
Differences in Soil Microbial Communities between Healthy and Diseased Lycium barbarum cv. Ningqi-5 Plants with Root Rot


Abstract
1. Introduction
2. Materials and Methods
2.1. Location Description
2.2. Sample Collection
2.3. Sample Processing
2.4. DNA Extraction and PCR Amplification
2.5. Illumina MiSeq Sequencing
2.6. Statistical Analysis
3. Results
3.1. Microbial Alpha Diversity of Soil of Ningqi-5
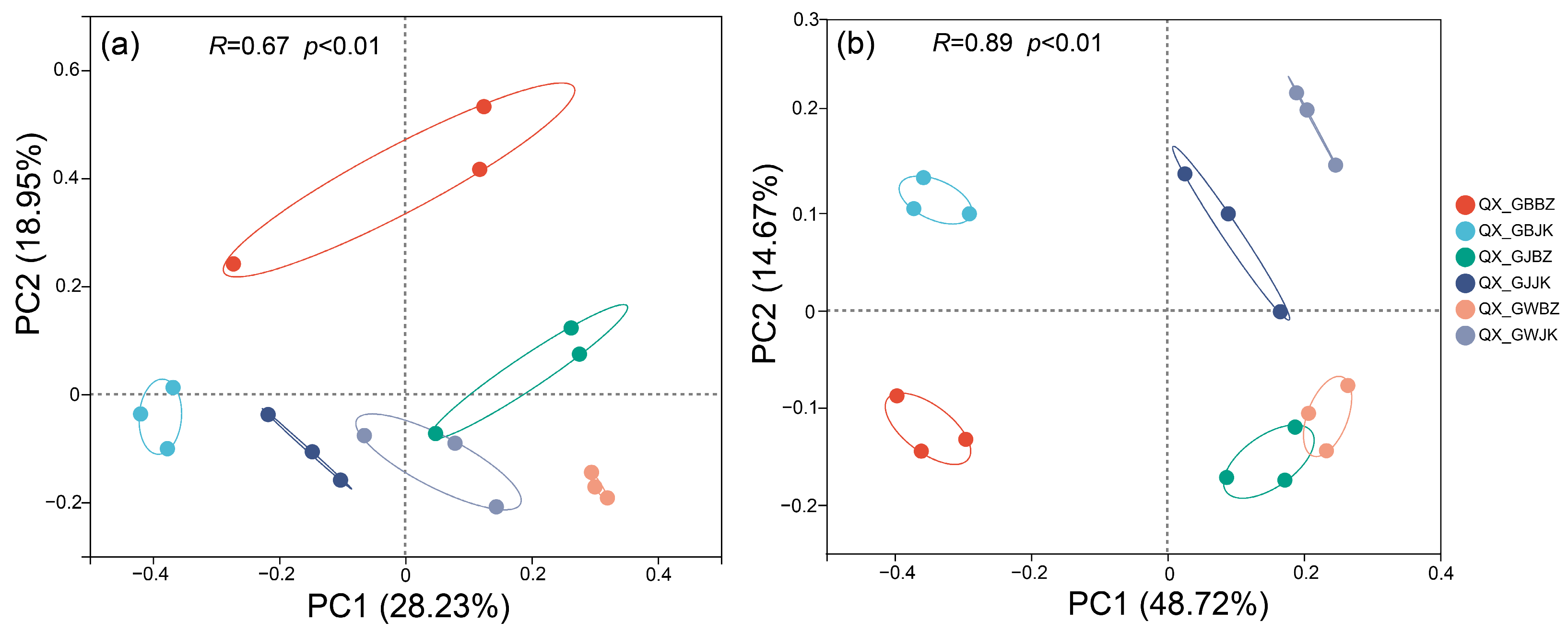
3.2. Principal Coordinate Analysis Based on OTU Level
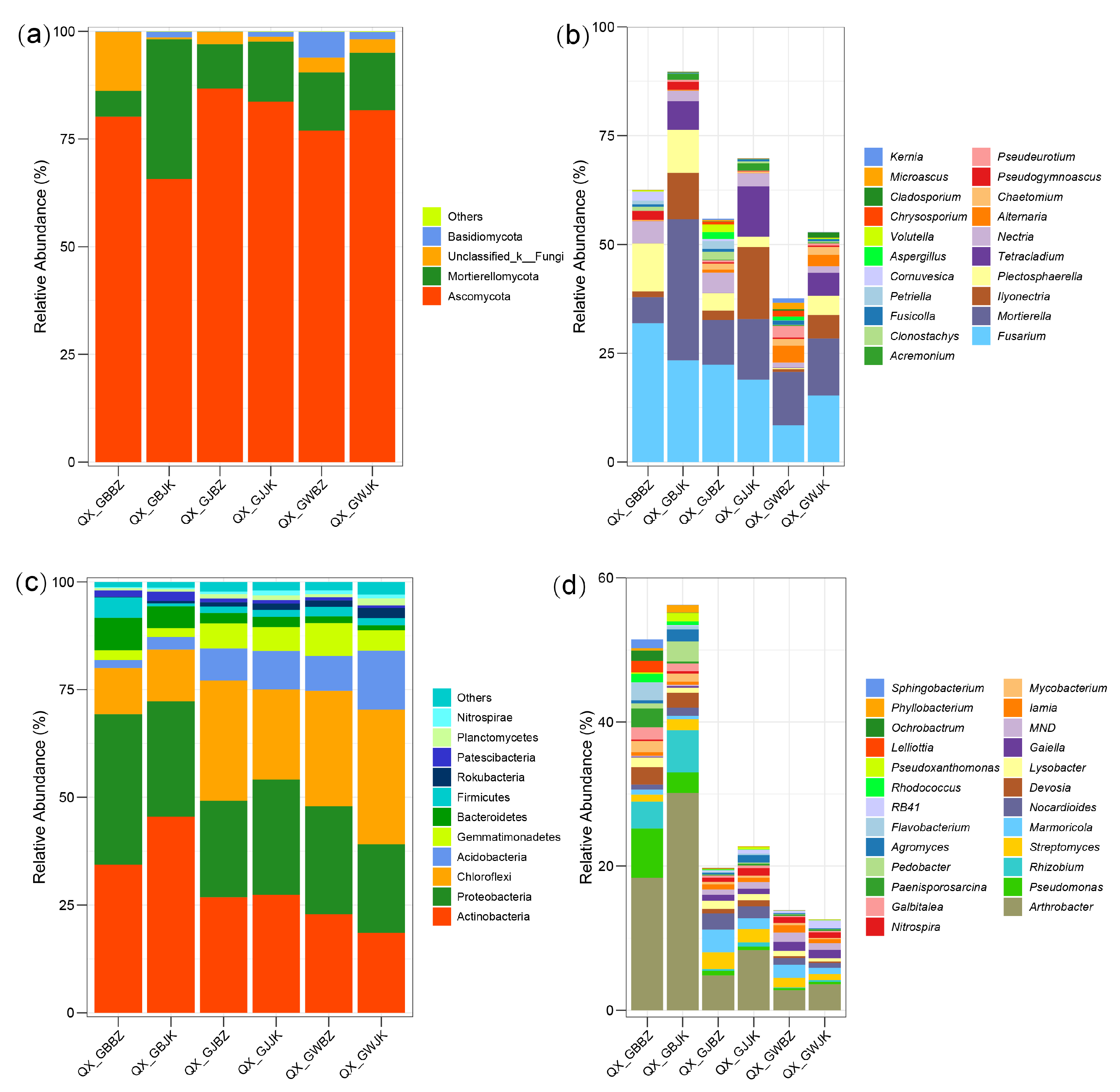
3.3. Analysis of Microbial Community Composition
3.4. Significant Difference between Each Group
3.5. Prediction of Microbial Function
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Tian, L.; Ou, J.; Sun, X.; Miao, Y.; Pei, J.; Zhao, L.; Huang, L. The discovery of pivotal fungus and major determinant factor shaping soil microbial community composition associated with rot root of American ginseng. Plant Signal. Behav. 2021, 16, 1952372. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhang, D.; Wen, H.; Wang, Q.; Peng, C.; Gao, J. Soil fungal biodiversity and pathogen identification of rotten disease in Aconitum carmichaelii (Fuzi) roots. PLoS ONE 2018, 13, e0205891. [Google Scholar] [CrossRef] [PubMed]
- Song, X.H.; Wang, Y.; Li, L.Y.; Tan, J. Research on bacteria microecology in root rot rhizosphere soil of Coptis chinensis produced in Shizhu city. Zhongguo J. Chin. Mater. Med. 2017, 42, 1304–1311. [Google Scholar]
- Bardgett, R.D.; van der Putten, W.H. Belowground biodiversity and ecosystem functioning. Nature 2014, 515, 505–511. [Google Scholar] [CrossRef]
- Wagg, C.; Bender, S.F.; Widmer, F.; van der Heijden, M.G. Soil biodiversity and soil community composition determine ecosystem multifunctionality. Proc. Natl. Acad. Sci. USA 2014, 111, 5266–5270. [Google Scholar] [CrossRef] [PubMed]
- Ritpitakphong, U.; Falquet, L.; Vimoltust, A.; Berger, A.; Métraux, J.P.; L’Haridon, F. The microbiome of the leaf surface of Arabidopsis protects against a fungal pathogen. New Phytol. 2016, 210, 1033–1043. [Google Scholar] [CrossRef]
- Haney, C.H.; Samuel, B.S.; Bush, J.; Ausubel, F.M. Associations with rhizosphere bacteria can confer an adaptive advantage to plants. Nat. Plants 2015, 1, 15051. [Google Scholar] [CrossRef]
- Usyskin-Tonne, A.; Hadar, Y.; Yermiyahu, U.; Minz, D. Elevated CO(2) and nitrate levels increase wheat root-associated bacterial abundance and impact rhizosphere microbial community composition and function. ISME J. 2021, 15, 1073–1084. [Google Scholar] [CrossRef]
- Müller, D.B.; Vogel, C.; Bai, Y.; Vorholt, J.A. The Plant Microbiota: Systems-Level Insights and Perspectives. Annu. Rev. Genet. 2016, 50, 211–234. [Google Scholar] [CrossRef]
- Gonzalez, A.; King, A.; Robeson, M.S., 2nd; Song, S.; Shade, A.; Metcalf, J.L.; Knight, R. Characterizing microbial communities through space and time. Curr. Opin. Biotechnol. 2012, 23, 431–436. [Google Scholar] [CrossRef]
- İnceoğlu, Ö.; Falcão Salles, J.; van Elsas, J.D. Soil and cultivar type shape the bacterial community in the potato rhizosphere. Microb. Ecol. 2012, 63, 460–470. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.; Bauer, R.; Melchart, D.; Xiao, P.G.; Staudinger, A. Chromatographic Fingerprint Analysis of Herbal Medicines; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Chen, J.Z.; Lu, X.; Hu, Y.Q.; Guo, H.H.; Ma, X.L.; Guo, X.; Jiang, Z.B.; Wang, F. Research progress on chemical constituents and pharmacological studies on root bark of Lycium barbarum. China J. Chin. Mater. Med. 2021, 46, 3066–3075. [Google Scholar]
- Masci, A.; Carradori, S.; Casadei, M.A.; Paolicelli, P.; Petralito, S.; Ragno, R.; Cesa, S. Lycium barbarum polysaccharides: Extraction, purification, structural characterisation and evidence about hypoglycaemic and hypolipidaemic effects. A review. Food Chem. 2018, 254, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Guo, S.; Zhang, F.; Yan, H.; Qian, D.W.; Shang, E.X.; Wang, H.Q.; Duan, J.A. Nutritional components characterization of Goji berries from different regions in China. J. Pharm. Biomed. Anal. 2021, 195, 113859. [Google Scholar] [CrossRef]
- Cassileth, B. Lycium (Lycium barbarum). Oncology 2010, 24, 1353. [Google Scholar]
- Qian, D.; Zhao, Z.Y.; Ma, S.; Yang, G.; Zhong, J.Y.; Zang, C.X. Analysis of characteristics and problems of international trade of wolfberry in China. Zhongguo Zhong Yao Za Zhi = China J. Chin. Mater. Med 2019, 44, 2880–2885. [Google Scholar]
- Bai, L.; Li, X.; Cao, Y.; Song, Z.; Ma, K.; Fan, Y.; Ma, M. Fusarium culmorum and Fusarium equiseti causing root rot Disease on Lycium barbarum (Goji Berry) in China. Plant Dis. 2020, 104, 3066–3067. [Google Scholar] [CrossRef]
- Tyler, B.M. Phytophthora sojae: Root rot pathogen of soybean and model oomycete. Mol. Plant Pathol. 2007, 8, 1–8. [Google Scholar] [CrossRef]
- Nyandoro, R.; Chang, K.F.; Hwang, S.F.; Ahmed, H.U.; Turnbull, G.D.; Strelkov, S.E. Management of root rot of soybean in Alberta with fungicide seed treatments and genetic resistance. Can. J. Plant Sci. 2019, 99, 499–509. [Google Scholar] [CrossRef]
- Li, M.J.; Pan, X.; Liu, Z.; Wu, L.J.; Zhang, Y.; Wang, Q. First Report of Root Rot Caused by Fusarium equiseti on Ginseng (Panax ginseng) in China. Plant Dis. 2020, 104, 2294. [Google Scholar] [CrossRef]
- Fromme, F.D.; Thomas, H.E. The root-rot disease of the apple in virginia. Science 1917, 45, 93. [Google Scholar] [CrossRef] [PubMed]
- Reglinski, T.; Spiers, T.M.; Dick, M.A.; Taylor, J.T.; Gardner, J. Management of phytophthora root rot in radiata pine seedlings. Plant Pathol. 2009, 58, 723–730. [Google Scholar] [CrossRef]
- Fang, X.L.; Phillips, D.; Li, H.; Sivasithamparam, K.; Barbetti, M.J. Severity of crown and root diseases of strawberry and associated fungal and oomycete pathogens in Western Australia. Australas. Plant Pathol. 2011, 40, 109–119. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Wu, Z.; Hao, Z.; Zeng, Y.; Guo, L.; Huang, L.; Chen, B. Molecular characterization of microbial communities in the rhizosphere soils and roots of diseased and healthy Panax notoginseng. Antonie Van Leeuwenhoek 2015, 108, 1059–1074. [Google Scholar] [CrossRef]
- Shi, S.; Richardson, A.E.; O’Callaghan, M.; DeAngelis, K.M.; Jones, E.E.; Stewart, A.; Firestone, M.K.; Condron, L.M. Effects of selected root exudate components on soil bacterial communities. FEMS Microbiol. Ecol. 2011, 77, 600–610. [Google Scholar] [CrossRef]
- Bais, H.P.; Weir, T.L.; Perry, L.G.; Gilroy, S.; Vivanco, J.M. The role of root exudates in rhizosphere interactions with plants and other organisms. Annu. Rev. Plant Biol. 2006, 57, 233–266. [Google Scholar] [CrossRef]
- Hartmann, A.; Schmid, M.; van Tuinen, D.; Berg, G. Plant-driven selection of microbes. Plant Soil 2009, 321, 235–257. [Google Scholar] [CrossRef]
- Gilroy, S.; Jones, D.L. Through form to function: Root hair development and nutrient uptake. Trends Plant Sci. 2000, 5, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Lei, F.; Fu, J.; Zhou, R.; Wang, D.; Zhang, A.; Ma, W.; Zhang, L. Chemotactic response of Ginseng bacterial soft-rot to Ginseng root exudates. Saudi J. Biol. Sci. 2017, 24, 1620–1625. [Google Scholar] [CrossRef] [PubMed]
- Tkacz, A.; Cheema, J.; Chandra, G.; Grant, A.; Poole, P.S. Stability and succession of the rhizosphere microbiota depends upon plant type and soil composition. ISME J. 2015, 9, 2349–2359. [Google Scholar] [CrossRef]
- Kong, H.G.; Kim, B.K.; Song, G.C.; Lee, S.; Ryu, C.M. Aboveground Whitefly Infestation-Mediated Reshaping of the Root Microbiota. Front. Microbiol. 2016, 7, 1314. [Google Scholar] [CrossRef]
- Berendsen, R.L.; Vismans, G.; Yu, K.; Song, Y.; de Jonge, R.; Burgman, W.P.; Burmølle, M.; Herschend, J.; Bakker, P.; Pieterse, C.M.J. Disease-induced assemblage of a plant-beneficial bacterial consortium. ISME J. 2018, 12, 1496–1507. [Google Scholar] [CrossRef]
- Vives-Peris, V.; de Ollas, C.; Gómez-Cadenas, A.; Pérez-Clemente, R.M. Root exudates: From plant to rhizosphere and beyond. Plant Cell Rep. 2020, 39, 3–17. [Google Scholar] [CrossRef]
- Bertrand, H.; Nalin, R.; Bally, R.; Cleyet-Marel, J.C. Isolation and identification of the most efficient plant growth-promoting bacteria associated with canola (Brassica napus). Biol. Fertil. Soils 2001, 33, 152–156. [Google Scholar] [CrossRef]
- Bakker, M.G.; Schlatter, D.C.; Otto-Hanson, L.; Kinkel, L.L. Diffuse symbioses: Roles of plant-plant, plant-microbe and microbe-microbe interactions in structuring the soil microbiome. Mol. Ecol. 2014, 23, 1571–1583. [Google Scholar] [CrossRef]
- Wen, Z.; Zeng, Z.; Ren, F.; Asiegbu, F.O. The Conifer Root and Stem Rot Pathogen (Heterobasidion parviporum): Effectome Analysis and Roles in Interspecific Fungal Interactions. Microorganisms 2019, 7, 658. [Google Scholar] [CrossRef] [PubMed]
- Beimforde, C.; Feldberg, K.; Nylinder, S.; Rikkinen, J.; Tuovila, H.; Dörfelt, H.; Gube, M.; Jackson, D.J.; Reitner, J.; Seyfullah, L.J.; et al. Estimating the Phanerozoic history of the Ascomycota lineages: Combining fossil and molecular data. Mol. Phylogenet. Evol. 2014, 78, 386–398. [Google Scholar] [CrossRef]
- Chittem, K.; Mathew, F.M.; Gregoire, M.; Lamppa, R.S.; Chang, Y.W.; Markell, S.G.; Bradley, C.A.; Barasubiye, T.; Goswami, R.S. Identification and characterization of Fusarium spp. associated with root rots of field pea in North Dakota. Eur. J. Plant Pathol. 2015, 143, 641–649. [Google Scholar] [CrossRef]
- Arias, M.M.; Leandro, L.F.; Munkvold, G.P. Aggressiveness of Fusarium species and impact of root infection on growth and yield of soybeans. Phytopathology 2013, 103, 822–832. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Chen, L.; Redmile-Gordon, M.; Zhang, J.; Zhang, C.; Ning, Q.; Li, W. Mortierella elongata’s roles in organic agriculture and crop growth promotion in a mineral soil. Land Degrad. Dev. 2018, 29, 1642–1651. [Google Scholar] [CrossRef]
- Han, L.; Zhou, X.; Zhao, Y.; Wu, L.; Ping, X.; He, Y.; Peng, S.; He, X.; Du, Y. First report of Plectosphaerella plurivora causing root rot disease in Panax notoginseng in China. J. Phytopathol. 2020, 168, 375–379. [Google Scholar] [CrossRef]
- Alam, M.W.; Malik, A.; Rehman, A.; Sarwar, M.; Mehboob, S. First report of potato wilt caused by Plectosphaerella cucumerina in Pakistan. J. Plant Pathol. 2021, 103, 687. [Google Scholar] [CrossRef]
- Little, A.E.; Robinson, C.J.; Peterson, S.B.; Raffa, K.F.; Handelsman, J. Rules of engagement: Interspecies interactions that regulate microbial communities. Annu. Rev. Microbiol. 2008, 62, 375–401. [Google Scholar] [CrossRef]
- Wei, Z.; Yang, T.; Friman, V.P.; Xu, Y.; Shen, Q.; Jousset, A. Trophic network architecture of root-associated bacterial communities determines pathogen invasion and plant health. Nat. Commun. 2015, 6, 8413. [Google Scholar] [CrossRef]
- Wang, G.; Bei, S.; Li, J.; Bao, X.; Zhang, J.; Schultz, P.A.; Li, H.; Li, L.; Zhang, F.; Bever, J.D.; et al. Soil microbial legacy drives crop diversity advantage: Linking ecological plant-soil feedback with agricultural intercropping. J. Appl. Ecol. 2021, 58, 496–506. [Google Scholar] [CrossRef]
- Peay, K.G.; Garbelotto, M.; Bruns, T.D. Evidence of dispersal limitation in soil microorganisms: Isolation reduces species richness on mycorrhizal tree islands. Ecology 2010, 91, 3631–3640. [Google Scholar] [CrossRef] [PubMed]
- Lekberg, Y.; Schnoor, T.; Kjoller, R.; Gibbons, S.M.; Hansen, L.H.; Al-Soud, W.A.; Sorensen, S.J.; Rosendahl, S. 454-sequencing reveals stochastic local reassembly and high disturbance tolerance within arbuscular mycorrhizal fungal communities. J. Ecol. 2012, 100, 151–160. [Google Scholar] [CrossRef]
- Wang, J.; Shen, J.; Wu, Y.; Tu, C.; Soininen, J.; Stegen, J.C.; He, J.; Liu, X.; Zhang, L.; Zhang, E. Phylogenetic beta diversity in bacterial assemblages across ecosystems: Deterministic versus stochastic processes. ISME J. 2013, 7, 1310–1321. [Google Scholar] [CrossRef] [PubMed]
- Partida-Martinez, L.P.; Hertweck, C. Pathogenic fungus harbours endosymbiotic bacteria for toxin production. Nature 2005, 437, 884–888. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, M.; Frey, B.; Mayer, J.; Mäder, P.; Widmer, F. Distinct soil microbial diversity under long-term organic and conventional farming. ISME J. 2015, 9, 1177–1194. [Google Scholar] [CrossRef]
- Lupatini, M.; Korthals, G.W.; de Hollander, M.; Janssens, T.K.; Kuramae, E.E. Soil Microbiome Is More Heterogeneous in Organic Than in Conventional Farming System. Front. Microbiol. 2016, 7, 2064. [Google Scholar] [CrossRef]
- Lori, M.; Symnaczik, S.; Mäder, P.; De Deyn, G.; Gattinger, A. Organic farming enhances soil microbial abundance and activity-A meta-analysis and meta-regression. PLoS ONE 2017, 12, e0180442. [Google Scholar] [CrossRef]
- Kong, H.G.; Song, G.C.; Ryu, C.M. Inheritance of seed and rhizosphere microbial communities through plant-soil feedback and soil memory. Environ. Microbiol. Rep. 2019, 11, 479–486. [Google Scholar] [CrossRef]
- Hartman, K.; van der Heijden, M.G.A.; Wittwer, R.A.; Banerjee, S.; Walser, J.C.; Schlaeppi, K. Correction to: Cropping practices manipulate abundance patterns of root and soil microbiome members paving the way to smart farming. Microbiome 2020, 8, 66. [Google Scholar] [CrossRef]
- van der Heijden, M.G.; de Bruin, S.; Luckerhoff, L.; van Logtestijn, R.S.; Schlaeppi, K. A widespread plant-fungal-bacterial symbiosis promotes plant biodiversity, plant nutrition and seedling recruitment. ISME J. 2016, 10, 389–399. [Google Scholar] [CrossRef]
- Wardle, D.A.; Bardgett, R.D.; Klironomos, J.N.; Setälä, H.; van der Putten, W.H.; Wall, D.H. Ecological linkages between aboveground and belowground biota. Science 2004, 304, 1629–1633. [Google Scholar] [CrossRef] [PubMed]
- Wardle, D.A. The influence of biotic interactions on soil biodiversity. Ecol. Lett. 2006, 9, 870–886. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Chen, Y.; Lin, Z.; Tuo, Y.; Li, H.; Wang, Y. Differences in Soil Microbial Community Composition Between Suppressive and Root Rot-Conducive in Tobacco Fields. Curr. Microbiol. 2021, 78, 624–633. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, J.E.; Kent, A.D.; Brisson, V.L.; Gaudin, A.C.M. Agricultural management and plant selection interactively affect rhizosphere microbial community structure and nitrogen cycling. Microbiome 2019, 7, 146. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rhizoplane | Rhizosphere | Root Zone | |
|---|---|---|---|
| Diseased plants | QX_GBBZ (QX111, QX112, QX113) | QX_GJBZ (QX121, QX122, QX123) | QX_GWBZ (QX131, QX132, QX133) |
| Healthy plants | QX_GBJK (QX114, QX115, QX116) | QX_GJJK (QX124, QX125, QX126) | QX_GWBZ (QX134, QX135, QX136) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, C.; An, Y.; Du, Z.; Gao, H.; Su, J.; Xu, C. Differences in Soil Microbial Communities between Healthy and Diseased Lycium barbarum cv. Ningqi-5 Plants with Root Rot. Microorganisms 2023, 11, 694. https://doi.org/10.3390/microorganisms11030694
Jia C, An Y, Du Z, Gao H, Su J, Xu C. Differences in Soil Microbial Communities between Healthy and Diseased Lycium barbarum cv. Ningqi-5 Plants with Root Rot. Microorganisms. 2023; 11(3):694. https://doi.org/10.3390/microorganisms11030694
Chicago/Turabian StyleJia, Chenbo, Yingrui An, Zhongyu Du, Huihui Gao, Jianyu Su, and Chunyan Xu. 2023. "Differences in Soil Microbial Communities between Healthy and Diseased Lycium barbarum cv. Ningqi-5 Plants with Root Rot" Microorganisms 11, no. 3: 694. https://doi.org/10.3390/microorganisms11030694
APA StyleJia, C., An, Y., Du, Z., Gao, H., Su, J., & Xu, C. (2023). Differences in Soil Microbial Communities between Healthy and Diseased Lycium barbarum cv. Ningqi-5 Plants with Root Rot. Microorganisms, 11(3), 694. https://doi.org/10.3390/microorganisms11030694