
In Silico Analysis of a GH3 β-Glucosidase from Microcystis aeruginosa CACIAM 03

 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Organism and Amino Acid Sequence
2.2. In Silico Analysis of Primary and Secondary Structures
2.3. Identification of Conserved Motifs
2.4. ProtParam Analysis
2.5. Homology Modeling
2.6. Molecular Docking and Molecular Dynamics Simulations
2.7. Binding Free Energy Calculation
2.8. Extraction and β-Glucosidase Activity
3. Results
3.1. MaBgl3 Amino Acid Sequence
3.2. Identification of Conserved Motifs
3.3. Secondary Structure Analysis of MaBgl3
3.4. Prediction of Physicochemical Properties
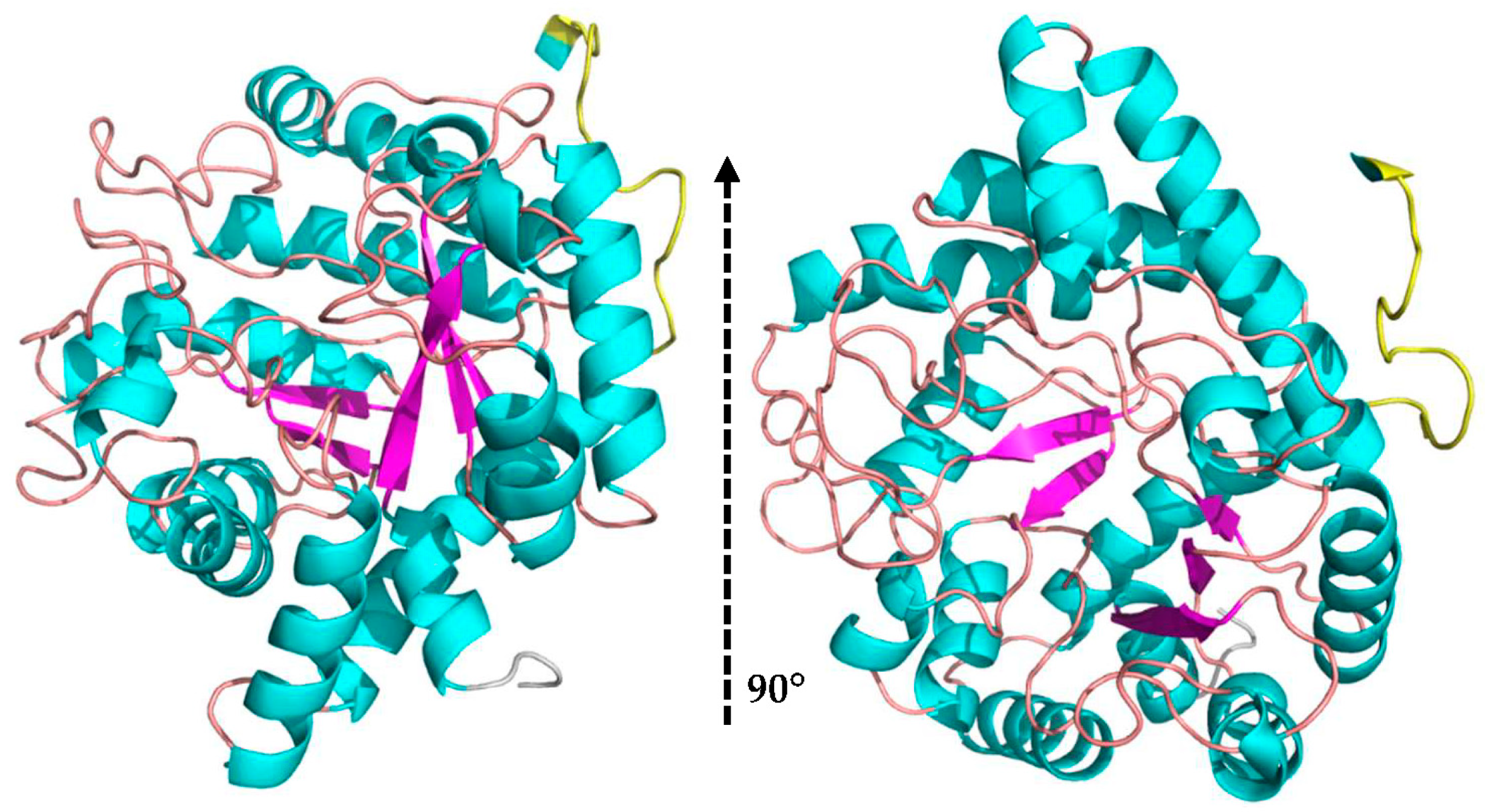
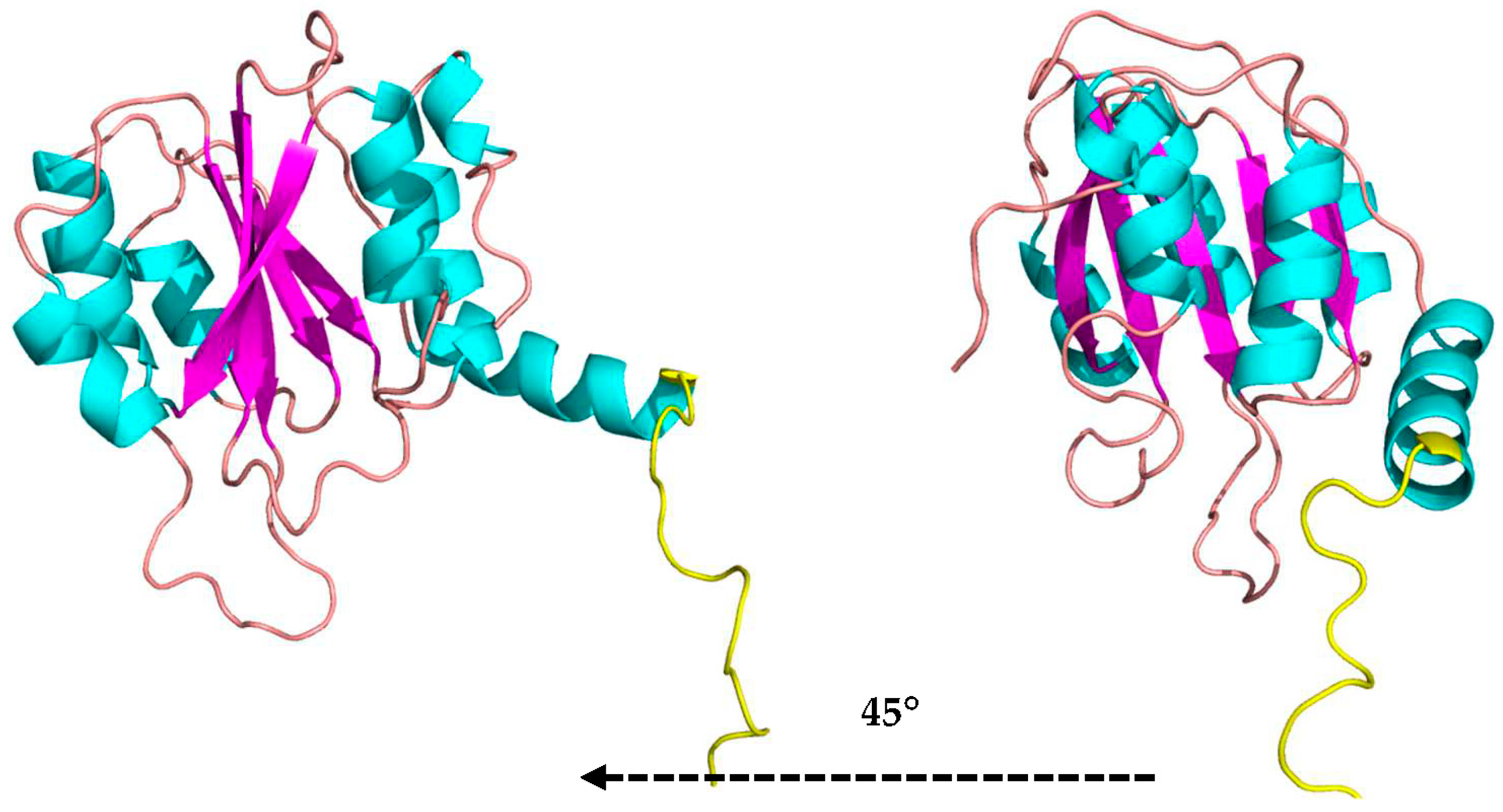
3.5. 3D Model of MaBgl3
3.6. Molecular Docking Study
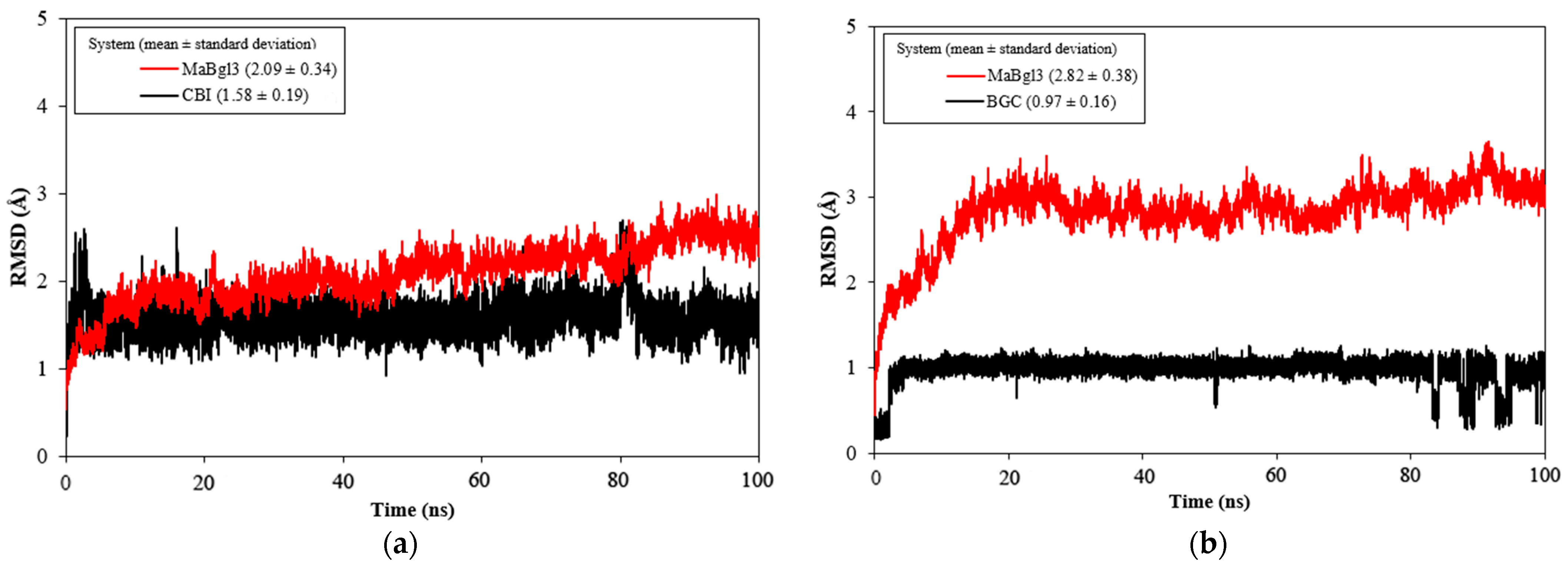
3.7. MD Simulation Analysis
3.8. Binding Free Energy Analysis
3.9. Validation of Enzyme Activity using Lab Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Demay, J.; Bernard, C.; Reinhardt, A.; Marie, B. Natural Products from Cyanobacteria: Focus on Beneficial Activities. Mar. Drugs. 2019, 17, 320. [Google Scholar] [CrossRef]
- Gaysina, L.; Saraf, A.; Singh, P. Cyanobacteria in Diverse Habitats. In Cyanobacteria: From Basic Science to Applications, 1st ed.; Misha, A., Tiwari, D., Rai, A., Eds.; Academic Press: Cambridge, MA, USA, 2018; Volume 1, pp. 1–28. [Google Scholar]
- Sant′Anna, C.; Teresade, P.; Werner, V.; Dogo, C.; Rios, F. Review of toxic species of Cyanobacteria in Brazil. Algol. Stud. 2008, 126, 251–265. [Google Scholar] [CrossRef]
- Falconer, I.R. Cyanobacterial Toxins of Drinking Water Supplies, 1st ed.; T&F Group: Boca Raton, FL, USA, 2005; pp. 1–296. [Google Scholar]
- Welker, M.; Dittmann, E.; Von Döhren., H. Chapter Two–Cyanobacteria as a Source of Natural Products. In Methods of Enzymology; Hopwood, D., Ed.; Academic Press: Cambridge, MA, USA, 2012; Volume 517, pp. 23–46. [Google Scholar]
- Mazur-Marzec, H.; Fidor, A.; Ceglowsla, M.; Wieczerzak, E.; Kropidłowska, M.; Goua, M.; Macaskill, J.; Edwards, C. Cyanopeptolins with trypsin and chymotrypsin inhibitory activity from the cyanobacterium Nostoc edaphichum CCNP1411. Mar. Drugs. 2018, 16, 220. [Google Scholar] [CrossRef] [PubMed]
- Segato, F.; Damásio, A.; de Lucas, R.; Squina, F.; Prade, R. Genomics Review of Holocellulose Deconstruction by Aspergilli. Microbiol. Mol. Biol. Rev. 2014, 78, 588–613. [Google Scholar] [CrossRef]
- Brasil, B.; de Siqueira, F.; Salum, T.; Zanette, C.; Spier, M. Microalgae and cyanobacteria as enzyme biofactories. Algal Res. 2017, 25, 76–89. [Google Scholar] [CrossRef]
- Hati, S.; Vij, S.; Singh, B.; Mandal, S. β-Glucosidase activity and bioconversion of isoflavones during fermentation of soymilk. J. Sci. Food. Agric. 2015, 95, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, R.; Singh, S.; Shukla, P.; Nain, L. Novel cold temperature active β-glucosidase from Pseudomonas lutea BG8 suitable for simultaneous saccharification and fermentation. RSC Adv. 2014, 4, 58108–58115. [Google Scholar] [CrossRef]
- Singh, G.; Verma, A.; Kumar, V. Catalytic properties, functional attributes and industrial applications of β-glucosidases. 3 Biotech 2016, 6, 3. [Google Scholar] [CrossRef]
- De Giuseppe, P.; Souza, T.; Souza, F.; Zanphorlin, L.; Machado, C.; Ward, R.; Jorge, J.; Furriel, R.; Murakami, M. Structural basis for glucose tolerance in GH1 β-glucosidase. ACSDAD 2014, 70, 1631–1639. [Google Scholar] [CrossRef]
- Ravanal, M.; Rosa, L.; Eyzaguirre, J. A-L-arabinofuranosidase 3 from Penicillium purpurogenum (ABF3): Potential application in the enhancement of wine flavor and heterologous expression of the enzyme. Food Chem. 2012, 134, 888–893. [Google Scholar] [CrossRef]
- Lee, W.; Nan, H.; Kim, H.; Jin, Y. Simultaneous saccharification and fermentation by engineered Saccharomyces cerevisiae without supplementing extracellular β-glucosidase. J. Biotechnol. 2013, 167, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Qin, Y.; Cao, Q.; Liu, G.; Li, J.; Li, Z.; Zhao, J.; Qu, Y. Promotional extracellular lignocellulolytic enzymes production by restraining the intracellular β-glucosidase in Penicillium decumbens. Bioresour. Technol. 2013, 137, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Ugwuanyi, O.; Harvey, L.; McNeil, B. Linamarase activities in Bacillus spp. Responsible for thermophilic aerobic digestion of agricultural wastes for animal nutrition. Waste Manag. 2007, 27, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Henrissat, B.; Romeu, A. Families, superfamilies and subfamilies of glycosyl hydrolases. Biochem. J. 1995, 311, 350–351. [Google Scholar] [CrossRef] [PubMed]
- Cantarel, B.; Coutinho, P.; Rancurel, C.; Bernard, T.; Lombard, V.; Henrissat, B. The Carbohydrate-Active EnZymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids. Res. 2009, 37, 233–238. [Google Scholar] [CrossRef]
- Castro, W.; Lima, A.; Moraes, P.; Siqueira, A.S.; Aguiar, D.C.F.; Baraúna, A.R.F.; Martins, L.C.; Fuzii, H.T.; de Lima, C.P.S.; Vianez-Júnior, J.L.S.G.; et al. Draft Genome Sequence of Microcystis aeruginosa CACIAM03, a Cyanobacterium Isolated from na Amazonian Freshwater. Genome Announc. 2016, 4, 1299–1316. [Google Scholar] [CrossRef]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids. Res. 2020, 48, 265–268. [Google Scholar] [CrossRef]
- Letunic, I.; Khedkar, S.; Bork, P. SMART: Recent updates, new developments and status in 2020. Nucleic Acids. Res. 2020, 49, 458–460. [Google Scholar] [CrossRef]
- Geourjon, C.; Deléage, G. SOPMA: Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput. Appl. Biosci. 1995, 11, 681–684. [Google Scholar] [CrossRef]
- Garnier, J.; Gibrat, J.; Robson, B. GOR method for predicting protein secondary structure from amino acid sequence. Meth. Enzymol. 1996, 266, 540–553. [Google Scholar]
- Kumar, A. CFSSP: Chou and Fasman Secondary Structure Prediction server. Wide Spec. 2013, 1, 15–19. [Google Scholar]
- Altschul, S.; Gish, W.; Miller, W.; Myers, E.; Lipman, D. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Thompson, J.; Higgins, D.; Gibson, T. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids. Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef] [PubMed]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids. Res. 2014, 42, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Hoogland, C.; Gattiker, A.; Duvaud, S.; Wilkins, M.; Appel, R.; Bairoch, A. Protein identification and Analysis Tools on the ExPASy Server. In The Proteomics Protocols Handbook, 1st ed.; Walker, J., Ed.; Humana Press: Totowa, NJ, USA, 2005; pp. 571–607. [Google Scholar]
- Berman, H.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.; Weissig, H.; Shindyalov, I.; Bourne, P. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using Modeller. Curr. Protoc. Bioinform. 2016, 54, 1–5. [Google Scholar] [CrossRef]
- Williams, C.; Headd, J.; Moriarty, N.; Prisant, M.G.; Videau., L.L.; Deis, L.N.; Verma, V.; Keedy, D.A.; Hintze, B.J.; Chen, V.B.; et al. MolProbity: More and better reference data for improved all-atom structure validation. Prot. Sci. 2018, 27, 293–315. [Google Scholar] [CrossRef]
- Eisenberg, D.; Lüthy, R.; Bowie, J. VERIFY 3D: Assessment of protein models with three-dimensional profiles. Meth. Enzym. 1997, 277, 396–404. [Google Scholar]
- Colovos, C.; Yeates, T. Verification of protein structures: Patterns of nonbonded atomic interaction. Protein Sci. 1993, 9, 1511–1519. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Substance Record for SID 823266, BGC, Source: NCBI Structure. Available online: https://pubchem.ncbi.nlm.nih.gov/substance/823266 (accessed on 14 May 2022).
- Guedes, I.; Costa, L.; dos Santos, K.; Karl, A.L.M.; Rocha, G.K.; Teixeira, L.M.; Galheigo, M.M.; Medeiros, V.; Krempser, E.; Custódio, F.L.; et al. Drug design and repurposing with DockThor-VS web server focusing on SARS-CoV-2 therapeutic targets and their non-synonym variants. Sci. Rep. 2021, 11, 5543. [Google Scholar] [CrossRef]
- Laskowski, R.; Swindells, M. LigPlot+: Multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011, 51, 2778–2786. [Google Scholar] [CrossRef]
- Dolinsky, T.; Czodrowski, P.; Li, H.; Nielsen, J.; Jensen, J.; Kleber, G.; Baker, N. PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007, 35, 522–525. [Google Scholar] [CrossRef]
- Case, D.; Cheatham, T.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.; Onuffrey, A.; Simmerling, C.; Wang, B.; Woods, R. “The Amber biomolecular simulation programs”. J. Computat. Chem. 2005, 26, 1668–1688. [Google Scholar] [CrossRef]
- Maier, J.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and Dynamics of the TIP3P, SPC, and SPC/E Water Models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Siqueira, A.; Lima, A.; Aguiar, D.; Santos, A.; Gonçalves Vianez Júnior, J.L.d.S.; Gonçalves, E.C. Genomic screening of new putative antiviral lectins from Amazonian cyanobacteria based on a bioinformatics approach. Proteins 2018, 86, 1047–1054. [Google Scholar] [CrossRef]
- PyMOL by Schrödinger. Available online: https//www.pymol.org/pymol (accessed on 23 August 2021).
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the MM/PBSA and MM/GBSA methods. The accuracy of binding free energy calculations based on molecular dynamics simulations. J. Chem. Inf. Model. 2011, 51, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Sulea, T.; Vivcharuk, V.; Corbeil, C.; Deprez, C.; Purisima, E. Assessment of Solvated Interaction Energy Function for Ranking Antibody-Antigen Binding Affinities. J. Chem. Inf. Model. 2016, 56, 1292–1303. [Google Scholar] [CrossRef] [PubMed]
- Sulea, T.; Cui, Q.; Purisima, E. Solvated Interaction Energy (SIE) for Scoring Protein-Ligand Binding Affinities. Methods Mol. Biol. 2012, 819, 295–303. [Google Scholar] [PubMed]
- Luthy, R.; Bowie, J.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef]
- Qin, Z.; Xiao, Y.; Yang, X.; Mesters, J.; Yang, S.; Jiang, Z. A unique GCN5-related glucosamine N-acetyltransferase region exist in the fungal multi-domain glycoside hydrolase family 3 β-N-acetyglucosaminidase. Sci. Rep. 2015, 5, 18292. [Google Scholar] [CrossRef] [PubMed]
- Pozzo, T.; Pasten, J.; Karlsson, E.; Logan, D. Structural and functional analyses of beta-glucosidase 3B from Thermotoga neapolitana: A thermostable tree-domain representative of glycoside hydrolase 3. J. Mol. Biol. 2010, 397, 724–739. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.; Vocadlo, D.; Mah, M.; Rupitz, K.; Stoll, D.; Warren, R.A.J.; Withers, S.G. Characterization of β-N-acetylhexosaminidase and β-N-acetylglucosaminidase/β-glucosidase from Cellulomonas fimi. J. FEBS 2006, 273, 2929–2941. [Google Scholar] [CrossRef] [PubMed]
- Stierand, K.; Maaß, P.; Rarey, M. Molecular Complexes at a Glance: Automated Generation of two-dimensional Complex Diagrams. Bioinformatics 2006, 22, 1710–1716. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Sheng, X.; Liu, Y. Insight into the catalytic mechanism of N-acetylglucosaminidase glycoside hydrolase from Bacillus subtilis: A QM/MM study. Org. Biomol. Chem. 2016, 14, 3432–3442. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Yang, J.; Bocola, M.; Schwaneberg, U.; Zhu, L. Loop Engineering Reveals the importance of Active-Site-Decoring Loops and Gating Residue in Substrate Affinity Modulaton of Arginine Deiminase (an Anti-Tumor Enzyme). Biochem. Biophys. Res. Commun. 2018, 499, 233–238. [Google Scholar] [CrossRef]
- Martínez, L. Automatic Identification of Mobile and Rigid Substructures in Molecular Dynamics Simulations and Fractional Structural Fluctuation Analysis. PLoS ONE 2015, 10, e0119264. [Google Scholar] [CrossRef]
- Genhenden, S.; Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert. Opin. Drug. Discov. 2015, 5, 449–461. [Google Scholar] [CrossRef]
- Wang, C.; Greene, D.; Xiao, L.; Qi, R.; Luo, R. Recent Developments and Applications of the MMPBSA Method. Front. Mol. Biosci. 2018, 4, 87. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Kumari, R.; Kumar, R.; Lynn, A. G-mmpbsa–a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Info. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Lee, R.; Hrmova, M.; Burton, R.; Lahnstein, J.; Fincher, G. Bifunctional family 3 glycoside hydrolases from barley with alpha-L-arabinofuranosidase and beta-D-xylosidase activity. Characterization, primary structures, and COOH-terminal processing. J. Biol. Chem. 2003, 278, 5377–5387. [Google Scholar] [CrossRef] [PubMed]
- Nishida, V.; De Oliveira, R.; Brugnari, T.; Correa, R.C.G.; Peralta, R.A.; Castoldi, R.; de Souza, C.G.M.; Bracht, A.; Peralta, R.M. Immobilization of Aspergillus awamori β-glucosidase on commercial gelatin: An inexpensive and efficient process. Int. J. Biol. Macromol. 2018, 111, 1206–1213. [Google Scholar] [CrossRef] [PubMed]
- Pereira, J.; Marques, N.; Rodrigues, A.; Oliveira, T.; Boscolo, M.; Da-Silva, R.; Gomes, E.; Bocchini-Martins, D. Thermophilic fungi as new sources for production of cellulases and xynalases with potential use in sugarcane bagasse saccharification. J. Appl. Microbiol. 2015, 118, 928–939. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, K.; Megyeri, L.; Szakacs, G.; Kubicek, C.; Galbe, M.; Zachi, G. Trichoderma atroviridae mutants with enhanced production of cellulose and β-glucosidase on pretreated willow. Enzym. Microbiol. Technol. 2008, 43, 48–55. [Google Scholar] [CrossRef]
- Saha, B.; Bothast, R. Production, purification, and characterization of a highly glucose-tolerant novel beta-glucosidase from Candida peltata. Appl. Environ. Microbiol. 1996, 62, 3165–3170. [Google Scholar] [CrossRef]
- Goyal, N.; Gupta, J.; Soni, S. A novel raw starch digesting thermostable α-amylase from Bacillus sp. 1-3 and its use in the direct hydrolysis of raw potato starch. Enzym. Microb. Technol. 2005, 37, 723–734. [Google Scholar] [CrossRef]
- Linding, R.; Jensen, L.; Diella, F.; Bork, P.; Gibson, T.; Russell, R. Protein disorder prediction: Implication for structural proteomics. Structure 2003, 11, 1453–1459. [Google Scholar] [CrossRef]
- Morris, A.; MacArthur, M.; Hutchinson, E.; Thornton, J. Stereochemical quality of protein structure coordinates. Proteins 1992, 12, 345–364. [Google Scholar] [CrossRef]
- MacDonald, S.; Blaukopf, M.; Withers, S. N-acetylglucosaminidases from CAZy family GH3 are really glycoside phosphorylases, thereby explaining their use of histidine as an acid/base catalyst in place of glutamic acid. J. Biol. Chem. 2015, 290, 4887–4895. [Google Scholar] [CrossRef]
- Mondon, M.; Hur, S.; Vadlamani, G.; Rodrigues, P.; Tsybina, P.; Oliver, A.; Mark, B.L.; Vocadlo, D.J.; Blériot, Y. Selective trihydroxyazepane NagZ inhibitors increase sensitivity of Pseudomonas aeruginosa to beta-lactams. Chem. Commun. 2013, 49, 10983–10985. [Google Scholar] [CrossRef] [PubMed]
- Litzinger, S.; Fischer, S.; Polzer, P.; Diederichs, K.; Welte, W.; Mayer, C. Structural and Kinetic Analysis of Bacillus subtilis N-Acetylglucosaminidase Reveals a Unique Asp-His Dyad Mechanism. J. Bio. Chem. 2010, 285, 35675–35684. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description | Protein Length (AA) | Molecular Mass (kDa) | Identity (%) | Access Number |
|---|---|---|---|---|
| β-glycosidase (Microcystis aeruginosa) * | 526 | 57.249 | 100 | OCY13127.1 |
| β-glycosidase GH3 (Microcystis aeruginosa) | 526 | 57.415 | 98.10 | WP_159250140.1 |
| β-glycosidase (Microcystis sp. 0824) | 526 | 57.283 | 98.10 | WP_108937562.1 |
| β-glycosidase (Microcystis sp. LEGE 00066) | 512 | 55.728 | 98.24 | WP_051048499.1 |
| β-glycosidase (Microcystis wesenbergii) | 512 | 55.750 | 97.85 | WP_199319088.1 |
| α-Helix | β-Sheet | Loop | ||||
|---|---|---|---|---|---|---|
| Servers | Residues | (%) | Residues | (%) | Residues | (%) |
| GOR IV | 209 | 39.4 | 76 | 14.5 | 241 | 45.8 |
| SopMA | 225 | 42.8 | 69 | 13.1 | 206 | 39.2 |
| CFSSP | 221 | 42 | 61 | 11.6 | 228 | 43.3 |
| Parameters | M. aeruginosa CACIAM 03 * | M. aeruginosa | Microcystis sp. 0824 | Microcystis sp. LEGE 00066 | M. wesenbergii |
|---|---|---|---|---|---|
| MW (kDa) | 57.25 | 57.42 | 57.28 | 55.73 | 55.75 |
| pI | 5.01 | 5.13 | 5.13 | 4.96 | 5.05 |
| R+ | 56 | 55 | 55 | 54 | 53 |
| R- | 39 | 40 | 40 | 37 | 37 |
| EC (M−1 cm−1) | 64.53 | 71.52 | 64.40 | 66,02 | 65.90 |
| II | 41.95 | 42.56 | 42.33 | 42.86 | 43.30 |
| Stability | Unstable | Unstable | Unstable | Unstable | Unstable |
| AI | 101.73 | 102.28 | 102.83 | 101.46 | 101.66 |
| GRAVY | −0.004 | −0.017 | −0.009 | −0.004 | −0.001 |
| Complex | Binding Free Energy | Total Energy | EvdW 1 Energy | Electrostatic Energy | Intermolecular Energy |
|---|---|---|---|---|---|
| MaBgl3–CBI | −7.10 | 24.29 | −10.78 | −34.85 | −45.66 |
| MaBgl3–BGC | −6.18 | −3.66 | −6.95 | −29.88 | −36.83 |
| Complex | Hydrogen Interactions | Hydrophobic Interaction |
|---|---|---|
| MaBgl3–CBI | Trp31, Asp81, Arg444 | Arg16, Tyr28, Leu53, His198, Ala271, Ile273, Met274 |
| MaBgl3–BGC | Asp81, Asp270, Phe443 | Ile51, Arg155, His198, Ala271, Arg444 |
| Components (kcal/mol) | Complexes | |
|---|---|---|
| MM–GBSA | MaBgl3–CBI | MaBgl3–BGC |
| 1 EvdW | −23.99 (±3.74) | −10.50 (±2.98) |
| 2 Eele | −80.98 (±9.78) | −64.74 (±6.26) |
| 3 Gsolv | 84.22 (±6.88) | 64.80 (±4.20) |
| 4 Enon-polar | −4.95 (±0.22) | −3.05 (±0.18) |
| 5 ΔGgas | −104.97 (±8.9) | −75.24 (±5.49) |
| 6 ΔGsolv | 79.28 (±6.79) | 61.76 (±4.16) |
| 7 ΔGbind | −25.69 (±4.69) | −13.48 (±2.89) |
| MM–PBSA | ||
| 1 EvdW | −23.99 (±3.73) | −10.50 (±2.98) |
| 2 Eele | −80.98 (±9.78) | −64.74 (±6.27) |
| 3 Gsolv | 95.58 (±8.96) | 66.86 (±4.59) |
| 4 Enon-polar | −5.20 (±0.17) | −3.33 (±0.14) |
| 5 ΔGgas | −104.97 (±8.90) | −75.29 (±5.49) |
| 6 ΔGsolv | 90.38 (±8.92) | 63.52 (±4.56) |
| 7 ΔGbind | −14.59 (±5.33) | −11.71 (±3.69) |
| SIE | ||
| 1 EvdW | −24.38 (±3.6) | −10.30 (±3.14) |
| 8 Ecoul | −35.86 (±4.12) | −28.81 (±2.88) |
| 9 ΔGR | 36.35 (±3.13) | 28.87 (±2.61) |
| 10 ΔGnonpol | −6.63 (±0.48) | −4.38 (±0.59) |
| 11 ΔGpol | −2.89 (±0.00) | −2.89 (±0.00) |
| 7 ΔGbind | −6.03 (±0.38) | −4.42 (±0.34) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serra, G.M.; Siqueira, A.S.; de Molfetta, F.A.; Santos, A.V.; Xavier, L.P. In Silico Analysis of a GH3 β-Glucosidase from Microcystis aeruginosa CACIAM 03. Microorganisms 2023, 11, 998. https://doi.org/10.3390/microorganisms11040998
Serra GM, Siqueira AS, de Molfetta FA, Santos AV, Xavier LP. In Silico Analysis of a GH3 β-Glucosidase from Microcystis aeruginosa CACIAM 03. Microorganisms. 2023; 11(4):998. https://doi.org/10.3390/microorganisms11040998
Chicago/Turabian StyleSerra, Gustavo Marques, Andrei Santos Siqueira, Fábio Alberto de Molfetta, Agenor Valadares Santos, and Luciana Pereira Xavier. 2023. "In Silico Analysis of a GH3 β-Glucosidase from Microcystis aeruginosa CACIAM 03" Microorganisms 11, no. 4: 998. https://doi.org/10.3390/microorganisms11040998
APA StyleSerra, G. M., Siqueira, A. S., de Molfetta, F. A., Santos, A. V., & Xavier, L. P. (2023). In Silico Analysis of a GH3 β-Glucosidase from Microcystis aeruginosa CACIAM 03. Microorganisms, 11(4), 998. https://doi.org/10.3390/microorganisms11040998