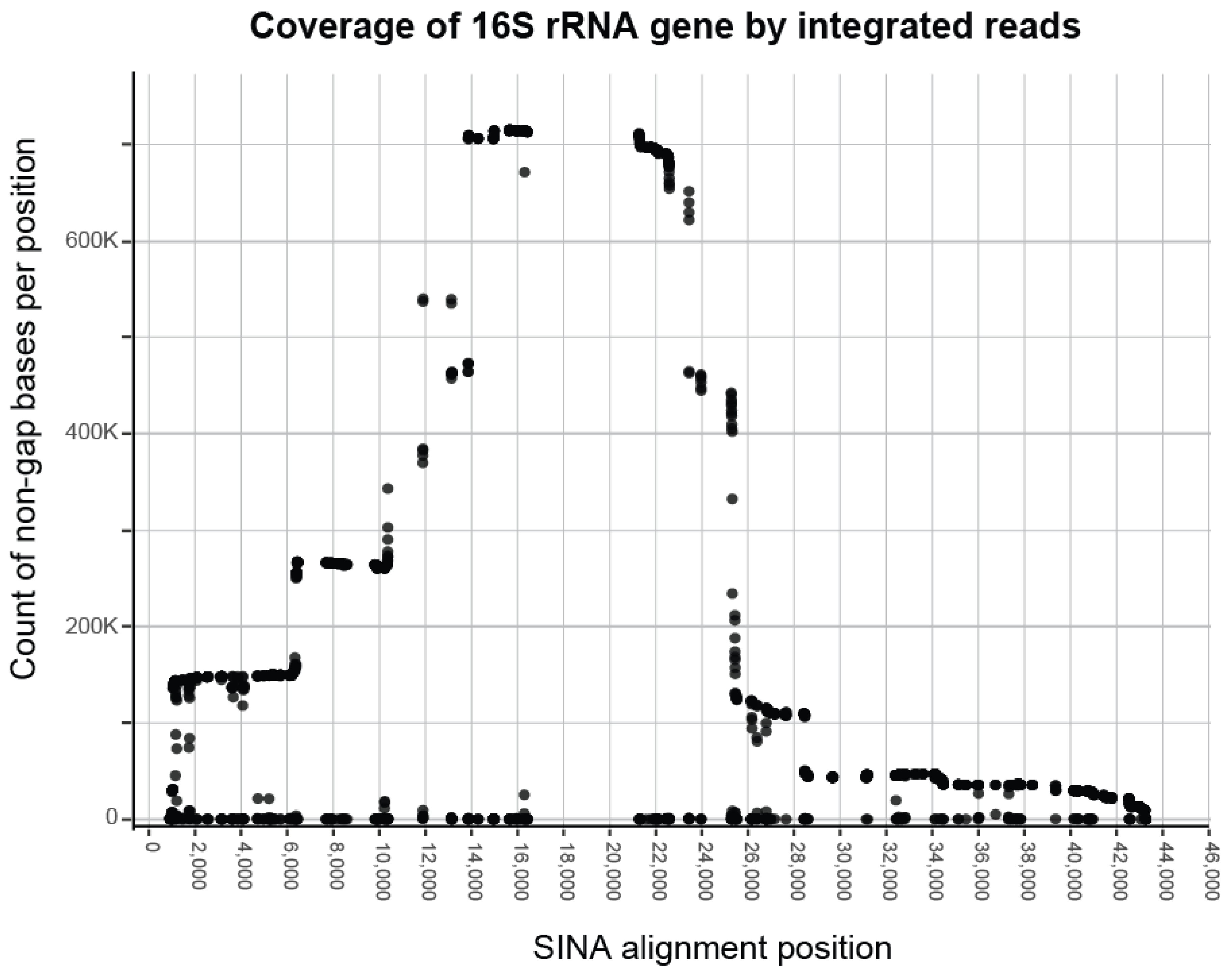
3.1. Processing Results
3.2. Analysis of Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Schauer, M.; Hahn, M.W. Diversity and phylogenetic affiliations of morphologically conspicuous large filamentous bacteria occurring in the pelagic zones of a broad spectrum of freshwater habitats. Appl. Environ. Microbiol. 2005, 71, 1931–1940. [Google Scholar] [CrossRef] [PubMed]
- Schauer, M.; Kamenik, C.; Hahn, M.W. Ecological differentiation within a cosmopolitan group of planktonic freshwater bacteria (SOL cluster, Saprospiraceae, Bacteroidetes). Appl. Environ. Microbiol. 2005, 71, 5900–5907. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.D. Lewinella agarilytica sp. nov., a novel marine bacterium of the phylum Bacteroidetes, isolated from beach sediment. Int. J. Syst. Evol. Microbiol. 2007, 57, 2814–2818. [Google Scholar] [CrossRef] [PubMed]
- Lian, J.; Steinert, G.; de Vree, J.; Meijer, S.; Heryanto, C.; Bosma, R.; Wijffels, R.H.; Barbosa, M.J.; Smidt, H.; Sipkema, D. Bacterial diversity in different outdoor pilot plant photobioreactor types during production of the microalga Nannochloropsis sp. CCAP211/78. Appl. Microbiol. Biotechnol. 2022, 106, 2235–2248. [Google Scholar] [CrossRef]
- Gerdts, G.; Brandt, P.; Kreisel, K.; Boersma, M.; Schoo, K.L.; Wichels, A. The microbiome of North Sea copepods. Helgol. Mar. Res. 2013, 67, 757–773. [Google Scholar] [CrossRef]
- Sung, H.R.; Lee, J.M.; Kim, M.; Shin, K.S. Lewinella xylanilytica sp. nov., a member of the family Saprospiraceae isolated from coastal seawater. Int. J. Syst. Evol. Microbiol. 2015, 65 Pt 10, 3433–3438. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.M.; Lee, K.; Cho, J.C. Lewinella antarctica sp. nov., a marine bacterium isolated from Antarctic seawater. Int. J. Syst. Evol. Microbiol. 2009, 59, 65–68. [Google Scholar] [CrossRef]
- Jung, Y.T.; Lee, J.S.; Yoon, J.H. Lewinella aquimaris sp. nov., isolated from seawater. Int. J. Syst. Evol. Microbiol. 2016, 66, 3989–3994. [Google Scholar] [CrossRef]
- Kim, I.; Chhetri, G.; Kim, J.; Kang, M.; Seo, T. Lewinella aurantiaca sp. nov., a carotenoid pigment-producing bacterium isolated from surface seawater. Int. J. Syst. Evol. Microbiol. 2020, 70, 6180–6187. [Google Scholar] [CrossRef]
- Park, S.; Won, S.M.; Yoon, J.H. Lewinella litorea sp. nov., isolated from marine sand. Int. J. Syst. Evol. Microbiol. 2020, 70, 246–250. [Google Scholar] [CrossRef]
- Kang, H.; Kim, H.; Joung, Y.; Joh, K. Lewinella maritima sp. nov., and Lewinella lacunae sp. nov., novel bacteria from marine environments. Int. J. Syst. Evol. Microbiol. 2017, 67, 3603–3609. [Google Scholar] [CrossRef] [PubMed]
- Hahn, M.W.; Schauer, M. ‘Candidatus Aquirestis calciphila’ and ‘Candidatus Haliscomenobacter calcifugiens’, filamentous, planktonic bacteria inhabiting natural lakes. Int. J. Syst. Evol. Microbiol. 2007, 57, 936–940. [Google Scholar] [CrossRef] [PubMed]
- Hosoya, S.; Arunpairojana, V.; Suwannachart, C.; Kanjana-Opas, A.; Yokota, A. Aureispira maritima sp. nov., isolated from marine barnacle debris. Int. J. Syst. Evol. Microbiol. 2007, 57, 1948–1951. [Google Scholar] [CrossRef] [PubMed]
- Hosoya, S.; Arunpairojana, V.; Suwannachart, C.; Kanjana-Opas, A.; Yokota, A. Aureispira marina gen. nov., sp. nov., a gliding, arachidonic acid-containing bacterium isolated from the southern coastline of Thailand. Int. J. Syst. Evol. Microbiol. 2006, 56, 2931–2935. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.; Li, Y.; Wang, G.; Chen, Y.; Lai, Q.; Chen, Z.; Zhang, J.; Liao, P.; Zhu, H.; Zheng, W.; et al. Phaeodactylibacter luteus sp. nov., isolated from the oleaginous microalga Picochlorum sp. Int. J. Syst. Evol. Microbiol. 2015, 65 Pt 8, 2666–2670. [Google Scholar] [CrossRef]
- Li, X.; Liu, Y.; Chen, Z.; Liu, L.Z.; Liu, Z.P. Membranicola marinus gen. nov., sp. nov., a new member of the family Saprospiraceae isolated from a biofilter in a recirculating aquaculture system. Int. J. Syst. Evol. Microbiol. 2016, 66, 1275–1280. [Google Scholar] [CrossRef]
- Chen, Z.; Lei, X.; Lai, Q.; Li, Y.; Zhang, B.; Zhang, J.; Zhang, H.; Yang, L.; Zheng, W.; Tian, Y.; et al. Phaeodactylibacter xiamenensis gen. nov., sp. nov., a member of the family Saprospiraceae isolated from the marine alga Phaeodactylum tricornutum. Int. J. Syst. Evol. Microbiol. 2014, 64 Pt 10, 3496–3502. [Google Scholar] [CrossRef]
- Jiang, X.; Ma, H.; Zhao, Q.L.; Yang, J.; Xin, C.Y.; Chen, B. Bacterial communities in paddy soil and ditch sediment under rice-crab co-culture system. AMB Express 2021, 11, 163. [Google Scholar] [CrossRef]
- Vortsepneva, E.; Chevaldonné, P.; Klyukina, A.; Naduvaeva, E.; Todt, C.; Zhadan, A.; Tzetlin, A.; Kublanov, I. Microbial associations of shallow-water Mediterranean marine cave Solenogastres (Mollusca). PeerJ 2021, 9, e12655. [Google Scholar] [CrossRef]
- Su, Y.; Lv, J.L.; Yu, M.; Ma, Z.H.; Xi, H.; Kou, C.L.; He, Z.C.; Shen, A.L. Long-term decomposed straw return positively affects the soil microbial community. J. Appl. Microbiol. 2020, 128, 138–150. [Google Scholar] [CrossRef]
- Yoon, J.; Katsuta, A.; Kasai, H. Rubidimonas crustatorum gen. nov., sp. nov., a novel member of the family Saprospiraceae isolated from a marine crustacean. Antonie van Leeuwenhoek 2012, 101, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.; Matsuo, Y.; Kasai, H.; Yokota, A. Portibacter lacus gen. nov., sp. nov., a new member of the family Saprospiraceae isolated from a saline lake. J. Gen. Appl. Microbiol. 2012, 58, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Kong, Y.; Thomsen, T.R.; Halkjær Nielsen, P. Identification and ecophysiological characterization of epiphytic protein-hydrolyzing Saprospiraceae (“Candidatus Epiflobacter” spp.) in activated sludge. Appl. Environ. Microbiol. 2008, 74, 2229–2238. [Google Scholar] [CrossRef]
- Van Veen, W.L.; Van Der Kooij, D.; Geuze, E.C.; Van der Vlies, A.W. Investigations on the sheathed bacterium Haliscomenobacter hydrossis gen. n., sp. n., isolated from activated sludge. Antonie van Leeuwenhoek 1973, 39, 207–216. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Masuzawa, N.; Kondo, K.; Nakanishi, Y.; Chida, S.; Uehara, D.; Katahira, M.; Takeda, M. A heterodimeric hyaluronate lyase secreted by the activated sludge bacterium Haliscomenobacter hydrossis. Biosci. Biotechnol. Biochem. 2023, 87, 256–266. [Google Scholar] [CrossRef]
- Kondrotaite, Z.; Valk, L.C.; Petriglieri, F.; Singleton, C.; Nierychlo, M.; Dueholm, M.K.; Nielsen, P.H. Diversity and Ecophysiology of the Genus OLB8 and Other Abundant Uncultured Saprospiraceae Genera in Global Wastewater Treatment Systems. Front. Microbiol. 2022, 13, 917553. [Google Scholar] [CrossRef]
- Speth, D.R.; Guerrero-Cruz, S.; Dutilh, B.E.; Jetten, M.S. Genome-based microbial ecology of anammox granules in a full-scale wastewater treatment system. Nat. Commun. 2016, 7, 11172. [Google Scholar] [CrossRef]
- Zeng, T.; Wang, L.; Zhang, X.; Song, X.; Li, J.; Yang, J.; Chen, S.; Zhang, J. Characterization of Microbial Communities in Wastewater Treatment Plants Containing Heavy Metals Located in Chemical Industrial Zones. Int. J. Environ. Res. Public Health 2022, 19, 6529. [Google Scholar] [CrossRef]
- Ma, K.; Li, X.; Bao, L.; Li, X.; Cui, Y. The performance and bacterial community shifts in an anaerobic-aerobic process treating purified terephthalic acid wastewater under influent composition variations and ambient temperatures. J. Clean. Prod. 2020, 276, 124190. [Google Scholar] [CrossRef]
- De Carluccio, M.; Sabatino, R.; Eckert, E.M.; Di Cesare, A.; Corno, G.; Rizzo, L. Co-treatment of landfill leachate with urban wastewater by chemical, physical and biological processes: Fenton oxidation preserves autochthonous bacterial community in the activated sludge process. Chemosphere 2023, 313, 137578. [Google Scholar] [CrossRef]
- Wang, D.; Tao, L.; Yang, J.; Xu, Z.; Yang, Q.; Zhang, Y.; Liu, X.; Liu, Q.; Huang, J. Understanding the interaction between triclocarban and denitrifiers. J. Hazard. Mater. 2021, 401, 123343. [Google Scholar] [CrossRef]
- Li, C.; Liu, X.; Du, M.; Yang, J.; Lu, Q.; Fu, Q.; Dandan, H.; Zhao, W.; Wang, D. Peracetic acid promotes biohydrogen production from anaerobic dark fermentation of waste activated sludge. Sci. Total Environ. 2022, 844, 156991. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, X.; Liu, L.; Lu, H.; Wang, L.; Tang, J. Effects of microplastics on greenhouse gas emissions and microbial communities in sediment of freshwater systems. J. Hazard. Mater. 2022, 435, 129030. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, L.; Hu, S.; Diao, Y.; Jin, X.; Jin, P.; Chen, C.; Wu, X.; Wang, X.C. Electro-dissolved ozone flotation (E-DOF) integrated anoxic/oxic membrane reactor for leachate treatment from a waste transfer station. Environ. Sci. Pollut. Res. 2022, 29, 55803–55815. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Tan, L.; Shen, Z.; Zhou, Y.; He, X.; Chen, X. Enhanced denitrification of dispersed swine wastewater using Ca(OH) 2-pretreated rice straw as a solid carbon source. Chemosphere 2022, 305, 135316. [Google Scholar] [CrossRef]
- Nielsen, P.H.; Mielczarek, A.T.; Kragelund, C.; Nielsen, J.L.; Saunders, A.M.; Kong, Y.; Hansen, A.A.; Vollertsen, J. A conceptual ecosystem model of microbial communities in enhanced biological phosphorus removal plants. Water Res. 2010, 44, 5070–5088. [Google Scholar] [CrossRef]
- Xu, X.J.; Wu, Y.N.; Xiao, Q.Y.; Xie, P.; Ren, N.Q.; Yuan, Y.X.; Lee, D.J.; Chen, C. Simultaneous removal of NOX and SO2 from flue gas in an integrated FGD-CABR system by sulfur cycling-mediated Fe (II) EDTA regeneration. Environ. Res. 2022, 205, 112541. [Google Scholar] [CrossRef] [PubMed]
- Hahnke, R.L.; Meier-Kolthoff, J.P.; García-López, M.; Mukherjee, S.; Huntemann, M.; Ivanova, N.N.; Woyke, T.; Kyrpides, N.C.; Klenk, H.P.; Göker, M. Genome-based taxonomic classification of Bacteroidetes. Front. Microbiol. 2016, 7, 2003. [Google Scholar] [CrossRef]
- García-López, M.; Meier-Kolthoff, J.P.; Tindall, B.J.; Gronow, S.; Woyke, T.; Kyrpides, N.C.; Hahnke, R.L.; Göker, M. Analysis of 1,000 type-strain genomes improves taxonomic classification of Bacteroidetes. Front. Microbiol. 2019, 10, 2083. [Google Scholar] [CrossRef]
- Huang, Z.; Hong, Q.; Lai, Q.; Zhang, Q. Portibacter marinus sp. nov., isolated from the sediment on the surface of plastics and proposal of a novel genus Neolewinella gen. nov. based on the genome-based phylogeny of the family Lewinellaceae. Int. J. Syst. Evol. Microbiol. 2022, 72, 005561. [Google Scholar] [CrossRef]
- Deshmukh, U.B.; Oren, A. Proposal of Membranihabitans gen. nov. as a replacement name for the illegitimate prokaryotic generic name Membranicola Li et al. 2016. Int. J. Syst. Evol. Microbiol. 2022, 72, 005576. [Google Scholar] [CrossRef] [PubMed]
- Gross, J. Über freilebende Spironemaceen. Mitteilungen Zoologischen Station Neapel 1911, 20, 188–203. [Google Scholar]
- Khan, S.T.; Fukunaga, Y.; Nakagawa, Y.; Harayama, S. Emended descriptions of the genus Lewinella and of Lewinella cohaerens, Lewinella nigricans and Lewinella persica, and description of Lewinella lutea sp. nov. and Lewinella marina sp. nov. Int. J. Syst. Evol. Microbiol. 2007, 57, 2946–2951. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Lagkouvardos, I.; Joseph, D.; Kapfhammer, M.; Giritli, S.; Horn, M.; Haller, D.; Clavel, T. IMNGS: A comprehensive open resource of processed 16S rRNA microbial profiles for ecology and diversity studies. Sci. Rep. 2016, 6, 33721. [Google Scholar] [CrossRef]
- Pruesse, E.; Peplies, J.; Glöckner, F.O. SINA: Accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 2012, 28, 1823–1829. [Google Scholar] [CrossRef] [PubMed]
- Kioukis, A.; Pourjam, M.; Neuhaus, K.; Lagkouvardos, I. Taxonomy Informed Clustering, an Optimized Method for Purer and More Informative Clusters in Diversity Analysis and Microbiome Profiling. Front. Bioinform. 2022, 2, 864597. [Google Scholar] [CrossRef]
- Asnicar, F.; Weingart, G.; Tickle, T.L.; Huttenhower, C.; Segata, N. Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ 2015, 3, e1029. [Google Scholar] [CrossRef]
- Béziat, N.S.; Duperron, S.; Halary, S.; Azede, C.; Gros, O. Bacterial ectosymbionts colonizing gills of two Caribbean mangrove crabs. Symbiosis 2022, 85, 105–114. [Google Scholar] [CrossRef]
- Lagkouvardos, I.; Weinmaier, T.; Lauro, F.M.; Cavicchioli, R.; Rattei, T.; Horn, M. Integrating metagenomic and amplicon databases to resolve the phylogenetic and ecological diversity of the Chlamydiae. ISME J. 2014, 8, 115–125. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Known | Predicted | Predicted (≥0.1%) | |
|---|---|---|---|
| Families | 3 | 1272 | 10 |
| Genera | 17 | 15,049 | 479 |
| Species | 32 | 118,062 | 1565 |
| Families | Known | Pred. | Pred. % | Pred. (≥0.1%) | Pred. % (≥0.1%) |
|---|---|---|---|---|---|
| Saprospiraceae | 11 | 4483 | 29.79% | 132 | 27.56% |
| Lewinellaceae | 3 | 5430 | 36.08% | 186 | 38.83% |
| Haliscomenobacteraceae | 3 | 3675 | 24.42% | 150 | 31.32% |
| Unknown families | NA | 1461 * | 9.71% | 11 ** | 2.30% |
| Families | Known | Pred. | Pred. % | Pred. (≥0.1%) | Pred. % (≥0.1%) |
|---|---|---|---|---|---|
| Saprospiraceae | 14 | 39,332 | 33.31% | 553 | 35.34% |
| Lewinellaceae | 13 | 42,921 | 36.36% | 564 | 36.04% |
| Haliscomenobacteraceae | 5 | 33,191 | 28.11% | 435 | 27.76% |
| Unknown families | NA | 2618 * | 2.22% | 13 ** | 0.83% |
| Known Families | Genera | Known | Pred. | Pred. % | Pred. (≥0.1%) | Pred. % (≥0.1%) |
|---|---|---|---|---|---|---|
| Saprospiraceae | Aquirestis | 1 | 3795 | 3.21% | 42 | 2.68% |
| Aureispira | 2 | 3655 | 3.10% | 58 | 3.71% | |
| Brachybacter | 1 | 3796 | 3.22% | 45 | 2.88% | |
| Defluviibacterium | 1 | 109 | 0.09% | 11 | 0.70% | |
| Membranihabitans | 1 | 3739 | 3.17% | 26 | 1.66% | |
| Opimibacter | 2 | 1907 | 1.62% | 47 | 3.00% | |
| Parvibacillus | 1 | 2697 | 2.28% | 43 | 2.75% | |
| Rubidimonas | 1 | 4 | 3.4 × 10−3% | 0 | 0.00% | |
| Saprospira | 1 | 17 | 0.01% | 0 | 0.00% | |
| Vicinibacter | 2 | 1261 | 1.07% | 35 | 2.24% | |
| Epiflobacter | 0 | 34 | 0.03% | 4 | 0.26% | |
| Unknown genera | NA | 18,318 * | 15.52% | 242 ** | 15.46% | |
| Lewinellaceae | Flavilitoribacter | 1 | 3366 | 2.85% | 81 | 5.18% |
| Lewinella | 1 | 3850 | 3.26% | 46 | 2.94% | |
| Neolewinella | 11 | 11,511 | 9.75% | 105 | 6.71% | |
| Unknown genera | NA | 24,194 *3 | 20.49% | 332 *4 | 21.21% | |
| Haliscomenobacteraceae | Haliscomenobacter | 1 | 2690 | 2.28% | 25 | 1.60% |
| Phaeodactylibacter | 2 | 2535 | 2.15% | 71 | 4.54% | |
| Portibacter | 2 | 1295 | 1.10% | 42 | 2.68% | |
| Unknown genera | NA | 26,671 *5 | 22.59% | 297 *6 | 18.98% |
| Family | Genus | Species | SOTU ID | # Positive Samples (Abund. ≥ 0.1%) | Dominant Environment | Maximum Abundance Environment | Maximum Abundance |
|---|---|---|---|---|---|---|---|
| eSaprospiraceae | Aquirestis | Aquirestis calciphila | SOTU89 | 5 | freshwater | freshwater | 4.37% |
| Aureispira | Aureispira marina | SOTU412 | 6 | saline water | saline water | 0.48% | |
| Aureispira | Aureispira maritima | SOTU411 | 7 | saline water | saline water | 3.41% | |
| Brachybacter | Brachybacter algidus | SOTU50 | 1 | bioreactor | bioreactor | 0.36% | |
| Defluviibacterium | Defluviibacterium haderslevense | SOTU203 | 2 | bioreactor | bioreactor | 0.56% | |
| Epiflobacter | Epiflobacter spp. | SOTU4123 | 6 | freshwater | freshwater | 0.56% | |
| Membranihabitans | Membranihabitans marinus | SOTU207 | 15 | host saline water | host saline water | 1.26% | |
| Opimibacter | Opimibacter skivensis | SOTU225 | 282 | soil | soil | 1.53% | |
| Parvibacillus | Parvibacillus calidus | SOTU68 | 1 | bioreactor | bioreactor | 0.11% | |
| Vicinibacter | Vicinibacter affinis/proximus | SOTU83 | 5 | bioreactor | bioreactor | 1.63% | |
| Lewinellaceae | Flavilitoribacter | Flavilitoribacter nigricans | SOTU290 | 8 | saline water | bioreactor | 0.22% |
| Lewinella | Lewinella cohaerens | SOTU209 | 10 | saline water | bioreactor | 1.39% | |
| Neolewinella | Neolewinella antarctica | SOTU384 | 2 | saline water | plant saline water | 0.88% | |
| Neolewinella | Neolewinella aquimaris | SOTU315 | 9 | saline water | saline water | 0.33% | |
| Neolewinella | Neolewinella aurantiaca | SOTU344 | 1 | plant saline water | plant saline water | 0.21% | |
| Neolewinella | Neolewinella lutea | SOTU309 | 7 | saline water | saline water | 4.84% | |
| Neolewinella | Neolewinella marina/litorea | SOTU85 | 7 | saline water | plant saline water | 0.61% | |
| Neolewinella | Neolewinella maritima | SOTU326 | 2 | saline water | saline water | 0.72% | |
| Neolewinella | Neolewinella persica/agarilytica | SOTU330 | 2 | saline water | saline water | 0.40% | |
| Neolewinella | Neolewinella xylanilytica | SOTU338 | 9 | saline water | saline water | 2.67% | |
| Haliscomenobacteraceae | Haliscomenobacter | Haliscomenobacter hydrossis | SOTU236 | 1 | saline water | saline water | 0.91% |
| Phaeodactylibacter | Phaeodactylibacter luteus | SOTU19 | 1 | freshwater | freshwater | 0.36% | |
| Phaeodactylibacter | Phaeodactylibacter xiamenensis | SOTU18 | 1 | saline water | saline water | 0.17% | |
| Portibacter | Portibacter lacus/marinus | SOTU416 | 2 | saline water | saline water | 0.17% |
| Family | Genus | Species | SOTU ID | # Positive Samples | # Positive Samples (Abundance ≥ 0.1%) | Environment of Prevalence | Max Abundance Environment | Max Abundance |
|---|---|---|---|---|---|---|---|---|
| Saprospiraceae | Opimibacter | Opimibacter skivensis | SOTU225 | 3066 | 282 | soil | soil | 1.53% |
| Lewinellaceae | Neolewinella | Unknown species | SOTU306 | 516 | 25 | saline water | saline water | 12.20% |
| Lewinellaceae | Neolewinella | Unknown species | SOTU307 | 420 | 20 | saline water | saline water | 1.82% |
| Lewinellaceae | Flavilitoribacter | Unknown species | SOTU4353 | 415 | 27 | saline water | saline water | 2.52% |
| Lewinellaceae | Neolewinella | Unknown species | SOTU321 | 408 | 9 | saline water | saline water | 1.43% |
| Saprospiraceae | Aureispira | Unknown species | SOTU414 | 333 | 20 | saline water | saline water | 0.90% |
| Lewinellaceae | Neolewinella | Unknown species | SOTU3384 | 332 | 31 | saline water | saline water | 1.03% |
| Saprospiraceae | GOTU74 | Unknown species | SOTU5520 | 327 | 14 | saline water | saline water | 2.43% |
| Lewinellaceae | Flavilitoribacter | Flavilitoribacter nigricans | SOTU290 | 315 | 8 | saline water | bioreactor | 0.22% |
| Saprospiraceae | Aquirestis | Unknown species | SOTU112 | 287 | 11 | freshwater | saline water | 0.31% |
| Haliscomenobacteraceae | Haliscomenobacter | Unknown species | SOTU277 | 285 | 21 | freshwater | freshwater | 0.45% |
| Saprospiraceae | GOTU946 | Unknown species | SOTU527 | 272 | 12 | soil | soil | 0.42% |
| Haliscomenobacteraceae | Phaeodactylibacter | Unknown species | SOTU11811 | 265 | 12 | saline water | saline water | 0.71% |
| Lewinellaceae | Neolewinella | Unknown species | SOTU310 | 263 | 13 | saline water | air | 0.39% |
| Lewinellaceae | Neolewinella | Neolewinella aquimaris | SOTU315 | 256 | 9 | saline water | saline water | 0.33% |
| Haliscomenobacteraceae | GOTU588 | Unknown species | SOTU4189 | 253 | 3 | saline water | saline water | 0.41% |
| Saprospiraceae | Aquirestis | Unknown species | SOTU187 | 246 | 2 | saline water | freshwater | 0.36% |
| Lewinellaceae | Flavilitoribacter | Unknown species | SOTU1854 | 243 | 10 | saline water | saline water | 0.35% |
| Lewinellaceae | Lewinella | Unknown species | SOTU220 | 234 | 4 | saline water | bioreactor | 7.92% |
| Haliscomenobacteraceae | Portibacter | Unknown species | SOTU10607 | 233 | 2 | saline water | saline water | 1.62% |
| Family | Genus | Species | SOTU ID | # Positive Samples | # Positive Samples (Abundance ≥ 0.1%) | Environment of Prevalence | Max. Abundance Environment | Max. Abundance |
|---|---|---|---|---|---|---|---|---|
| Saprospiraceae | Parvibacillus | Unknown species | SOTU51 | 230 | 25 | bioreactor | bioreactor | 28.02% |
| Saprospiraceae | Aureispira | Unknown species | SOTU413 | 71 | 1 | host saline water | host saline water | 22.60% |
| Saprospiraceae | GOTU1969 | Unknown species | SOTU83565 | 1 | 1 | bioreactor | bioreactor | 21.73% |
| Saprospiraceae | Parvibacillus | Unknown species | SOTU80 | 2 | 1 | bioreactor | bioreactor | 14.97% |
| Lewinellaceae | GOTU1025 | Unknown species | SOTU13902 | 27 | 1 | saline water | saline water | 12.90% |
| Saprospiraceae | GOTU42 | Unknown species | SOTU565 | 205 | 14 | bioreactor | bioreactor | 12.72% |
| Lewinellaceae | Neolewinella | Unknown species | SOTU306 | 516 | 25 | saline water | saline water | 12.20% |
| Haliscomenobacteraceae | Phaeodactylibacter | Unknown species | SOTU6 | 5 | 3 | saline water | saline water | 11.92% |
| Saprospiraceae | GOTU741 | Unknown species | SOTU7213 | 49 | 8 | saline water | saline water | 11.79% |
| Saprospiraceae | GOTU4577 | Unknown species | SOTU50336 | 6 | 1 | bioreactor | bioreactor | 10.01% |
| Saprospiraceae | Parvibacillus | Unknown species | SOTU57 | 11 | 2 | bioreactor | bioreactor | 9.33% |
| Lewinellaceae | Neolewinella | Unknown species | SOTU312 | 105 | 3 | saline water | saline water | 9.23% |
| Lewinellaceae | GOTU755 | Unknown species | SOTU14539 | 7 | 3 | bioreactor | bioreactor | 8.93% |
| Lewinellaceae | Lewinella | Unknown species | SOTU220 | 234 | 4 | saline water | bioreactor | 7.92% |
| Haliscomenobacteraceae | GOTU423 | Unknown species | SOTU42257 | 12 | 3 | saline water | saline water | 7.45% |
| Lewinellaceae | Neolewinella | Unknown species | SOTU14993 | 21 | 1 | freshwater | freshwater | 7.37% |
| Saprospiraceae | Brachybacter | Unknown species | SOTU31 | 32 | 7 | bioreactor | bioreactor | 7.31% |
| Haliscomenobacteraceae | GOTU2075 | Unknown species | SOTU32764 | 12 | 2 | freshwater | bioreactor | 7.12% |
| Lewinellaceae | GOTU6152 | Unknown species | SOTU72597 | 4 | 1 | bioreactor | bioreactor | 6.24% |
| Haliscomenobacteraceae | GOTU62 | Unknown species | SOTU4917 | 124 | 3 | saline water | saline water | 5.89% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mourgela, R.N.; Kioukis, A.; Pourjam, M.; Lagkouvardos, I. Large-Scale Integration of Amplicon Data Reveals Massive Diversity within Saprospirales, Mostly Originating from Saline Environments. Microorganisms 2023, 11, 1767. https://doi.org/10.3390/microorganisms11071767
Mourgela RN, Kioukis A, Pourjam M, Lagkouvardos I. Large-Scale Integration of Amplicon Data Reveals Massive Diversity within Saprospirales, Mostly Originating from Saline Environments. Microorganisms. 2023; 11(7):1767. https://doi.org/10.3390/microorganisms11071767
Chicago/Turabian StyleMourgela, Rafaila Nikola, Antonios Kioukis, Mohsen Pourjam, and Ilias Lagkouvardos. 2023. "Large-Scale Integration of Amplicon Data Reveals Massive Diversity within Saprospirales, Mostly Originating from Saline Environments" Microorganisms 11, no. 7: 1767. https://doi.org/10.3390/microorganisms11071767
APA StyleMourgela, R. N., Kioukis, A., Pourjam, M., & Lagkouvardos, I. (2023). Large-Scale Integration of Amplicon Data Reveals Massive Diversity within Saprospirales, Mostly Originating from Saline Environments. Microorganisms, 11(7), 1767. https://doi.org/10.3390/microorganisms11071767