

Metagenomic Sequencing of Positive Blood Culture Fluid for Accurate Bacterial and Fungal Species Identification: A Pilot Study

Abstract
1. Introduction
2. Materials and Methods
2.1. Specimen and Conventional Microbiology
2.2. Metagenomic Sequencing
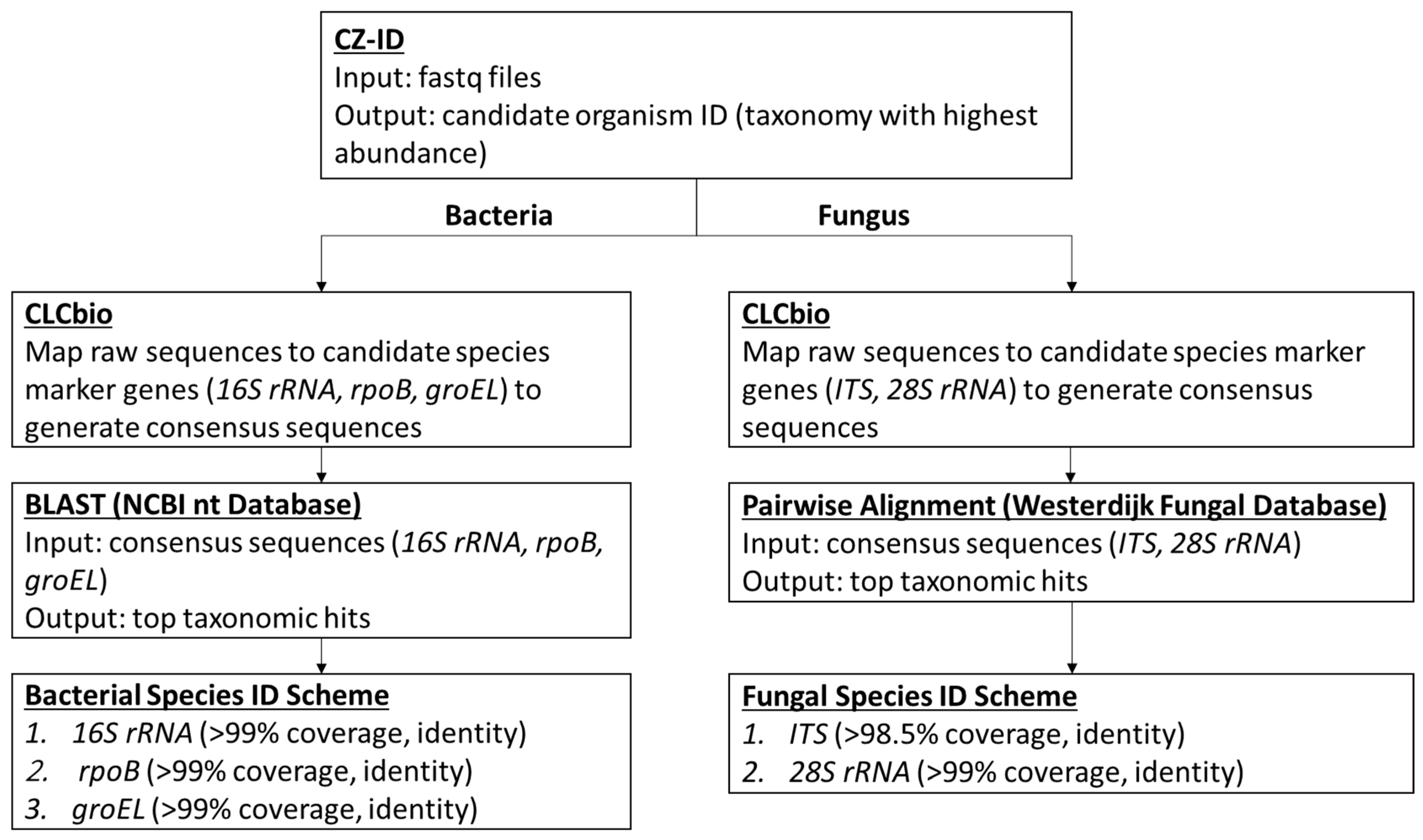
2.3. Bioinformatics Analysis
2.4. Clinical Utility Assessment and Study Ethics
3. Results
3.1. Quality Matrices and Bioinformatics Performance
3.2. Accuracy
3.3. Case Studies Demonstrating Clinical Utility
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peker, N.; Couto, N.; Sinha, B.; Rossen, J. Diagnosis of bloodstream infections from positive blood cultures and directly from blood samples: Recent developments in molecular approaches. Clin. Microbiol. Infect. 2018, 24, 944–955. [Google Scholar] [CrossRef] [PubMed]
- Rutanga, J.P.; Nyirahabimana, T. Clinical Significance of Molecular Diagnostic Tools for Bacterial Bloodstream Infections: A Systematic Review. Interdiscip. Perspect. Infect. Dis. 2016, 2016, 6412085. [Google Scholar] [CrossRef] [PubMed]
- Opota, O.; Croxatto, A.; Prod’Hom, G.; Greub, G. Blood culture-based diagnosis of bacteraemia: State of the art. Clin. Microbiol. Infect. 2015, 21, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Loonen, A.J.M.; Wolffs, P.F.G.; Bruggeman, C.A.; Brule, A.J.C.V.D. Developments for improved diagnosis of bacterial bloodstream infections. Eur. J. Clin. Microbiol. Infect. Dis. 2014, 33, 1687–1702. [Google Scholar] [CrossRef]
- Clinical and Laboratory Standards Institute (CLSI). Principles and Procedures for Blood Cultures; Approved Guideline; CLSI document M47-A; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2007. [Google Scholar]
- Weinstein, M.P.; Towns, M.L.; Quartey, S.M.; Mirrett, S.; Reimer, L.G.; Parmigiani, G.; Reller, L.B. The Clinical Significance of Positive Blood Cultures in the 1990s: A Prospective Comprehensive Evaluation of the Microbiology, Epidemiology, and Outcome of Bacteremia and Fungemia in Adults. Clin. Infect. Dis. 1997, 24, 584–602. [Google Scholar] [CrossRef]
- Kamau, E.; Maliksi, E.; Kwan, N.; Garner, O.B.; Yang, S. Catabacter hongkongensis bacteremia identified by direct metagenomic sequencing of positive blood culture fluid, first case report in the US. Anaerobe 2021, 71, 102421. [Google Scholar] [CrossRef]
- Price, T.K.; Realegeno, S.; Mirasol, R.; Tsan, A.; Chandrasekaran, S.; Garner, O.B.; Yang, S. Validation, Implementation, and Clinical Utility of Whole Genome Sequence-Based Bacterial Identification in the Clinical Microbiology Laboratory. J. Mol. Diagn. 2021, 23, 1468–1477. [Google Scholar] [CrossRef]
- Kalantar, K.L.; Carvalho, T.; De Bourcy, C.F.A.; Dimitrov, B.; Dingle, G.; Egger, R.; Han, J.; Holmes, O.B.; Juan, Y.-F.; King, R.; et al. IDseq—An open source cloud-based pipeline and analysis service for metagenomic pathogen detection and monitoring. Gigascience 2020, 9, giaa111. [Google Scholar] [CrossRef]
- Salem-Bango, Z.; Price, T.K.; Chan, J.L.; Chandrasekaran, S.; Garner, O.B.; Yang, S. Fungal Whole-Genome Sequencing for Species Identification: From Test Development to Clinical Utilization. J. Fungi 2023, 9, 183. [Google Scholar] [CrossRef]
- Xu, K.; Finn, L.E.; Geist, R.L.; Prestel, C.; Moulton-Meissner, H.; Kim, M.; Stacey, B.; McAllister, G.A.; Gable, P.; Kamali, T.; et al. Mycobacterium chimaera infections among cardiothoracic surgery patients associated with heater-cooler devices—Kansas and California, 2019. Infect. Control. Hosp. Epidemiol. 2022, 43, 1333–1338. [Google Scholar] [CrossRef]
- Shojaei, H.; Daley, C.; Gitti, Z.; Hashemi, A.; Heidarieh, P.; Moore, E.R.B.; Naser, A.D.; Russo, C.; van Ingen, J.; Tortoli, E. Mycobacterium iranicum sp. nov., a rapidly growing scotochromogenic species isolated from clinical specimens on three different continents. Int. J. Syst. Evol. Microbiol. 2013, 63 Pt 4, 1383–1389. [Google Scholar] [CrossRef]
- Lapierre, S.G.; Toro, A.; Drancourt, M. Mycobacterium iranicum bacteremia and hemophagocytic lymphohistiocytosis: A case report. BMC Res. Notes 2017, 10, 372. [Google Scholar] [CrossRef]
- Ranson, E.L.; Tsevat, R.K.; von Bredow, B.; Kamau, E.; Yang, S.; Prabaker, K.K. Catheter-Related Bloodstream Infection Caused by Mycolicibacterium iranicum, California, USA. Emerg. Infect. Dis. 2023, 29, 217–219. [Google Scholar] [CrossRef]
- Dubourg, G.; Raoult, D. Emerging methodologies for pathogen identification in positive blood culture testing. Expert Rev. Mol. Diagn. 2016, 16, 97–111. [Google Scholar] [CrossRef]
- Gyarmati, P.; Kjellander, C.; Aust, C.; Song, Y.; Öhrmalm, L.; Giske, C.G. Metagenomic analysis of bloodstream infections in patients with acute leukemia and therapy-induced neutropenia. Sci. Rep. 2016, 6, 23532. [Google Scholar] [CrossRef]
- Faria, M.; Conly, J.; Surette, M. The development and application of a molecular community profiling strategy to identify polymicrobial bacterial DNA in the whole blood of septic patients. BMC Microbiol. 2015, 15, 215. [Google Scholar] [CrossRef]
- Decuypere, S.; Meehan, C.J.; Van Puyvelde, S.; De Block, T.; Maltha, J.; Palpouguini, L.; Tahita, M.; Tinto, H.; Jacobs, J.; Deborggraeve, S. Diagnosis of Bacterial Bloodstream Infections: A 16S Metagenomics Approach. PLoS Negl. Trop. Dis. 2016, 10, e0004470. [Google Scholar] [CrossRef]
- Sabat, A.J.; van Zanten, E.; Akkerboom, V.; Wisselink, G.; van Slochteren, K.; de Boer, R.F.; Hendrix, R.; Friedrich, A.W.; Rossen, J.W.A.; Kooistra-Smid, A.M.D. Targeted next-generation sequencing of the 16S-23S rRNA region for culture-independent bacterial identification—Increased discrimination of closely related species. Sci. Rep. 2017, 7, 3434. [Google Scholar] [CrossRef]
- Couto, N.; Schuele, L.; Raangs, E.C.; Machado, M.P.; Mendes, C.I.; Jesus, T.F.; Chlebowicz, M.; Rosema, S.; Ramirez, M.; Carriço, J.A.; et al. Critical steps in clinical shotgun metagenomics for the concomitant detection and typing of microbial pathogens. Sci. Rep. 2018, 8, 13767. [Google Scholar] [CrossRef]
- Didelot, X.; Bowden, R.; Wilson, D.J.; Peto, T.E.A.; Crook, D.W. Transforming clinical microbiology with bacterial genome sequencing. Nat. Rev. Genet. 2012, 13, 601–612. [Google Scholar] [CrossRef]
- Laupland, K.B.; Valiquette, L. The changing culture of the microbiology laboratory. Can. J. Infect. Dis. Med. Microbiol. 2013, 24, 125–128. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Chen, Q.; Xiong, M.; Zhao, J.; Shen, S.; Chen, L.; Pan, Y.; Li, Z.; Li, Y. Clinical Performance of Nanopore Targeted Sequencing for Diagnosing Infectious Diseases. Microbiol. Spectr. 2022, 10, e0027022. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Rothman, R.E. PCR-based diagnostics for infectious diseases: Uses, limitations, and future applications in acute-care settings. Lancet Infect. Dis. 2004, 4, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.E.; Ellis, B.C.; Lee, R.; Stamper, P.D.; Zhang, S.X.; Carroll, K.C. Prospective Evaluation of a Matrix-Assisted Laser Desorption Ionization–Time of Flight Mass Spectrometry System in a Hospital Clinical Microbiology Laboratory for Identification of Bacteria and Yeasts: A Bench-by-Bench Study for Assessing the Impact on Time to Identification and Cost-Effectiveness. J. Clin. Microbiol. 2012, 50, 3301–3308. [Google Scholar] [CrossRef]
- Glaser, C.A.; Honarmand, S.; Anderson, L.J.; Schnurr, D.P.; Forghani, B.; Cossen, C.K.; Schuster, F.L.; Christie, L.J.; Tureen, J.H. Beyond Viruses: Clinical Profiles and Etiologies Associated with Encephalitis. Clin. Infect. Dis. 2006, 43, 1565–1577. [Google Scholar] [CrossRef]
- Schlaberg, R.; Chiu, C.Y.; Miller, S.; Procop, G.W.; Weinstock, G.; the Professional Practice Committee and Committee on Laboratory Practices of the American Society for Microbiology; the Microbiology Resource Committee of the College of American Pathologists. Validation of Metagenomic Next-Generation Sequencing Tests for Universal Pathogen Detection. Arch. Pathol. Lab. Med. 2017, 141, 776–786. [Google Scholar] [CrossRef]
- Simner, P.J.; Miller, S.; Carroll, K.C. Understanding the Promises and Hurdles of Metagenomic Next-Generation Sequencing as a Diagnostic Tool for Infectious Diseases. Clin. Infect. Dis. 2017, 66, 778–788. [Google Scholar] [CrossRef]
- Flygare, S.; Simmon, K.; Miller, C.; Qiao, Y.; Kennedy, B.; Di Sera, T.; Graf, E.H.; Tardif, K.D.; Kapusta, A.; Rynearson, S.; et al. Taxonomer: An interactive metagenomics analysis portal for universal pathogen detection and host mRNA expression profiling. Genome Biol. 2016, 17, 111. [Google Scholar] [CrossRef]
- Han, Y.; Jia, Z.; Shi, J.; Wang, W.; He, K. The active lung microbiota landscape of COVID-19 patients through the metatranscriptome data analysis. Bioimpacts 2022, 12, 139–146. [Google Scholar] [CrossRef]
- Ramachandran, P.S.; Ramesh, A.; Creswell, F.V.; Wapniarski, A.; Narendra, R.; Quinn, C.M.; Tran, E.B.; Rutakingirwa, M.K.; Bangdiwala, A.S.; Kagimu, E.; et al. Integrating central nervous system metagenomics and host response for diagnosis of tuberculosis meningitis and its mimics. Nat. Commun. 2022, 13, 1675. [Google Scholar] [CrossRef]
- Bohl, J.A.; Lay, S.; Chea, S.; Ahyong, V.; Parker, D.M.; Gallagher, S.; Fintzi, J.; Man, S.; Ponce, A.; Sreng, S.; et al. Discovering disease-causing pathogens in resource-scarce Southeast Asia using a global metagenomic pathogen monitoring system. Proc. Natl. Acad. Sci. USA 2022, 119, e2115285119. [Google Scholar] [CrossRef]
- Hogan, C.A.; Yang, S.; Garner, O.B.; Green, D.A.; Gomez, C.A.; Bard, J.D.; Pinsky, B.A.; Banaei, N. Clinical Impact of Metagenomic Next-Generation Sequencing of Plasma Cell-Free DNA for the Diagnosis of Infectious Diseases: A Multicenter Retrospective Cohort Study. Clin. Infect. Dis. 2021, 72, 239–245. [Google Scholar] [CrossRef]
- Babady, N.E. Clinical Metagenomics for Bloodstream Infections: Is the Juice Worth the Squeeze? Clin. Infect. Dis. 2021, 72, 246–248. [Google Scholar] [CrossRef]
- Larkin, P.M.; Lawson, K.L.; Contreras, D.A.; Le, C.Q.; Trejo, M.; Realegeno, S.; Hilt, E.E.; Chandrasekaran, S.; Garner, O.B.; Fishbein, G.A.; et al. Amplicon-Based Next-Generation Sequencing for Detection of Fungi in Formalin-Fixed, Paraffin-Embedded Tissues: Correlation with Histopathology and Clinical Applications. J. Mol. Diagn. 2020, 22, 1287–1293. [Google Scholar] [CrossRef]
- Culbreath, K.; Melanson, S.; Gale, J.; Baker, J.; Li, F.; Saebo, O.; Kommedal, O.; Contreras, D.; Garner, O.B.; Yang, S. Validation and Retrospective Clinical Evaluation of a Quantitative 16S rRNA Gene Metagenomic Sequencing Assay for Bacterial Pathogen Detection in Body Fluids. J. Mol. Diagn. 2019, 21, 913–923. [Google Scholar] [CrossRef]
- Pannaraj, P.S.; Li, F.; Cerini, C.; Bender, J.M.; Yang, S.; Rollie, A.; Adisetiyo, H.; Zabih, S.; Lincez, P.J.; Bittinger, K.; et al. Association Between Breast Milk Bacterial Communities and Establishment and Development of the Infant Gut Microbiome. JAMA Pediatr. 2017, 171, 647–654. [Google Scholar] [CrossRef]
- Tan, H.; Liu, T.; Yu, Y.; Tang, J.; Jiang, L.; Martin, F.M.; Peng, W. Morel Production Related to Soil Microbial Diversity and Evenness. Microbiol. Spectr. 2021, 9, e0022921. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, T.; Liu, L.; Chen, Y.; Tang, J.; Peng, W.; Tan, H. Application of the mushroom volatile 1-octen-3-ol to suppress a morel disease caused by Paecilomyces penicillatus. Appl. Microbiol. Biotechnol. 2022, 106, 4787–4799. [Google Scholar] [CrossRef]
- Hilt, E.E.; Ferrieri, P. Next Generation and Other Sequencing Technologies in Diagnostic Microbiology and Infectious Diseases. Genes 2022, 13, 1566. [Google Scholar] [CrossRef]


{kind=link}
| Sample ID # | Positive Blood Culture mNGS | Identification by Conventional Methods | Conventional Methods Used for Final Identification | |||||
|---|---|---|---|---|---|---|---|---|
| Identification | Marker Gene Pairwise Identity | |||||||
| 16S rRNA | rpoB | groEL | ITS Region | 28S rRNA | ||||
| * UCLA_467- 473, 562, 558 | Staphylococcus epidermidis | 100.00% | 100.00% | 100.00% | S. epidermidis | MALDI-TOF | ||
| UCLA_478 | Slackia exigua | 99.30% | n/a | n/a | S. exigua | MALDI-TOF | ||
| UCLA_499 | Desulfovibrio diazotrophicus | 99.7% | 100.0% | 99.9% | Desulfovibrio spp. | MALDI-TOF | ||
| UCLA_501 | Bacteroides dorei | 99.94% | 99.79% | 99.94% | B. dorei | MALDI-TOF | ||
| UCLA_503 | Bacteroides thetaiotaomicron | 99.96% | 99.97% | 100.00% | B. thetaiotaomicron | MALDI-TOF | ||
| UCLA_505 | Mycolicibacterium iranicum | 99.87% | 97.86% | 98.52% | Yellow pigmented rapid-growing Mycobacterium | Morphology and Stain | ||
| UCLA_510 | Proteus mirabilis | 100.00% | 100.00% | 100.00% | P. mirabilis | MALDI-TOF | ||
| UCLA_517 | Escherichia coli | 99.10% | 99.98% | 100.00% | E. coli | MALDI-TOF | ||
| UCLA_518 | Staphylococcus lugdunensis | 99.94% | 100.00% | 100.00% | S. lugdunensis | MALDI-TOF | ||
| UCLA_519 | Streptococcus intermedius | 99.94% | 99.05% | 98.34% | S. intermedius | MALDI-TOF | ||
| UCLA_520 | Clostridium butyricum | 100.00% | 99.97% | 99.88% | C. butyricum | MALDI-TOF | ||
| UCLA_524 | Mycobacterium intracellulare subsp. chimaera | 100.00% | 100.00% | 100.00% | M. avium complex | DNA Probe | ||
| UCLA_1007 | Mycobacterium avium subsp. hominissuis | 100.00% | 100.00% | 100.00% | M. avium complex | DNA Probe | ||
| UCLA_559 | Enterococcus faecium | 99.94% | 100.00% | 100.00% | E. faecium | MALDI-TOF | ||
| UCLA_560 | Fusobacterium nucleatum | 100.00% | 98.96% | 99.88% | F. nucleatum | MALDI-TOF | ||
| UCLA_561 | Staphylococcus haemolyticus | 99.81% | 99.47% | 98.40% | S. haemolyticus | MALDI-TOF | ||
| UCLA_496 | Candida tropicalis | 100% | n/p | C. tropicalis | MALDI-TOF | |||
| UCLA_497 | Candida glabrata | 100% | n/p | C. glabrata | MALDI-TOF | |||
| UCLA_498 | Fusarium proliferatum | 100% | n/p | F. proliferatum | MALDI-TOF | |||
| UCLA_500 | Clavispora lusitaniae | 100% | n/p | C. lusitaniae | MALDI-TOF | |||
| UCLA_502 | Candida albicans | 100% | n/p | C. albicans | MALDI-TOF | |||
| UCLA_596 | Aspergillus flavus-oryzae group | 100% | 100% | A. flavus-oryzae group | MALDI-TOF | |||
| Metric | Range | Average (SD) | QC Criteria |
|---|---|---|---|
| CZ-ID: Phred Quality Score (%) rPM (highest value) %id | 65.64–95.33 7534.8–601,308 83.7–100 | 88.3 (8.5) 280,248 (144,559) 98.8 (3.4) | >60% >5000 >80% |
| CLC Mapping to Marker Genes: Total Reads Average Coverage (×) 5× Coverage (%) | 1,215,048–12,781,338 18.7–352.8 99.2–100 | 2,853,970 (2,342,278) 109.0 (87.6) 99.9 (0.21) | >1,000,000 >10× >90% |
| 16S rRNA: Length Average Coverage Ambiguous Nucleotide (%) | 1513–1573 20.8–3943.7 0.0–0.9 | 1547.7 (13.5) 942.4 (901.0) 0.08 (0.21) | None >20× <1% |
| rpoB: Length Average Coverage Ambiguous Nucleotide (%) | 3552–4389 30.2–822.4 0.0–0.9 | 3717.7 (242.0) 176.6 (160.7) 0.05 (0.20) | None >20× <1% |
| groEL: Length Average Coverage Ambiguous Nucleotide (%) | 1620–1647 25.05–717.6 0.0–1.1 | 1627.0 (9.6) 157.6 (140.8) 0.05 (0.23) | None >20× <1% |
| Metric | Range | Average (SD) | QC Criteria |
|---|---|---|---|
| CZ-ID: Phred Quality Score (%) rPM (highest value) %id | 56.5–90.5 2981.9–619,105.8 96.4–99.8 | 80.6 (13.6) 225,922.8 (226,785.8) 98.8 (1.2) | >50% >2000 >80% |
| CLC Mapping to Marker Genes: Total Reads Average Coverage (×) 5× Coverage (%) | 2,385,524–4,777,322 5.9–1256.3 66.6–100 | 3,357,613.7 (779,513.2) 653.2 (556.9) 94.4 (13.7) | >1,000,000 >5× >60% |
| ITS: Length (nt) Average Coverage (×) | 341–2587 5.9–1256.3 | 880.7 (844.7) 727.7 (569.8) | None >5× |
| 28S (Aspergillus and Mucorales): Length (nt) Average Coverage (×) | 593 9.5 | 593 9.5 | None >5× |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamau, E.; Yang, S. Metagenomic Sequencing of Positive Blood Culture Fluid for Accurate Bacterial and Fungal Species Identification: A Pilot Study. Microorganisms 2023, 11, 1259. https://doi.org/10.3390/microorganisms11051259
Kamau E, Yang S. Metagenomic Sequencing of Positive Blood Culture Fluid for Accurate Bacterial and Fungal Species Identification: A Pilot Study. Microorganisms. 2023; 11(5):1259. https://doi.org/10.3390/microorganisms11051259
Chicago/Turabian StyleKamau, Edwin, and Shangxin Yang. 2023. "Metagenomic Sequencing of Positive Blood Culture Fluid for Accurate Bacterial and Fungal Species Identification: A Pilot Study" Microorganisms 11, no. 5: 1259. https://doi.org/10.3390/microorganisms11051259
APA StyleKamau, E., & Yang, S. (2023). Metagenomic Sequencing of Positive Blood Culture Fluid for Accurate Bacterial and Fungal Species Identification: A Pilot Study. Microorganisms, 11(5), 1259. https://doi.org/10.3390/microorganisms11051259