

Metagenomic Sequencing of Lloviu Virus from Dead Schreiber’s Bats in Bosnia and Herzegovina
 ,
,  , ,
, ,  , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
3. Results
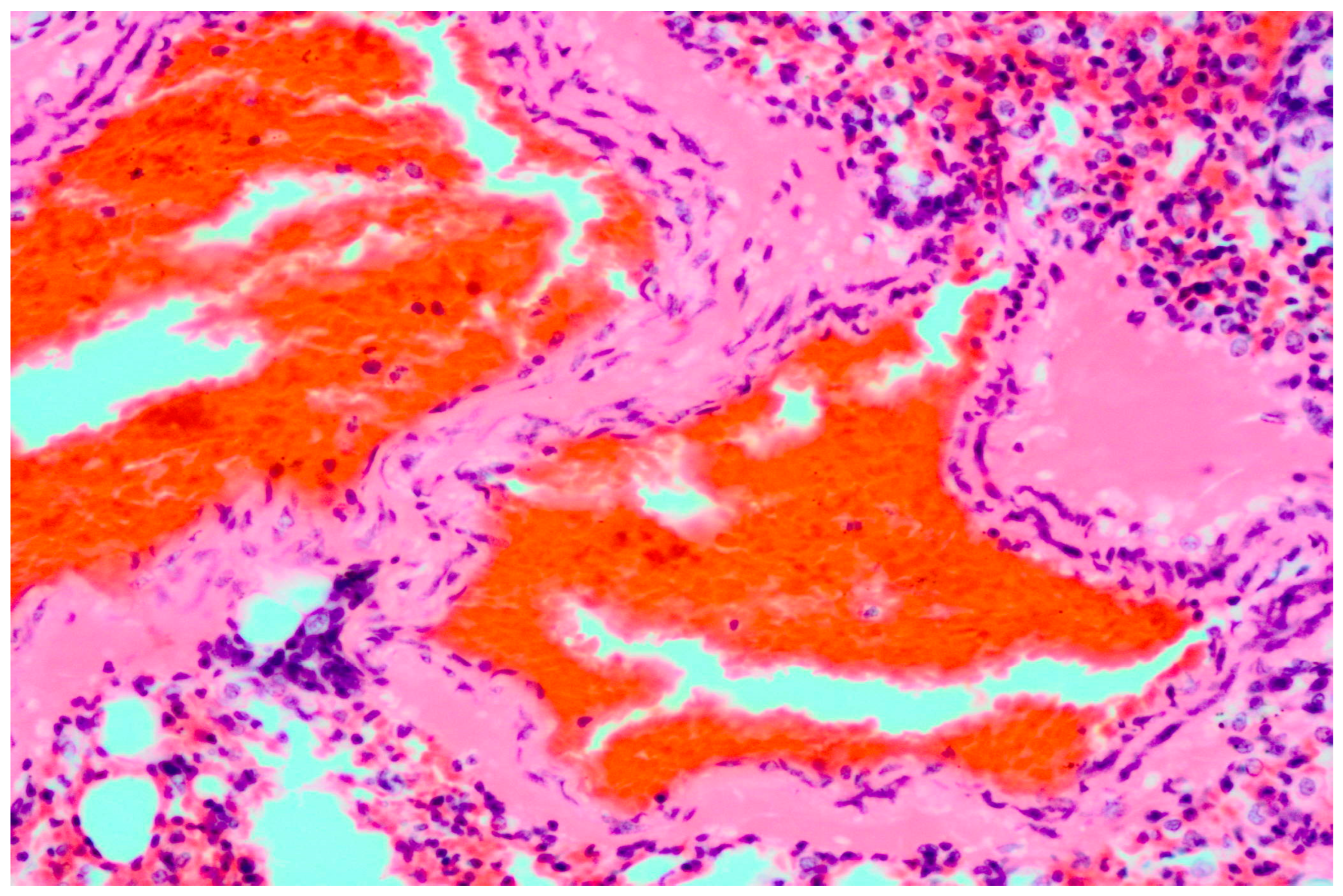
3.1. Pathology
3.2. Sequencing and Phylogenetic Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, J.; Anderson, D.E.; Halpin, K.; Hong, X.; Chen, H.; Walker, S.; Valdeter, S.; Van Der Heide, B.; Neave, M.J.; Bingham, J.; et al. A New Hendra Virus Genotype Found in Australian Flying Foxes. Virol. J. 2021, 18, 197. [Google Scholar] [CrossRef] [PubMed]
- Luby, S.P. The Pandemic Potential of Nipah Virus. Antivir. Res. 2013, 100, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Hayman, D.T.S. Biannual Birth Pulses Allow Filoviruses to Persist in Bat Populations. Proc. R. Soc. B 2015, 282, 20142591. [Google Scholar] [CrossRef]
- Koch, L.K.; Cunze, S.; Kochmann, J.; Klimpel, S. Bats as Putative Zaire Ebolavirus Reservoir Hosts and Their Habitat Suitability in Africa. Sci. Rep. 2020, 10, 14268. [Google Scholar] [CrossRef] [PubMed]
- Banyard, A.C.; Hayman, D.; Johnson, N.; McElhinney, L.; Fooks, A.R. Bats and Lyssaviruses. In Advances in Virus Research; Elsevier: Amsterdam, The Netherlands, 2011; Volume 79, pp. 239–289. ISBN 978-0-12-387040-7. [Google Scholar]
- Temmam, S.; Vongphayloth, K.; Baquero, E.; Munier, S.; Bonomi, M.; Regnault, B.; Douangboubpha, B.; Karami, Y.; Chrétien, D.; Sanamxay, D.; et al. Bat Coronaviruses Related to SARS-CoV-2 and Infectious for Human Cells. Nature 2022, 604, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Memish, Z.A.; Mishra, N.; Olival, K.J.; Fagbo, S.F.; Kapoor, V.; Epstein, J.H.; AlHakeem, R.; Durosinloun, A.; Al Asmari, M.; Islam, A.; et al. Middle East Respiratory Syndrome Coronavirus in Bats, Saudi Arabia. Emerg. Infect. Dis. 2013, 19, 1819–1823. [Google Scholar] [CrossRef] [PubMed]
- Jamiu, A.; Sabiu, S.; Unnisa, A.; Emran, T.B. Recent Outbreaks of Marburg Virus in Africa: An Urgent Call for Public Concern. Int. J. Surg. Glob. Health 2023, 6, e0151. [Google Scholar] [CrossRef]
- Negredo, A.; Palacios, G.; Vázquez-Morón, S.; González, F.; Dopazo, H.; Molero, F.; Juste, J.; Quetglas, J.; Savji, N.; De La Cruz Martínez, M.; et al. Discovery of an Ebolavirus-Like Filovirus in Europe. PLoS Pathog. 2011, 7, e1002304. [Google Scholar] [CrossRef]
- O’Shea, T.J.; Cryan, P.M.; Hayman, D.T.S.; Plowright, R.K.; Streicker, D.G. Multiple Mortality Events in Bats: A Global Review. Mammal. Rev. 2016, 46, 175–190. [Google Scholar] [CrossRef]
- Ramírez De Arellano, E.; Sanchez-Lockhart, M.; Perteguer, M.J.; Bartlett, M.; Ortiz, M.; Campioli, P.; Hernández, A.; Gonzalez, J.; Garcia, K.; Ramos, M.; et al. First Evidence of Antibodies Against Lloviu Virus in Schreiber’s Bent-Winged Insectivorous Bats Demonstrate a Wide Circulation of the Virus in Spain. Viruses 2019, 11, 360. [Google Scholar] [CrossRef]
- Kemenesi, G.; Kurucz, K.; Dallos, B.; Zana, B.; Földes, F.; Boldogh, S.; Görföl, T.; Carroll, M.W.; Jakab, F. Re-Emergence of Lloviu Virus in Miniopterus schreibersii Bats, Hungary, 2016. Emerg. Microbes Infect. 2018, 7, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Kemenesi, G.; Tóth, G.E.; Mayora-Neto, M.; Scott, S.; Temperton, N.; Wright, E.; Mühlberger, E.; Hume, A.J.; Suder, E.L.; Zana, B.; et al. Isolation of Infectious Lloviu Virus from Schreiber’s Bats in Hungary. Nat. Commun. 2022, 13, 1706. [Google Scholar] [CrossRef] [PubMed]
- Tóth, G.E.; Hume, A.J.; Suder, E.L.; Zeghbib, S.; Ábrahám, Á.; Lanszki, Z.; Varga, Z.; Tauber, Z.; Földes, F.; Zana, B.; et al. Isolation and Genome Characterization of Lloviu Virus from Italian Schreibers’s Bats. Sci. Rep. 2023, 13, 11310. [Google Scholar] [CrossRef] [PubMed]
- Takadate, Y.; Manzoor, R.; Saito, T.; Kida, Y.; Maruyama, J.; Kondoh, T.; Miyamoto, H.; Ogawa, H.; Kajihara, M.; Igarashi, M.; et al. Receptor-Mediated Host Cell Preference of a Bat-Derived Filovirus, Lloviu Virus. Microorganisms 2020, 8, 1530. [Google Scholar] [CrossRef] [PubMed]
- Hume, A.J.; Heiden, B.; Olejnik, J.; Suder, E.L.; Ross, S.; Scoon, W.A.; Bullitt, E.; Ericsson, M.; White, M.R.; Turcinovic, J.; et al. Recombinant Lloviu Virus as a Tool to Study Viral Replication and Host Responses. PLoS Pathog. 2022, 18, e1010268. [Google Scholar] [CrossRef]
- Feagins, A.R.; Basler, C.F. Lloviu Virus VP24 and VP35 Proteins Function as Innate Immune Antagonists in Human and Bat Cells. Virology 2015, 485, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Görföl, T.; Tóth, G.E.; Boldogh, S.A.; Jakab, F.; Kemenesi, G. Lloviu Virus in Europe Is an Emerging Disease of Concern. EcoHealth 2022, 19, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Chrzastek, K.; Lee, D.; Smith, D.; Sharma, P.; Suarez, D.L.; Pantin-Jackwood, M.; Kapczynski, D.R. Use of Sequence-Independent, Single-Primer-Amplification (SISPA) for Rapid Detection, Identification, and Characterization of Avian RNA Viruses. Virology 2017, 509, 159–166. [Google Scholar] [CrossRef]
- Greninger, A.L.; Naccache, S.N.; Federman, S.; Yu, G.; Mbala, P.; Bres, V.; Stryke, D.; Bouquet, J.; Somasekar, S.; Linnen, J.M.; et al. Rapid Metagenomic Identification of Viral Pathogens in Clinical Samples by Real-Time Nanopore Sequencing Analysis. Genome Med. 2015, 7, 99. [Google Scholar] [CrossRef]
- Borges, V.; Pinheiro, M.; Pechirra, P.; Guiomar, R.; Gomes, J.P. INSaFLU: An Automated Open Web-Based Bioinformatics Suite “from-Reads” for Influenza Whole-Genome-Sequencing-Based Surveillance. Genome Med. 2018, 10, 46. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Feng, Y.; Zhang, H.; Xu, L.; Yang, W.; Zhang, Y.; Li, X.; Tu, C. Filovirus RNA in Fruit Bats, China. Emerg. Infect. Dis. 2015, 21, 1675–1677. [Google Scholar] [CrossRef] [PubMed]
- Mühlberger, E. Filovirus Replication and Transcription. Future Virol. 2007, 2, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Varsani, A.; Krupovic, M. Family Genomoviridae: 2021 Taxonomy Update. Arch. Virol. 2021, 166, 2911–2926. [Google Scholar] [CrossRef] [PubMed]
- Šimić, I.; Zorec, T.M.; Lojkić, I.; Krešić, N.; Poljak, M.; Cliquet, F.; Picard-Meyer, E.; Wasniewski, M.; Zrnčić, V.; Ćukušić, A.; et al. Viral Metagenomic Profiling of Croatian Bat Population Reveals Sample and Habitat Dependent Diversity. Viruses 2020, 12, 891. [Google Scholar] [CrossRef] [PubMed]
- Doutre, G.; Philippe, N.; Abergel, C.; Claverie, J.-M. Genome Analysis of the First Marseilleviridae Representative from Australia Indicates That Most of Its Genes Contribute to Virus Fitness. J. Virol. 2014, 88, 14340–14349. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.; Freuling, C.M.; Müller, T.; Wegelt, A.; Kooi, E.A.; Rasmussen, T.B.; Voller, K.; Marston, D.A.; Fooks, A.R.; Beer, M.; et al. Molecular Double-Check Strategy for the Identification and Characterization of European Lyssaviruses. J. Virol. Methods 2014, 203, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Shuey, M.M.; Drees, K.P.; Lindner, D.L.; Keim, P.; Foster, J.T. Highly Sensitive Quantitative PCR for the Detection and Differentiation of Pseudogymnoascus Destructans and Other Pseudogymnoascus Species. Appl. Environ. Microbiol. 2014, 80, 1726–1731. [Google Scholar] [CrossRef]
- Martin, B.; Hoenen, T.; Canard, B.; Decroly, E. Filovirus Proteins for Antiviral Drug Discovery: A Structure/Function Analysis of Surface Glycoproteins and Virus Entry. Antivir. Res. 2016, 135, 1–14. [Google Scholar] [CrossRef]
- Jun, S.-R.; Leuze, M.R.; Nookaew, I.; Uberbacher, E.C.; Land, M.; Zhang, Q.; Wanchai, V.; Chai, J.; Nielsen, M.; Trolle, T.; et al. Ebolavirus Comparative Genomics. FEMS Microbiol. Rev. 2015, 39, 764–778. [Google Scholar] [CrossRef]
- Kondoh, T.; Letko, M.; Munster, V.J.; Manzoor, R.; Maruyama, J.; Furuyama, W.; Miyamoto, H.; Shigeno, A.; Fujikura, D.; Takadate, Y.; et al. Single-Nucleotide Polymorphisms in Human NPC1 Influence Filovirus Entry into Cells. J. Infect. Dis. 2018, 218, S397–S402. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus Family | The Total Number of Reads | Source(s) of Reads (%) | ||
|---|---|---|---|---|
| Lungs with Pathohistological Changes | Lungs without Pathohistological Changes | Guano | ||
| Filoviridae | 68,749 | 100.00% | 0.00% | 0.00% |
| Flaviviridae | 23,562 | 0.00% | 0.00% | 100.00% |
| Microviridae | 7025 | 0.00% | 0.00% | 100.00% |
| Myoviridae | 5751 | 0.00% | 0.00% | 100.00% |
| Genomoviridae | 5644 | 10.64% | 0.00% | 89.35% |
| Baculoviridae | 3987 | 0.00% | 0.00% | 100.00% |
| Coronaviridae | 2410 | 20.29% | 24.86% | 54.85% |
| Reoviridae | 2402 | 37.51% | 46.36% | 16.13% |
| Podoviridae | 1743 | 27.71% | 29.95% | 42.34% |
| Papillomaviridae | 1721 | 0.01% | 0.05% | 99.94% |
| Picornaviridae | 1701 | 45.11% | 0.00% | 54.89% |
| Iflaviviridae | 1623 | 0.00% | 0.00% | 100.00% |
| Syphoviridae | 1520 | 26.49% | 28.35% | 45.19% |
| Parvoviridae | 1377 | 42.98% | 38.97% | 18.05% |
| Paramyxoviridae | 1068 | 0.13% | 0.07% | 99.80% |
| Marseilleviridae | 906 | 16.27% | 21.04% | 62.69% |
| Retroviridae | 743 | 18.87% | 0.00% | 81.13% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goletic, S.; Goletic, T.; Omeragic, J.; Supic, J.; Kapo, N.; Nicevic, M.; Skapur, V.; Rukavina, D.; Maksimovic, Z.; Softic, A.; et al. Metagenomic Sequencing of Lloviu Virus from Dead Schreiber’s Bats in Bosnia and Herzegovina. Microorganisms 2023, 11, 2892. https://doi.org/10.3390/microorganisms11122892
Goletic S, Goletic T, Omeragic J, Supic J, Kapo N, Nicevic M, Skapur V, Rukavina D, Maksimovic Z, Softic A, et al. Metagenomic Sequencing of Lloviu Virus from Dead Schreiber’s Bats in Bosnia and Herzegovina. Microorganisms. 2023; 11(12):2892. https://doi.org/10.3390/microorganisms11122892
Chicago/Turabian StyleGoletic, Sejla, Teufik Goletic, Jasmin Omeragic, Jovana Supic, Naida Kapo, Melisa Nicevic, Vedad Skapur, Dunja Rukavina, Zinka Maksimovic, Adis Softic, and et al. 2023. "Metagenomic Sequencing of Lloviu Virus from Dead Schreiber’s Bats in Bosnia and Herzegovina" Microorganisms 11, no. 12: 2892. https://doi.org/10.3390/microorganisms11122892
APA StyleGoletic, S., Goletic, T., Omeragic, J., Supic, J., Kapo, N., Nicevic, M., Skapur, V., Rukavina, D., Maksimovic, Z., Softic, A., & Alic, A. (2023). Metagenomic Sequencing of Lloviu Virus from Dead Schreiber’s Bats in Bosnia and Herzegovina. Microorganisms, 11(12), 2892. https://doi.org/10.3390/microorganisms11122892