
Insights into the Genomic Potential of a Methylocystis sp. from Amazonian Floodplain Sediments
 , , and
, , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sediment Sampling and DNA Extraction
2.2. Metagenomic Sequencing and Bioinformatic Analysis
3. Results and Discussion
3.1. Metagenome-Assembled Genome from Amazonian Floodplain Sediments
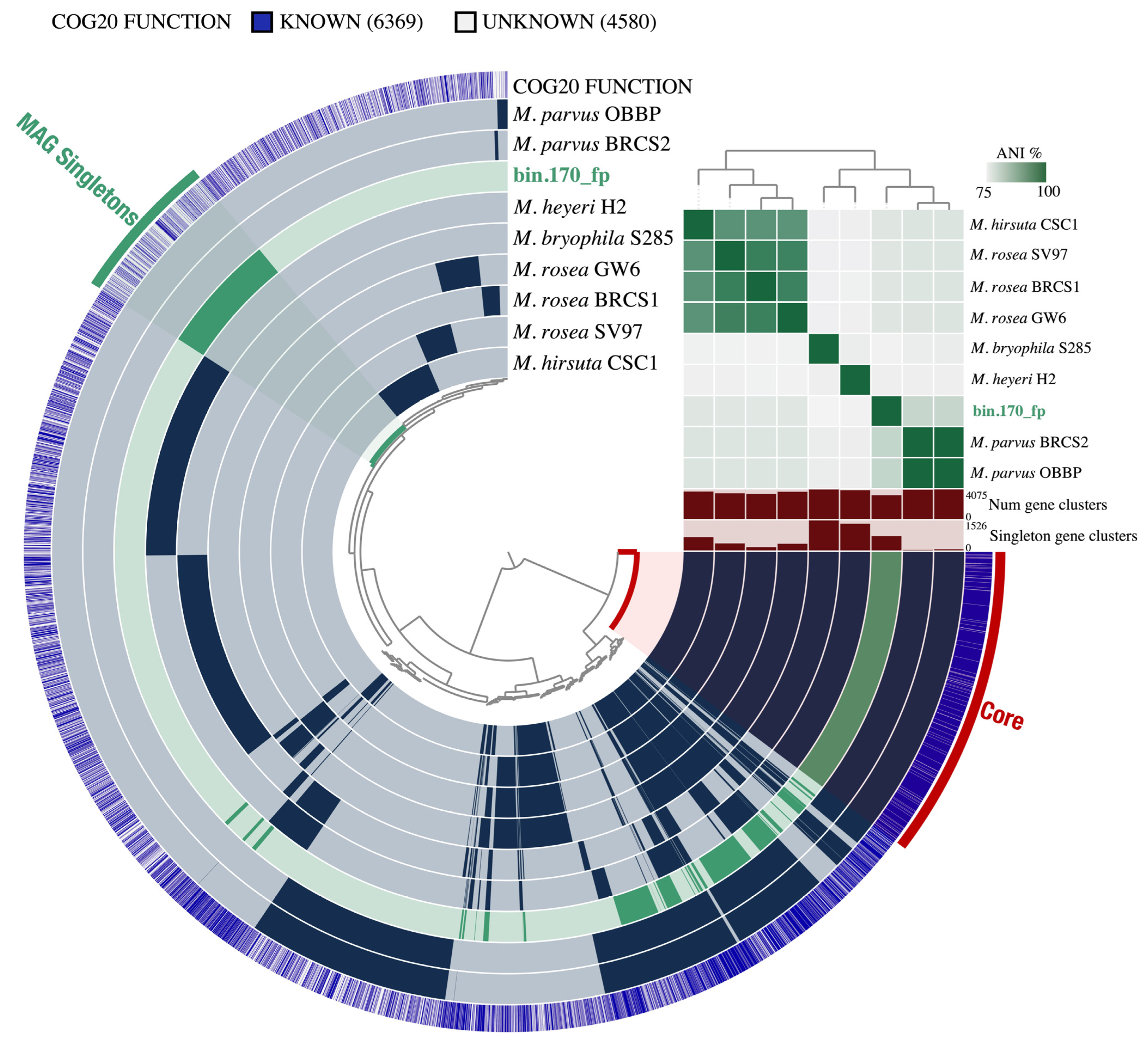
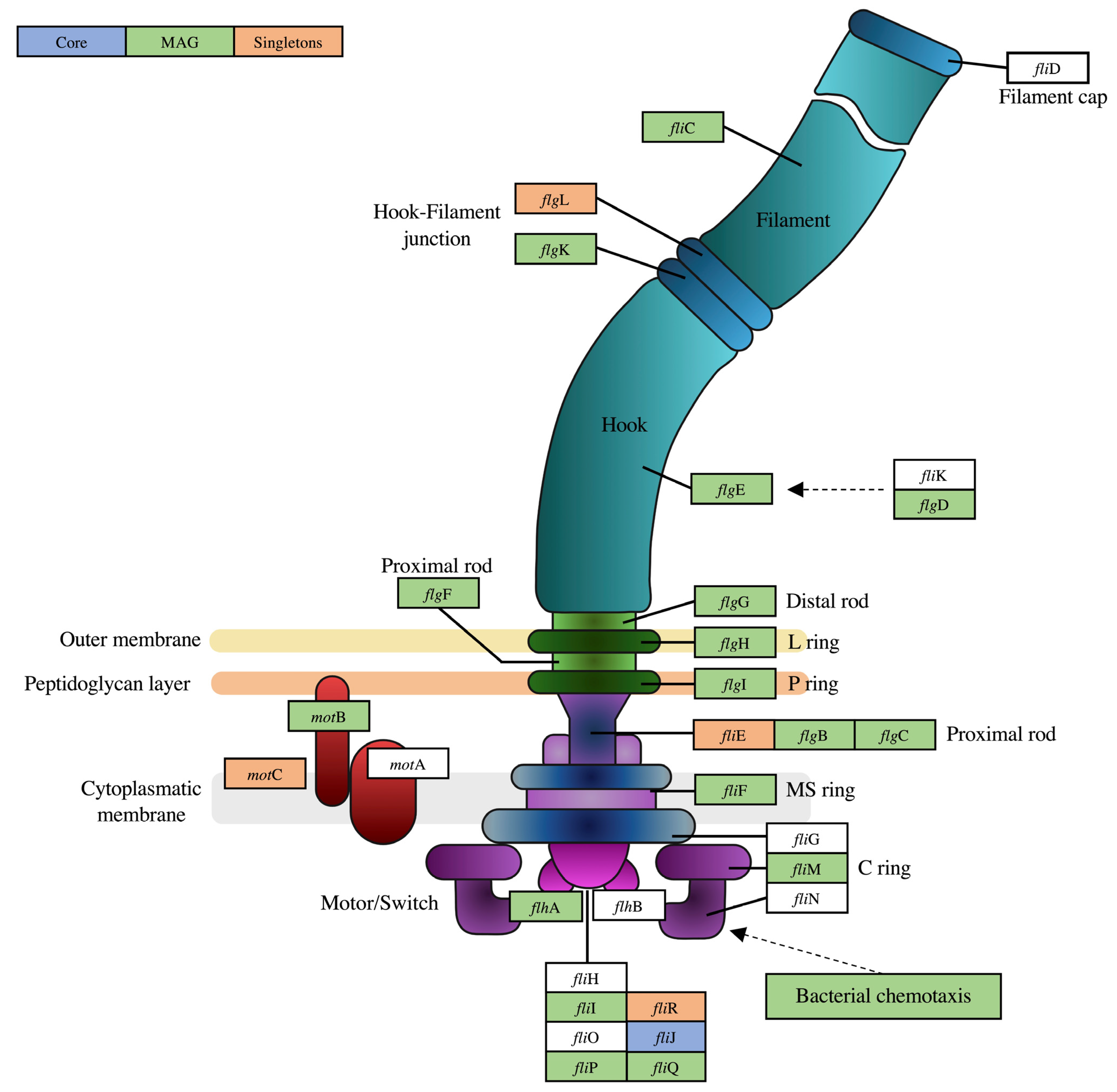
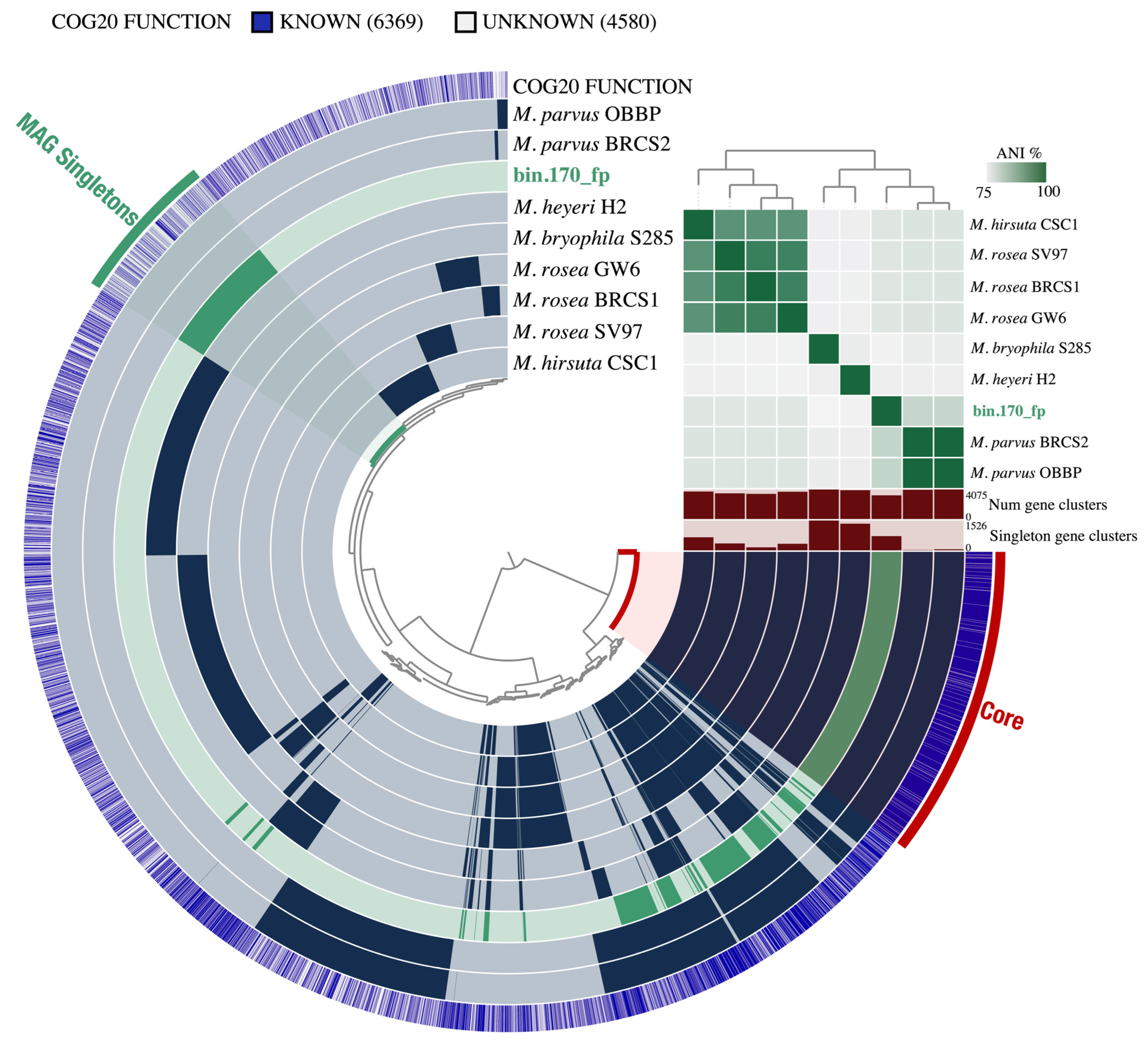
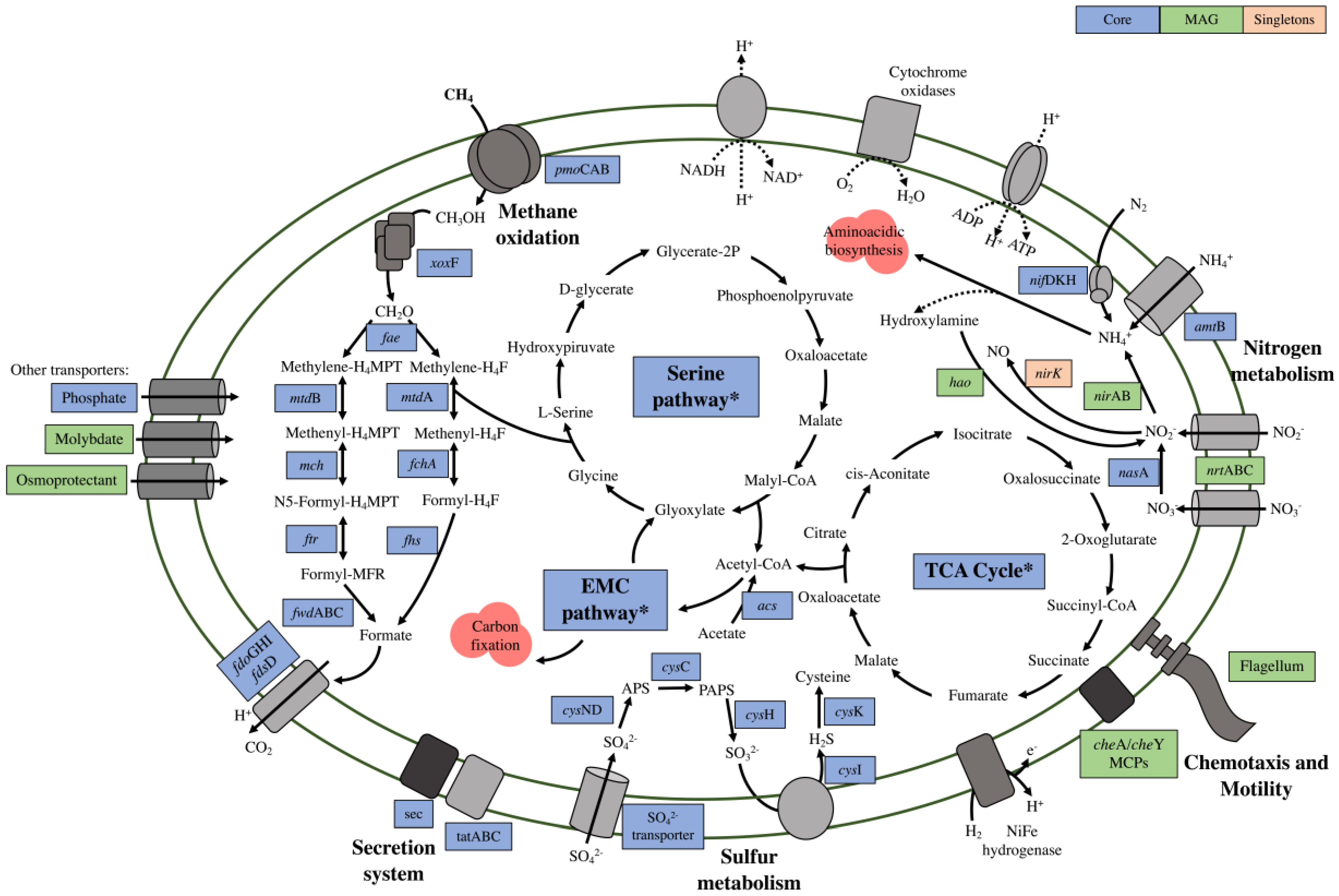
3.2. Functional Annotation and Pan-Genomic Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Turner, A.J.; Frankenberg, C.; Kort, E.A. Interpreting contemporary trends in atmospheric methane. Proc. Natl. Acad. Sci. USA 2019, 116, 2805–2813. [Google Scholar] [CrossRef] [PubMed]
- Angel, R.; Claus, P.; Conrad, R. Methanogenic archaea are globally ubiquitous in aerated soils and become active under wet anoxic conditions. ISME J. 2012, 6, 847–862. [Google Scholar] [CrossRef] [PubMed]
- Evans, P.N.; Boyd, J.A.; Leu, A.O.; Woodcroft, B.J.; Parks, D.H.; Hugenholtz, P.; Tyson, G.W. An evolving view of methane metabolism in the Archaea. Nat. Rev. Microbiol. 2019, 17, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Hess, L.L.; Melack, J.M.; Affonso, A.G.; Barbosa, C.; Gastil-Buhl, M.; Novo, E.M.L.M. Wetlands of the Lowland Amazon Basin: Extent, Vegetative Cover, and Dual-season Inundated Area as Mapped with JERS-1 Synthetic Aperture Radar. Wetlands 2015, 35, 745–756. [Google Scholar] [CrossRef]
- Pangala, S.R.; Enrich-Prast, A.; Basso, L.S.; Peixoto, R.B.; Bastviken, D.; Hornibrook, E.R.C.; Gatti, L.V.; Marotta, H.; Calazans, L.S.B.; Sakuragui, C.M.; et al. Large emissions from floodplain trees close the Amazon methane budget. Nature 2017, 552, 230–234. [Google Scholar] [CrossRef]
- Gedney, N.; Huntingford, C.; Comyn-Platt, E.; Wiltshire, A. Significant feedbacks of wetland methane release on climate change and the causes of their uncertainty. Environ. Res. Lett. 2019, 14, 084027. [Google Scholar] [CrossRef]
- Barbosa, P.M.; Melack, J.M.; Amaral, J.H.F.; MacIntyre, S.; Kasper, D.; Cortés, A.; Farjalla, V.F.; Forsberg, B.R. Dissolved methane concentrations and fluxes to the atmosphere from a tropical floodplain lake. Biogeochemistry 2020, 148, 129–151. [Google Scholar] [CrossRef]
- Basso, L.S.; Marani, L.; Gatti, L.V.; Miller, J.B.; Gloor, M.; Melack, J.; Cassol, H.L.G.; Tejada, G.; Domingues, L.G.; Arai, E.; et al. Amazon methane budget derived from multi-year airborne observations highlights regional variations in emissions. Commun. Earth Environ. 2021, 2, 246. [Google Scholar] [CrossRef]
- Dean, J.F.; Middelburg, J.J.; Röckmann, T.; Aerts, R.; Blauw, L.G.; Egger, M.; Jetten, M.S.M.; de Jong, A.E.E.; Meisel, O.H.; Rasigraf, O.; et al. Methane Feedbacks to the Global Climate System in a Warmer World. Rev. Geophys. 2018, 56, 207–250. [Google Scholar] [CrossRef]
- Haque, M.F.U.; Xu, H.J.; Colin Murrell, J.; Crombie, A. Facultative methanotrophs—Diversity, genetics, molecular ecology and biotechnological potential: A mini-review. Microbiology 2020, 166, 894–908. [Google Scholar] [CrossRef]
- Ho, A.; Kerckhof, F.M.; Luke, C.; Reim, A.; Krause, S.; Boon, N.; Bodelier, P.L.E. Conceptualizing functional traits and ecological characteristics of methane-oxidizing bacteria as life strategies. Environ. Microbiol. Rep. 2013, 5, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Knief, C. Diversity and habitat preferences of cultivated and uncultivated aerobic methanotrophic bacteria evaluated based on pmoA as molecular marker. Front. Microbiol. 2015, 6, 1346. [Google Scholar] [CrossRef] [PubMed]
- Padilla, C.C.; Bristow, L.A.; Sarode, N.; Garcia-Robledo, E.; Gómez Ramírez, E.; Benson, C.R.; Bourbonnais, A.; Altabet, M.A.; Girguis, P.R.; Thamdrup, B.; et al. NC10 bacteria in marine oxygen minimum zones. ISME J. 2016, 10, 2067–2071. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, G.V.M.; Oliveira, L.C.; Barros, D.J.; Bento, M.S.; Neu, V.; Toppa, R.H.; Carmoae, J.B.; Navarrete, A.A. Methane emission suppression in flooded soil from Amazonia. Chemosphere 2020, 250, 126263. [Google Scholar] [CrossRef]
- Leu, A.O.; Cai, C.; McIlroy, S.J.; Southam, G.; Orphan, V.J.; Yuan, Z.; Hu, S.; Tyson, G.W. Anaerobic methane oxidation coupled to manganese reduction by members of the Methanoperedenaceae. ISME J. 2020, 14, 1030–1041. [Google Scholar] [CrossRef]
- Su, G.; Zopfi, J.; Yao, H.; Steinle, L.; Niemann, H.; Lehmann, M.F. Manganese/iron-supported sulfate-dependent anaerobic oxidation of methane by archaea in lake sediments. Limnol. Oceanogr. 2019, 65, 863–875. [Google Scholar] [CrossRef]
- Gontijo, J.B.; Paula, F.S.; Venturini, A.M.; Yoshiura, C.A.; Borges, C.D.; Moura, J.M.S.; Bohannan, B.J.M.; Nüsslein, K.; Rodrigues, J.L.M.; Tsai, S.M. Not just a methane source: Amazonian floodplain sediments harbour a high diversity of methanotrophs with different metabolic capabilities. Mol. Ecol. 2021, 30, 2560–2572. [Google Scholar] [CrossRef]
- Bowman, J.P. Methylocystis. Bergey’s Man. Syst. Archaea Bact. 2015, 1–7. [Google Scholar] [CrossRef]
- Venturini, A.M.; Nakamura, F.M.; Gontijo, J.B.; da França, A.G.; Yoshiura, C.A.; Mandro, J.A.; MuiTsai, S. Robust DNA protocols for tropical soils. Heliyon 2020, 6, e03830. [Google Scholar] [CrossRef]
- Venturini, A.M.; Gontijo, J.B.; da França, A.G.; Moura, J.M.; Nüsslein, K.; Bohannan, B.J.; Rodrigues, J.L.M.; Tsai, S.M. Metagenomes from Eastern Brazilian Amazonian Floodplains in the Wet and Dry Seasons. Microbiol. Resour. Announc. 2020, e0043222. [Google Scholar] [CrossRef]
- Arkin, A.P.; Cottingham, R.W.; Henry, C.S.; Harris, N.L.; Stevens, R.L.; Maslov, S.; Dehal, P.; Ware, D.; Perez, F.; Canon, S.; et al. KBase: The United States department of energy systems biology knowledgebase. Nat. Biotechnol. 2018, 36, 566–569. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data [Online]. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 10 May 2022).
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Wu, Y.W.; Simmons, B.A.; Singer, S.W. MaxBin 2.0: An automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 2016, 32, 605–607. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef]
- Sieber, C.M.K.; Probst, A.J.; Sharrar, A.; Thomas, B.C.; Hess, M.; Tringe, S.G.; Banfield, J.F. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 2018, 3, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Bowers, R.M.; Kyrpides, N.C.; Stepanauskas, R.; Harmon-Smith, M.; Doud, D.; Reddy, T.B.K.; Schulz, F.; Jarett, J.; Rivers, A.R.; Eloe-Fadrosh, E.A.; et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 2017, 35, 725–731. [Google Scholar] [CrossRef]
- Chaumeil, P.A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the genome taxonomy database. Bioinformatics 2020, 36, 1925–1927. [Google Scholar] [CrossRef]
- Rodriguez-R, L.M.; Gunturu, S.; Harvey, W.T.; Rosselló-Mora, R.; Tiedje, J.M.; Cole, J.R.; Konstantinidis, K.T. The Microbial Genomes Atlas (MiGA) webserver: Taxonomic and gene diversity analysis of Archaea and Bacteria at the whole genome level. Nucleic Acids Res. 2018, 46, W282–W288. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Wilks, C.; Antonescu, V.; Charles, R. Scaling read aligners to hundreds of threads on general-purpose processors. Bioinformatics 2019, 35, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Shaffer, M.; Borton, M.A.; McGivern, B.B.; Zayed, A.A.; La Rosa, S.L.; Solden, L.M.; Liu, P.; Narrowe, A.B.; Rodríguez-Ramos, J.; Bolduc, B.; et al. DRAM for distilling microbial metabolism to automate the curation of microbiome function. Nucleic Acids Res. 2020, 48, 8883–8900. [Google Scholar] [CrossRef]
- Eren, A.M.; Esen, Ö.C.; Quince, C.; Vineis, J.H.; Morrison, H.G.; Sogin, M.L.; Delmont, T.O. Anvi’o: An advanced analysis and visualization platform for ‘omics data. PeerJ 2015, 3, e1319. [Google Scholar] [CrossRef]
- Galperin, M.Y.; Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Expanded Microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 2015, 43, D261–D269. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef]
- Parks, D.H.; Rinke, C.; Chuvochina, M.; Chaumeil, P.A.; Woodcroft, B.J.; Evans, P.N.; Hugenholtz, P.; Tyson, G.W. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2017, 2, 1533–1542. [Google Scholar] [CrossRef] [PubMed]
- Nelson, W.C.; Tully, B.J.; Mobberley, J.M. Biases in genome reconstruction from metagenomic data. PeerJ 2020, 8, e10119. [Google Scholar] [CrossRef]
- Whittenbury, R.; Phillips, K.C.; Wilkinson, J.F. Enrichment, isolation and some properties of methane-utilizing bacteria. Microbiology 1970, 61, 205–218. [Google Scholar] [CrossRef]
- Jung, G.Y.; Rhee, S.K.; Han, Y.S.; Kim, S.J. Genomic and physiological properties of a facultative methane-oxidizing bacterial strain of Methylocystis sp. From a wetland. Microorganisms 2020, 8, 1719. [Google Scholar] [CrossRef]
- Bento, M.d.S.; Barros, D.J.; Araújo, M.G.d.S.; Da Róz, R.; Carvalho, G.A.; do Carmo, J.B.; Toppa, R.H.; Neu, V.; Forsberg, B.R.; Bodelier, P.L.E.; et al. Active methane processing microbes and the disproportionate role of NC10 phylum in methane mitigation in Amazonian floodplains. Biogeochemistry 2021, 156, 293–317. [Google Scholar] [CrossRef]
- Han, D.; Dedysh, S.N.; Liesack, W. Unusual Genomic Traits Suggest Methylocystis bryophila S285 to Be Well Adapted for Life in Peatlands. Genome Biol. Evol. 2018, 10, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Cruz, S.; Vaksmaa, A.; Horn, M.A.; Niemann, H.; Pijuan, M.; Ho, A. Methanotrophs: Discoveries, Environmental Relevance, and a Perspective on Current and Future Applications. Front. Microbiol. 2021, 12, 678057. [Google Scholar] [CrossRef] [PubMed]
- Junk, W.J.; Piedade, M.T.F.; Schöngart, J.; Cohn-Haft, M.; Adeney, J.M.; Wittmann, F. A classification of major naturally-occurring Amazonian lowland wetlands. Wetlands 2011, 31, 623–640. [Google Scholar] [CrossRef]
- Taubert, M.; Grob, C.; Howat, A.M.; Burns, O.J.; Dixon, J.L.; Chen, Y.; Murrell, J.C. XoxF encoding an alternative methanol dehydrogenase is widespread in coastal marine environments. Environ. Microbiol. 2015, 17, 3937–3948. [Google Scholar] [CrossRef] [PubMed]
- Crombie, A.T. The effect of lanthanum on growth and gene expression in a facultative methanotroph. Environ. Microbiol. 2021, 24, 596–613. [Google Scholar] [CrossRef]
- Belova, S.E.; Baani, M.; Suzina, N.E.; Bodelier, P.L.; Liesack, W.; Dedysh, S.N. Acetate utilization as a survival strategy of peat-inhabiting Methylocystis spp. Environ. Microbiol. Rep. 2011, 3, 36–46. [Google Scholar] [CrossRef]
- Oshkin, I.Y.; Miroshnikov, K.K.; Grouzdev, D.S.; Dedysh, S.N. Pan-genome-based analysis as a framework for demarcating two closely related methanotroph genera Methylocystis and Methylosinus. Microorganisms 2020, 8, 768. [Google Scholar] [CrossRef]
- Bordel, S.; Rodríguez, Y.; Hakobyan, A.; Rodríguez, E.; Lebrero, R.; Muñoz, R. Genome scale metabolic modeling reveals the metabolic potential of three Type II methanotrophs of the genus Methylocystis. Metab. Eng. 2019, 54, 191–199. [Google Scholar] [CrossRef]
- Campbell, M.A.; Nyerges, G.; Kozlowski, J.A.; Poret-Peterson, A.T.; Stein, L.Y.; Klotz, M.G. Model of the molecular basis for hydroxylamine oxidation and nitrous oxide production in methanotrophic bacteria. FEMS Microbiol. Lett. 2011, 322, 82–89. [Google Scholar] [CrossRef]
- Hanson, R.S.; Hanson, T.E. Methanotrophic bacteria. Microbiol. Rev. 1996, 60, 439–471. [Google Scholar] [CrossRef] [PubMed]
- Versantvoort, W.; Pol, A.; MJetten, M.S.; van Niftrik, L.; Reimann, J.; Kartal, B.; den Camp, H.J.M.O. Multiheme hydroxylamine oxidoreductases produce NO during ammonia oxidation in methanotrophs. Proc. Natl. Acad. Sci. USA 2020, 117, 24459–24463. [Google Scholar] [CrossRef] [PubMed]
- Nyerges, G.; Stein, L.Y. Ammonia cometabolism and product inhibition vary considerably among species of methanotrophic bacteria. FEMS Microbiol. Lett. 2009, 297, 131–136. [Google Scholar] [CrossRef]
- Stein, L.Y.; Klotz, M.G. Nitrifying and denitrifying pathways of methanotrophic bacteria. Biochem. Soc. Trans. 2011, 39, 1826–1831. [Google Scholar] [CrossRef] [PubMed]
- Bordel, S.; Rodríguez, E.; Muñoz, R. Genome sequence of Methylocystis hirsuta CSC1, a polyhydroxyalkanoate producing methanotroph. Microbiol. Open 2019, 8, e00771. [Google Scholar] [CrossRef] [PubMed]
- Salah Ud-Din, A.I.M.; Roujeinikova, A. Methyl-accepting chemotaxis proteins: A core sensing element in prokaryotes and archaea. Cell. Mol. Life Sci. 2017, 74, 3293–3303. [Google Scholar] [CrossRef]
- Ortega, Á.; Zhulin, I.B.; Krell, T. Sensory Repertoire of Bacterial Chemoreceptors. Microbiol. Mol. Biol. Rev. 2017, 81, e00033-17. [Google Scholar] [CrossRef]
- Minamino, T.; Imada, K. The bacterial flagellar motor and its structural diversity. Trends Microbiol. 2015, 23, 267–274. [Google Scholar] [CrossRef]
- Nakamura, S.; Minamino, T. Flagella-driven motility of bacteria. Biomolecules 2019, 9, 279. [Google Scholar] [CrossRef]
- Oshkin, I.Y.; Miroshnikov, K.K.; Danilova, O.V.; Hakobyan, A.; Liesack, W.; Dedysh, S.N. Thriving in wetlands: Ecophysiology of the spiral- shaped methanotroph Methylospira mobilis as revealed by the complete genome sequence. Microorganisms 2019, 7, 683. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Ishiguro-Watanabe, M.; Tanabe, M. KEGG: Integrating viruses and cellular organisms. Nucleic Acids Res. 2021, 49, D545–D551. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gontijo, J.B.; Paula, F.S.; Venturini, A.M.; Mandro, J.A.; Bodelier, P.L.E.; Tsai, S.M. Insights into the Genomic Potential of a Methylocystis sp. from Amazonian Floodplain Sediments. Microorganisms 2022, 10, 1747. https://doi.org/10.3390/microorganisms10091747
Gontijo JB, Paula FS, Venturini AM, Mandro JA, Bodelier PLE, Tsai SM. Insights into the Genomic Potential of a Methylocystis sp. from Amazonian Floodplain Sediments. Microorganisms. 2022; 10(9):1747. https://doi.org/10.3390/microorganisms10091747
Chicago/Turabian StyleGontijo, Júlia B., Fabiana S. Paula, Andressa M. Venturini, Jéssica A. Mandro, Paul L. E. Bodelier, and Siu M. Tsai. 2022. "Insights into the Genomic Potential of a Methylocystis sp. from Amazonian Floodplain Sediments" Microorganisms 10, no. 9: 1747. https://doi.org/10.3390/microorganisms10091747
APA StyleGontijo, J. B., Paula, F. S., Venturini, A. M., Mandro, J. A., Bodelier, P. L. E., & Tsai, S. M. (2022). Insights into the Genomic Potential of a Methylocystis sp. from Amazonian Floodplain Sediments. Microorganisms, 10(9), 1747. https://doi.org/10.3390/microorganisms10091747