
Epidermal Microbiomes of Leopard Sharks (Triakis semifasciata) Are Consistent across Captive and Wild Environments
 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Materials and Methods
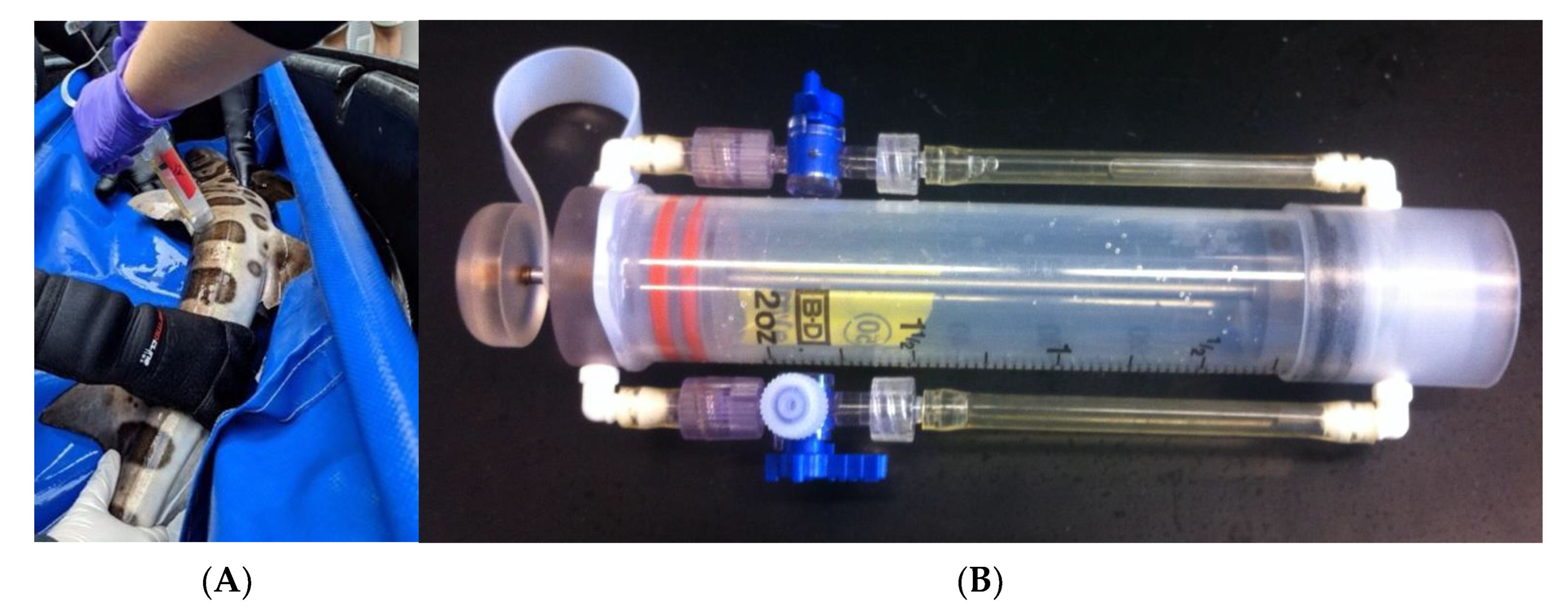
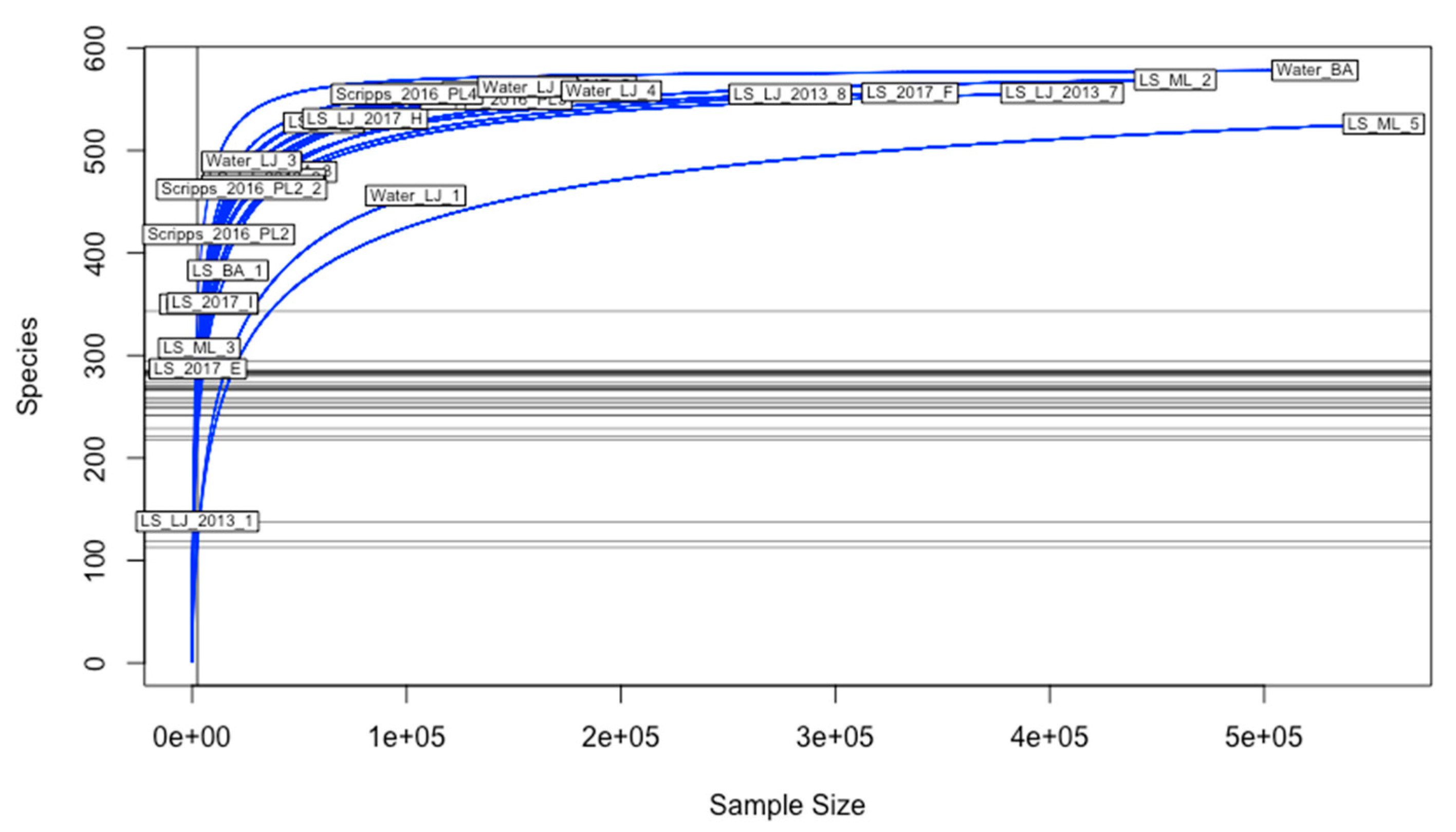
2.1. Sampling of Metagenomes
2.2. DNA Extraction and Metagenome Sequencing and Annotation
2.3. Assembly and Annotation of Metagenome-Assembled Genomes
2.4. Statistical Analyses
3. Results
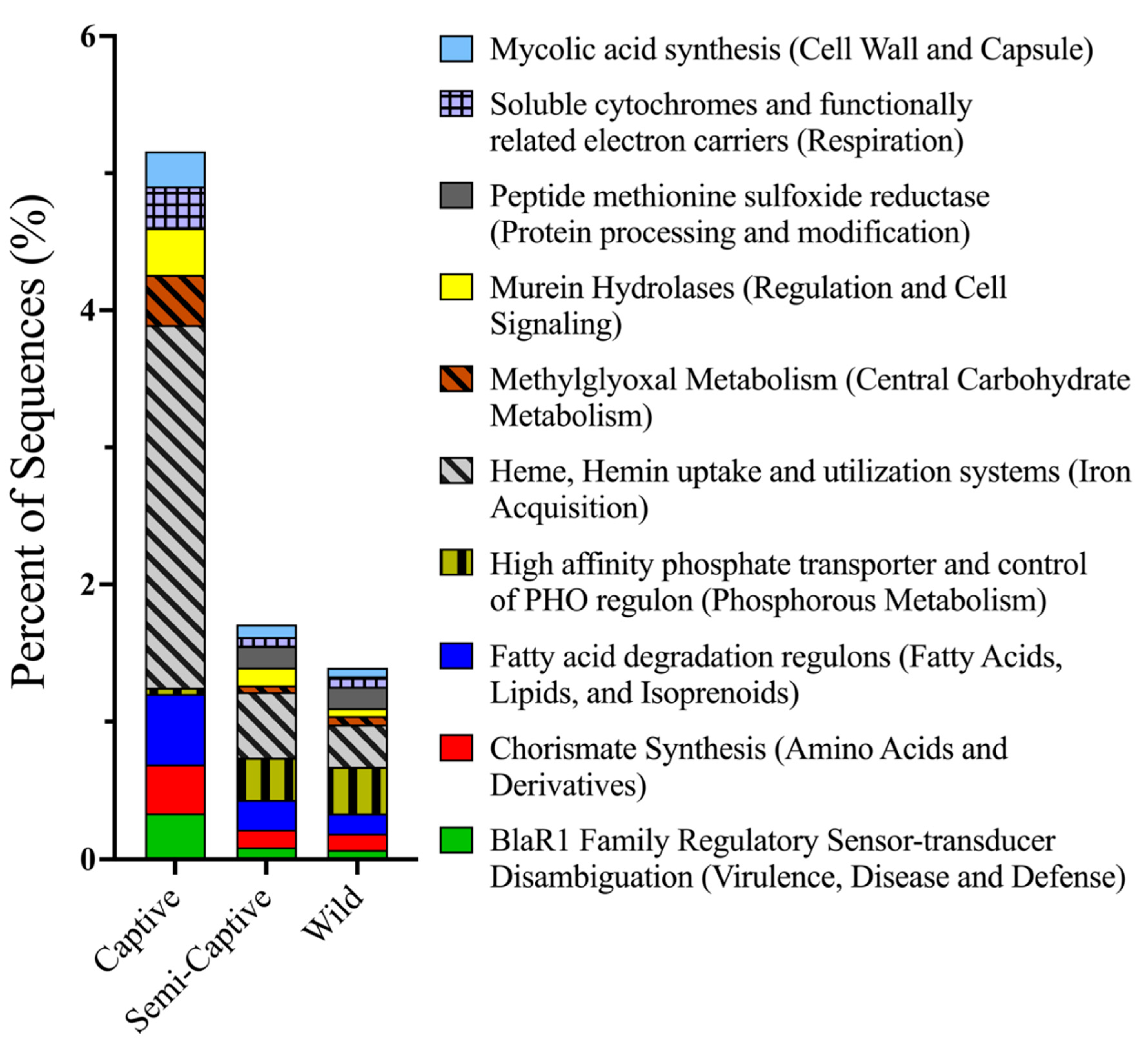
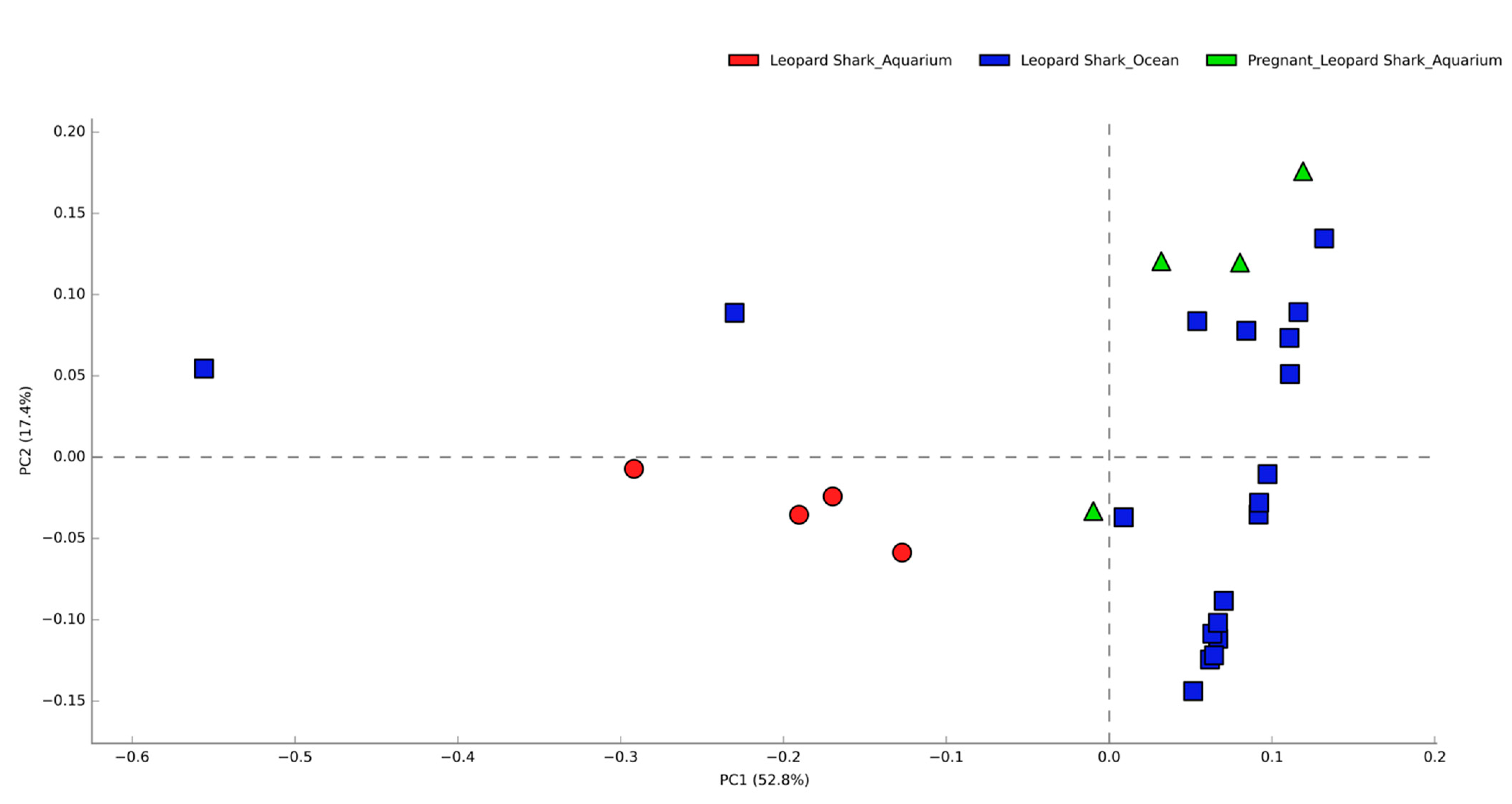
3.1. Comparisons of Functional Gene Potentials of T. semifasciata Epidermal Microbiomes across Environments
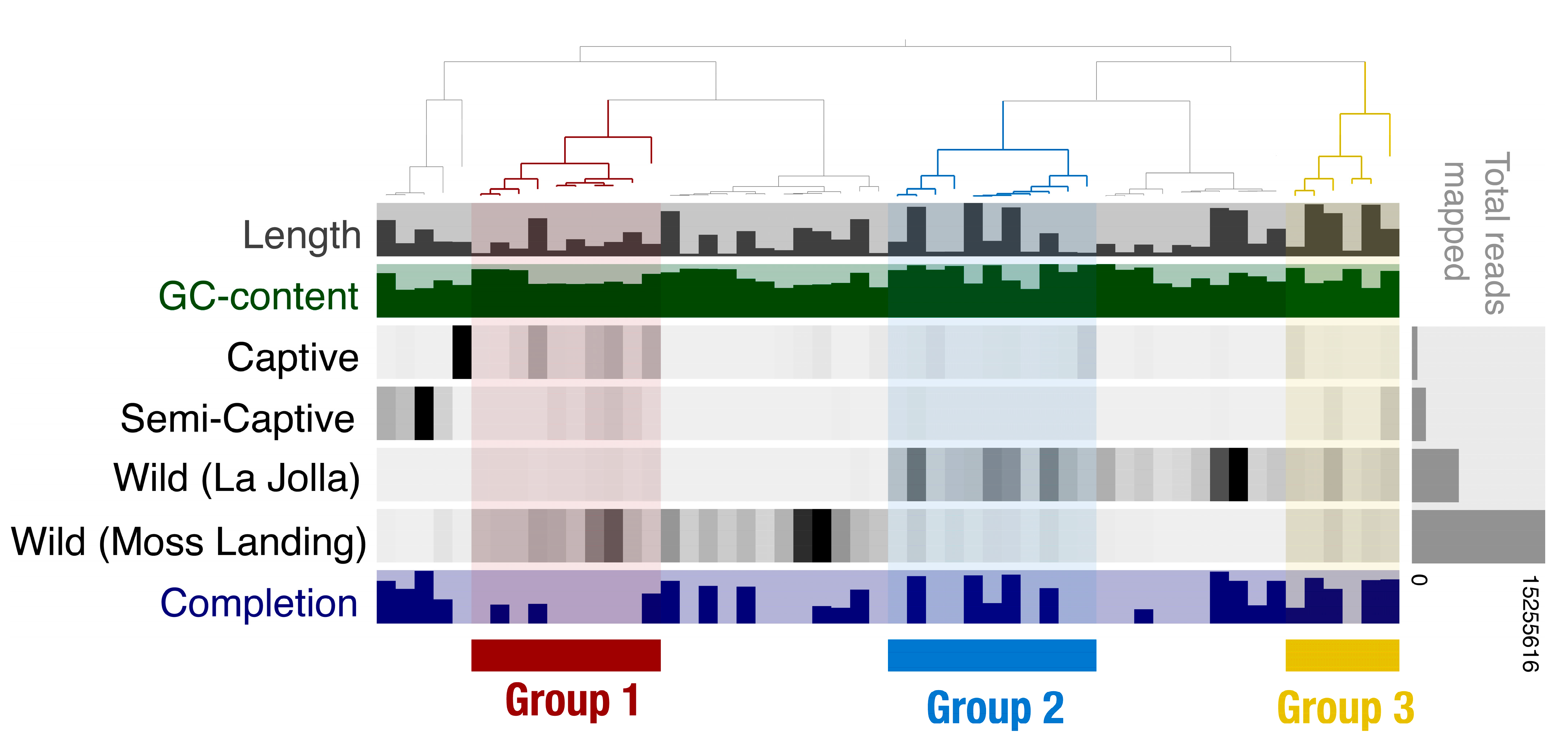
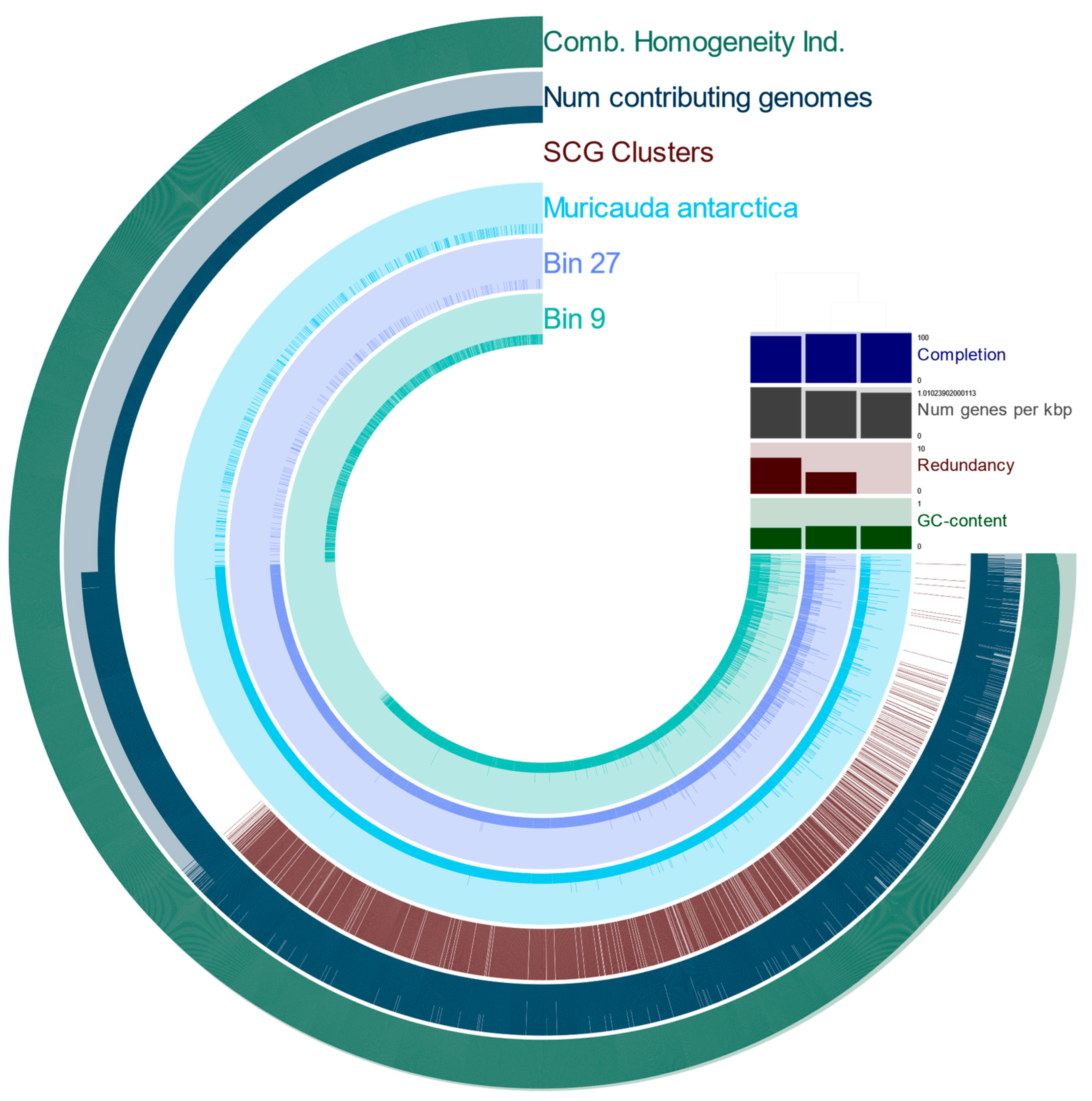
3.2. Metagenome-Assembled Genomes Constructed from Microbial Communities Associated with T. semifasciata
4. Discussion
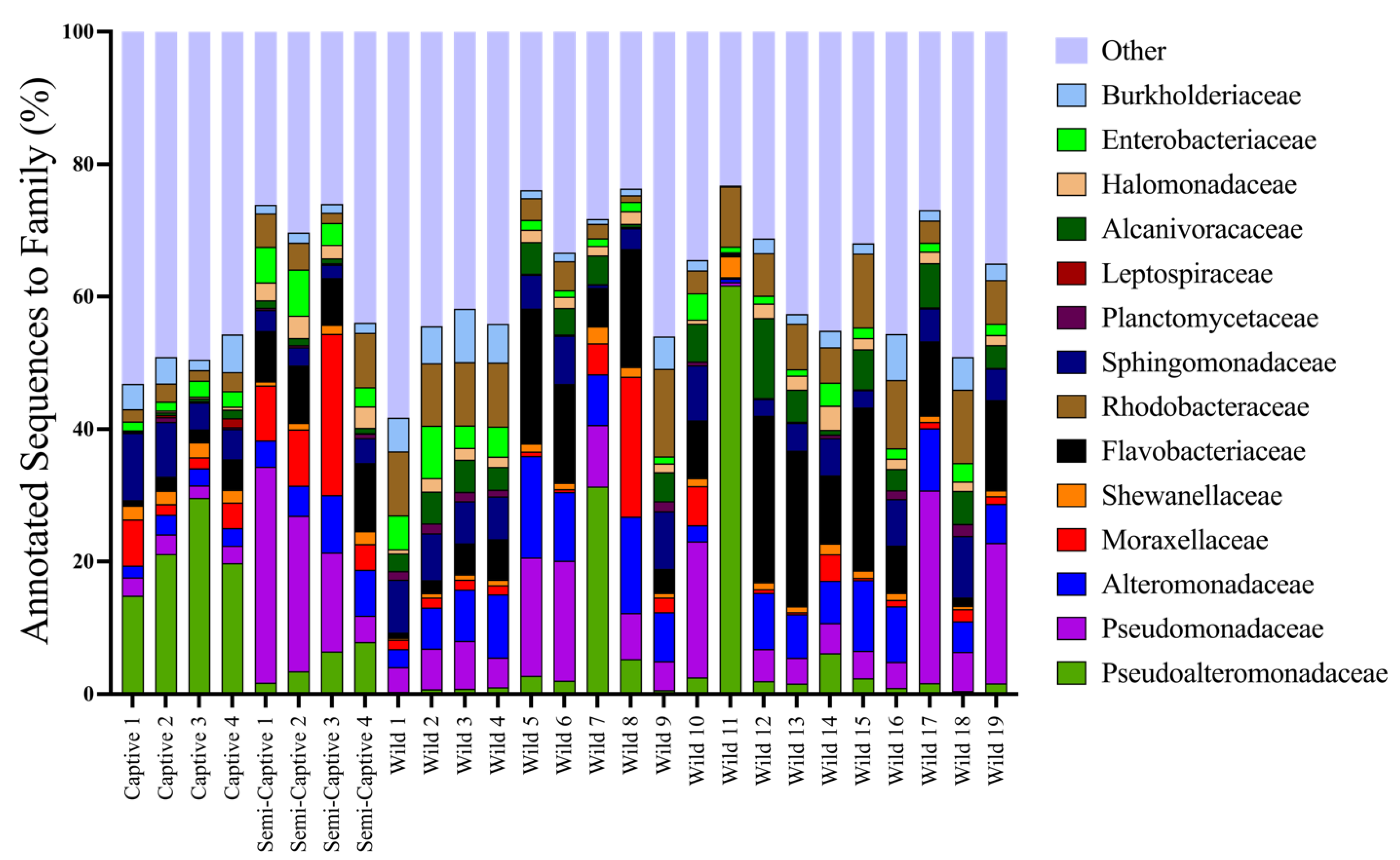
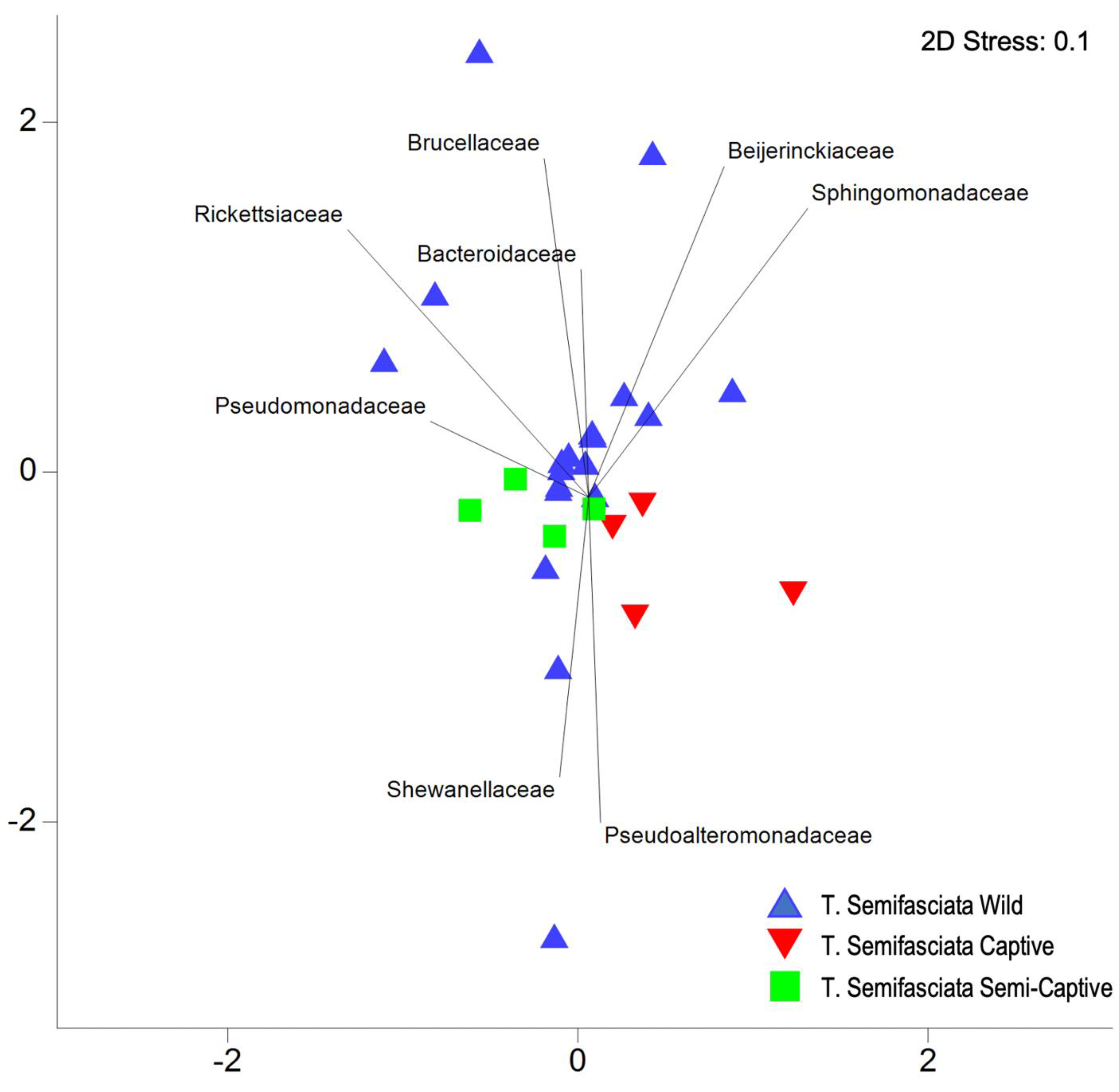
4.1. Epidermal Microbiome Taxonomic Structure as a Product of Captivity Duration
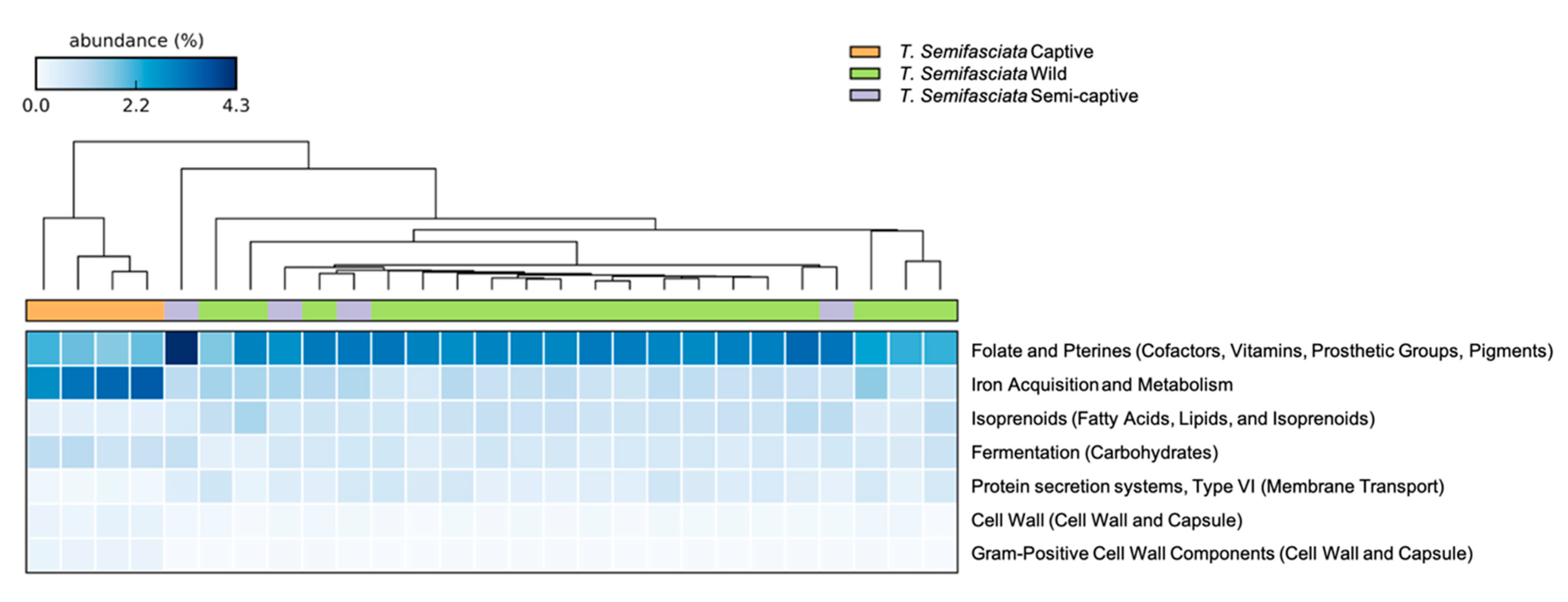
4.2. Metabolic Potentials of Captive Shark Microbiomes Reflect Environmental Conditions
4.3. MAGs reveal Novel, Constant Microbial Associations with T. semifasciata
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Triakis semifasciata Environment | Size | Mean | Std. Error |
|---|---|---|---|
| Captive | 4 | 7.03 | 1.07 |
| Semi-Captive | 4 | 6.29 | 0.24 |
| Wild | 19 | 10.1 | 1.34 |
| Family Level | |||
|---|---|---|---|
| Groups | T (test) | P (perm) | Permutations |
| Wild 1, Captive | 1.88 | 0.029 | 35 |
| Wild 2, Captive | 1.40 | 0.082 | 35 |
| Wild 3, Captive | 1.76 | 0.037 | 35 |
| Wild 4, Captive | 1.71 | 0.027 | 35 |
| Wild 5, Captive | 0.95 | 0.573 | 35 |
| Wild 6, Captive | 1.88 | 0.025 | 35 |
| Wild 7, Captive | 1.79 | 0.025 | 35 |
| Wild 8, Captive | 1.26 | 0.131 | 35 |
| Wild 9, Captive | 1.81 | 0.036 | 35 |
| Wild 10, Captive | 1.33 | 0.064 | 35 |
| Genus Level | |||
| Groups | t | P (perm) | perms |
| Wild 1, Captive | 1.59 | 0.025 | 35 |
| Wild 2, Captive | 1.46 | 0.037 | 35 |
| Wild 3, Captive | 1.75 | 0.032 | 35 |
| Wild 4, Captive | 1.52 | 0.023 | 35 |
| Wild 5, Captive | 0.96 | 0.565 | 35 |
| Wild 6, Captive | 1.63 | 0.029 | 35 |
| Wild 7, Captive | 1.64 | 0.03 | 35 |
| Wild 8, Captive | 1.24 | 0.076 | 35 |
| Wild 9, Captive | 1.63 | 0.025 | 35 |
| Wild 10, Captive | 1.39 | 0.061 | 35 |
References
- Bosch, T.C.G.; McFall-Ngai, M.J. Metaorganisms as the new frontier. Zoology 2011, 114, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Doane, M.P.; Haggerty, J.M.; Kacev, D.; Papudeshi, B.; Dinsdale, E.A. The skin microbiome of the common thresher shark (Alopias vulpinus) has low taxonomic and gene function β-diversity. Environ. Microbiol. Rep. 2017, 9, 357–373. [Google Scholar] [CrossRef]
- Doane, M.P.; Morris, M.M.; Papudeshi, B.; Allen, L.; Pande, D.; Haggerty, J.M.; Johri, S.; Turnlund, A.C.; Peterson, M.; Kacev, D.; et al. The skin microbiome of elasmobranchs follows phylosymbiosis, but in teleost fishes, the microbiomes converge. Microbiome 2020, 8, 93. [Google Scholar] [CrossRef] [PubMed]
- Lima, L.F.; Weissman, M.; Reed, M.; Papudeshi, B.; Alker, A.T.; Morris, M.M.; Edwards, R.A.; de Putron, S.J.; Vaidya, N.K.; Dinsdale, E.A. Modeling of the Coral Microbiome: The Influence of Temperature and Microbial Network. MBio 2020, 11, e02691-19. [Google Scholar] [CrossRef]
- Doane, M.P.; Johnson, C.J.; Johri, S.; Kerr, E.N.; Morris, M.M.; Desantiago, R.; Turnlund, A.C.; Goodman, A.; Mora, M.; Lima, L.F.O.; et al. The Epidermal Microbiome Within an Aggregation of Leopard Sharks (Triakis semifasciata) Has Taxonomic Flexibility with Gene Functional Stability Across Three Time-points. Microb. Ecol. 2022. [Google Scholar] [CrossRef]
- Montenegro, D.; Astudillo-García, C.; Hickey, T.; Lear, G. A non-invasive method to monitor marine pollution from bacterial DNA present in fish skin mucus. Environ. Pollut. 2020, 263, 114438. [Google Scholar] [CrossRef] [PubMed]
- Retallack, H.; Okihiro, M.S.; Britton, E.; Van Sommeran, S.; Derisi, J.L. Metagenomic next-generation sequencing reveals miamiensis avidus (Ciliophora: Scuticociliatida) in the 2017 epizootic of leopard sharks (Triakis semifasciata) in san francisco bay, California, USA. J. Wildl. Dis. 2019, 55, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Jeffree, R.A.; Warnau, M.; Teyssié, J.L.; Markich, S.J. Comparison of the bioaccumulation from seawater and depuration of heavy metals and radionuclides in the spotted dogfish Scyliorhinus canicula (Chondrichthys) and the turbot Psetta maxima (Actinopterygii: Teleostei). Sci. Total Environ. 2006, 368, 839–852. [Google Scholar] [CrossRef]
- Chiarello, M.; Villéger, S.; Bouvier, C.; Auguet, J.C.; Bouvier, T. Captive bottlenose dolphins and killer whales harbor a species-specific skin microbiota that varies among individuals. Sci. Rep. 2017, 7, 15269. [Google Scholar] [CrossRef] [PubMed]
- Gruber, D.F.; Loew, E.R.; Deheyn, D.D.; Akkaynak, D.; Gaffney, J.P.; Smith, W.L.; Davis, M.P.; Stern, J.H.; Pieribone, V.A.; Sparks, J.S. Biofluorescence in Catsharks (Scyliorhinidae): Fundamental Description and Relevance for Elasmobranch Visual Ecology. Sci. Rep. 2016, 6, 24751. [Google Scholar] [CrossRef] [PubMed]
- Kalmijn, A.J. Electric and Magnetic Field Detection in Elasmobranch Fishes; American Association for the Advancement of Science: Washington, DC, USA, 2019; Volume 218, pp. 916–918. [Google Scholar]
- Meyer, W.; Seegers, U. Basics of skin structure and function in elasmobranchs: A review. J. Fish Biol. 2012, 80, 1940–1967. [Google Scholar] [CrossRef]
- Bechert, D.W.; Bruse, M.; Hage, W. Experiments with three-dimensional riblets as an idealized model of shark skin. Exp. Fluids 2000, 28, 403–412. [Google Scholar] [CrossRef]
- Storo, R.; Easson, C.; Shivji, M.; Lopez, J.V. Microbiome Analyses Demonstrate Specific Communities Within Five Shark Species. Front. Microbiol. 2021, 12, 605285. [Google Scholar] [CrossRef] [PubMed]
- Pogoreutz, C.; Gore, M.A.; Perna, G.; Millar, C.; Nestler, R.; Ormond, R.F.; Clarke, C.R.; Voolstra, C.R. Similar bacterial communities on healthy and injured skin of black tip reef sharks. Anim. Microbiome 2019, 1, 9. [Google Scholar] [CrossRef] [PubMed]
- Egan, S.; Schreier, H.J.; Costa, R.; Oxley, A.P.A.; Legrand, T.P.R.A.; Catalano, S.R.; Wos-Oxley, M.L.; Stephens, F.; Landos, M.; Bansemer, M.S.; et al. The Inner Workings of the Outer Surface: Skin and Gill Microbiota as Indicators of Changing Gut Health in Yellowtail Kingfish. Front. Microbiol 2018, 8, 2664. [Google Scholar] [CrossRef]
- Antwis, R.E.; Haworth, R.L.; Engelmoer, D.J.P.; Ogilvy, V.; Fidgett, A.L.; Preziosi, R.F. Ex situ diet influences the bacterial community associated with the skin of red-eyed tree frogs (Agalychnis callidryas). PLoS ONE 2014, 9, e85563. [Google Scholar] [CrossRef] [PubMed]
- Ross, A.A.; Rodrigues Hoffmann, A.; Neufeld, J.D. The skin microbiome of vertebrates. Microbiome 2019, 7, 79. [Google Scholar] [CrossRef] [PubMed]
- Haggerty, J.M.; Dinsdale, E.A. Distinct biogeographical patterns of marine bacterial taxonomy and functional genes. Glob. Ecol. Biogeogr. 2017, 26, 177–190. [Google Scholar] [CrossRef]
- Kearns, P.J.; Bowen, J.L.; Tlusty, M.F. The skin microbiome of cow-nose rays (Rhinoptera bonasus) in an aquarium touch-tank exhibit. Zoo Biol. 2017, 36, 226–230. [Google Scholar] [CrossRef]
- Nosal, A.P.; Cartamil, D.C.; Long, J.W.; Lührmann, M.; Wegner, N.C.; Graham, J.B. Demography and movement patterns of leopard sharks (Triakis semifasciata) aggregating near the head of a submarine canyon along the open coast of southern California, USA. Environ. Biol. Fishes 2013, 96, 865–878. [Google Scholar] [CrossRef]
- Nosal, A.P.; Caillat, A.; Kisfaludy, E.K.; Royer, M.A.; Wegner, N.C. Aggregation behavior and seasonal philopatry in male and female leopard sharks Triakis semifasciata along the open coast of southern California, USA. Mar. Ecol. Prog. Ser. 2014, 499, 157–175. [Google Scholar] [CrossRef]
- Minich, J.J.; Morris, M.M.; Brown, M.; Doane, M.; Edwards, M.S.; Michael, T.P.; Dinsdale, E.A. Elevated temperature drives kelp microbiome dysbiosis, while elevated carbon dioxide induces water microbiome disruption. PLoS ONE 2018, 13, e0192772. [Google Scholar] [CrossRef]
- Perry, C.T.; Pratte, Z.A.; Clavere-Graciette, A.; Ritchie, K.B.; Hueter, R.E.; Newton, A.L.; Fischer, G.C.; Dinsdale, E.A.; Doane, M.P.; Wilkinson, K.A.; et al. Elasmobranch microbiomes: Emerging patterns and implications for host health and ecology. Anim. Microbiome 2021, 3, 61. [Google Scholar] [CrossRef]
- Dinsdale, E.A.; Pantos, O.; Smriga, S.; Edwards, R.A.; Angly, F.; Wegley, L.; Hatay, M.; Hall, D.; Brown, E.; Haynes, M.; et al. Microbial Ecology of Four Coral Atolls in the Northern Line Islands. PLoS ONE 2008, 3, e1584. [Google Scholar] [CrossRef] [PubMed]
- Bruce, T.; Meirelles, P.M.; Garcia, G.; Paranhos, R.; Rezende, C.E.; de Moura, R.L.; Filho, R.F.; Coni, E.O.C.; Vasconcelos, A.T.; Filho, G.; et al. Abrolhos bank reef health evaluated by means of water quality, microbial diversity, benthic cover, and fish biomass data. PLoS ONE 2012, 7, e36687. [Google Scholar] [CrossRef] [PubMed]
- Dinsdale, E.A.; Edwards, R.A.; Bailey, B.A.; Tuba, I.; Akhter, S.; McNair, K.; Schmieder, R.; Apkarian, N.; Creek, M.; Guan, E.; et al. Multivariate analysis of functional metagenomes. Front. Genet. 2013, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef]
- Overbeek, R.; Olson, R.; Pusch, G.D.; Olsen, G.J.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Parrello, B.; Shukla, M.; et al. The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 2014, 42, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Walsh, K.; Haggerty, J.M.; Doane, M.P.; Hansen, J.J.; Morris, M.M.; Moreira, A.P.B.; de Oliveira, L.; Leomil, L.; Garcia, G.D.; Thompson, F.; et al. Aura-biomes are present in the water layer above coral reef benthic macro-organisms. PeerJ 2017, 5, e3666. [Google Scholar] [CrossRef]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Corrigendum: Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 1211. [Google Scholar] [CrossRef] [PubMed]
- Luz Calle, M. Statistical analysis of metagenomics data. Genom. Inform. 2019, 17, e6. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Waste Not, Want Not: Why Rarefying Microbiome Data Is Inadmissible. PLoS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019, 2019, e7359. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Papudeshi, B.; Haggerty, J.M.; Doane, M.; Morris, M.M.; Walsh, K.; Beattie, D.T.; Pande, D.; Zaeri, P.; Silva, G.G.Z.; Thompson, F.; et al. Optimizing and evaluating the reconstruction of Metagenome-assembled microbial genomes. BMC Genomics 2017, 18, 915. [Google Scholar] [CrossRef]
- Parks, D.H.; Rinke, C.; Chuvochina, M.; Chaumeil, P.-A.A.; Woodcroft, B.J.; Evans, P.N.; Hugenholtz, P.; Tyson, G.W. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat. Microbiol. 2017, 2, 1533–1542. [Google Scholar] [CrossRef]
- Robbins, S.J.; Singleton, C.M.; Chan, C.X.; Messer, L.F.; Geers, A.U.; Ying, H.; Baker, A.; Bell, S.C.; Morrow, K.M.; Ragan, M.A.; et al. A genomic view of the reef-building coral Porites lutea and its microbial symbionts. Nat. Microbiol. 2019, 4, 2090–2100. [Google Scholar] [CrossRef]
- Nayfach, S.; Roux, S.; Seshadri, R.; Udwary, D.; Varghese, N.; Schulz, F.; Wu, D.; Paez-Espino, D.; Chen, I.M.; Huntemann, M.; et al. A genomic catalog of Earth’s microbiomes. Nat. Biotechnol. 2020. [Google Scholar] [CrossRef]
- Silva, G.G.Z.; Cuevas, D.A.; Dutilh, B.E.; Edwards, R.A. FOCUS: An alignment-free model to identify organisms in metagenomes using non-negative least squares. PeerJ 2014, 2, e425. [Google Scholar] [CrossRef] [PubMed]
- Wattam, A.R.; Abraham, D.; Dalay, O.; Disz, T.L.; Driscoll, T.; Gabbard, J.L.; Gillespie, J.J.; Gough, R.; Hix, D.; Kenyon, R.; et al. PATRIC, the bacterial bioinformatics database and analysis resource. Nucleic Acids Res. 2014, 42, D581–D591. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.H.; Ha, S.M.; Lim, J.; Kwon, S.; Chun, J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 2017, 110, 1281–1286. [Google Scholar] [CrossRef]
- Chun, J.; Rainey, F.A. Integrating genomics into the taxonomy and systematics of the Bacteria and Archaea. Int. J. Syst. Evol. Microbiol. 2014, 64, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Bowers, R.M.; Kyrpides, N.C.; Stepanauskas, R.; Harmon-Smith, M.; Doud, D.; Reddy, T.B.K.; Schulz, F.; Jarett, J.; Rivers, A.R.; Eloe-Fadrosh, E.A.; et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 2017, 35, 725–731. [Google Scholar] [CrossRef]
- Eren, A.M.; Esen, O.C.; Quince, C.; Vineis, J.H.; Morrison, H.G.; Sogin, M.L.; Delmont, T.O. Anvi’o: An advanced analysis and visualization platformfor ’omics data. PeerJ 2015, 2015, e1319. [Google Scholar] [CrossRef] [PubMed]
- Death, R. Margalef’s Index. In Encyclopedia of Ecology, Five-Volume Set; Elsevier Inc.: Amsterdam, The Netherlands, 2008; pp. 2209–2210. ISBN 9780080914565. [Google Scholar]
- Pielou, E.C. The measurement of diversity in different types of biological collections. J. Theor. Biol. 1966, 13, 131–144. [Google Scholar] [CrossRef]
- Chao, A.; Shen, T.J. S Index of Diversity When There Are Unseen Species in Sample. Environ. Ecol. Stat. 2003, 10, 429–443. [Google Scholar] [CrossRef]
- Clarke, K.R. Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 1993, 18, 117–143. [Google Scholar] [CrossRef]
- Anderson, M.J. Permutational Multivariate Analysis of Variance (PERMANOVA). In Wiley StatsRef: Statistics Reference Online; John and Wiley and Sons: Hoboken, NJ, USA, 2017. [Google Scholar] [CrossRef]
- Clarke, K.R.; Gorley, R.; Sommerfield, P.J.; Warwick, R.M. Change in Marine Communities—Statistical Analysis; Primer-e Ltd.: Ivybridge, UK, 2014. [Google Scholar]
- Goldford, J.E.; Lu, N.; Bajić, D.; Estrela, S.; Tikhonov, M.; Sanchez-Gorostiaga, A.; Segrè, D.; Mehta, P.; Sanchez, A. Emergent simplicity in microbial community assembly. Science 2018, 361, 469–474. [Google Scholar] [CrossRef] [PubMed]
- Kruskal, J.B. Multidimensional scaling by optimizing goodness of fit to a nonmetric hypothesis. Psychometrika 1964, 29, 1–27. [Google Scholar] [CrossRef]
- Anderson, M.J. Distance-based tests for homogeneity of multivariate dispersions. Biometrics 2006, 62, 245–253. [Google Scholar] [CrossRef]
- Clarke, K.R.; Gorley, R.N. PERMANOVA+ Primer V7: User Manual; Prim. Ltd.: Plymouth, UK, 2015; p. 93. [Google Scholar]
- Dinsdale, E.A.; Edwards, R.A.; Hall, D.; Angly, F.; Breitbart, M.; Brulc, J.M.; Furlan, M.; Desnues, C.; Haynes, M.; Li, L.; et al. Functional metagenomic profiling of nine biomes. Nature 2008, 452, 629. [Google Scholar] [CrossRef]
- Franzosa, E.A.; Morgan, X.C.; Segata, N.; Waldron, L.; Reyes, J.; Earl, A.M.; Giannoukos, G.; Boylan, M.R.; Ciulla, D.; Gevers, D.; et al. Relating the metatranscriptome and metagenome of the human gut. Proc. Natl. Acad. Sci. USA 2014, 111, E2329–E2338. [Google Scholar] [CrossRef]
- Gilbert, B.; Bennett, J.R. Partitioning variation in ecological communities: Do the numbers add up? J. Appl. Ecol. 2010, 47, 1071–1082. [Google Scholar] [CrossRef]
- Mason, N.W.H.; Richardson, S.J.; Peltzer, D.A.; de Bello, F.; Wardle, D.A.; Allen, R.B. Changes in coexistence mechanisms along a long-term soil chronosequence revealed by functional trait diversity. J. Ecol. 2012, 100, 678–689. [Google Scholar] [CrossRef]
- Parks, D.H.; Beiko, R.G. Identifying biologically relevant differences between metagenomic communities. Bioinformatics 2010, 26, 715–721. [Google Scholar] [CrossRef]
- Levin, D.; Raab, N.; Pinto, Y.; Rothschild, D.; Zanir, G.; Godneva, A.; Mellul, N.; Futorian, D.; Gal, D.; Leviatan, S.; et al. Diversity and functional landscapes in the microbiota of animals in the wild. Science 2021, 372, eabb5352. [Google Scholar] [CrossRef] [PubMed]
- Arun, A.B.; Chen, W.M.; Lai, W.A.; Chao, J.H.; Rekha, P.D.; Shen, F.T.; Singh, S.; Young, C.C. Muricauda lutaonensis sp. nov., a moderate thermophile isolated from a coastal hot spring. Int. J. Syst. Evol. Microbiol. 2009, 59, 2738–2742. [Google Scholar] [CrossRef] [PubMed]
- Sriswasdi, S.; Yang, C.C.; Iwasaki, W. Generalist species drive microbial dispersion and evolution. Nat. Commun. 2017, 8, 1162. [Google Scholar] [CrossRef]
- Kinnula, H.; Mappes, J.; Sundberg, L.R. Coinfection outcome in an opportunistic pathogen depends on the inter-strain interactions. BMC Evol. Biol. 2017, 17, 77. [Google Scholar] [CrossRef]
- Bernardet, J.F.; Nakagawa, Y.; Holmes, B. Proposed minimal standards for describing new taxa of the family Flavobacteriaceae and emended description of the family. Int. J. Syst. Evol. Microbiol. 2002, 52, 1049–1070. [Google Scholar] [CrossRef]
- Gray, D.A.; Dugar, G.; Gamba, P.; Strahl, H.; Jonker, M.J.; Hamoen, L.W. Extreme slow growth as alternative strategy to survive deep starvation in bacteria. Nat. Commun. 2019, 10, 890. [Google Scholar] [CrossRef]
- Bruns, A.; Rohde, M.; Berthe-Corti, L. Muricauda ruestringensis gen. nov., sp. nov., a facultatively anaerobic, appendaged bacterium from German North Sea intertidal sediment. Int. J. Syst. Evol. Microbiol. 2001, 51, 1997–2006. [Google Scholar] [CrossRef]
- Weber, J.; Kayser, A.; Rinas, U. Metabolic flux analysis of Escherichia coli in glucose-limited continuous culture. II. Dynamic response to famine and feast, activation of the methylglyoxal pathway and oscillatory behaviour. Microbiology 2005, 151, 707–716. [Google Scholar] [CrossRef]
- Gunn, J.S.; Murphy, E.R.; Johnson, J.G.; Richard, K.L.; Kelley, B.R. Heme Uptake and Utilization by Gram-Negative Bacterial Pathogens. Front. Cell. Infect. Microbiol. 2019, 1, 81. [Google Scholar] [CrossRef]
- Moak, M.; Molineux, I.J. Peptidoglycan hydrolytic activities associated with bacteriophage virions. Mol. Microbiol. 2004, 51, 1169–1183. [Google Scholar] [CrossRef]
- Chen, Y.; Wei, D.; Wang, Y.; Zhang, X. The role of interactions between bacterial chaperone, aspartate aminotransferase, and viral protein during virus infection in high temperature environment: The interactions between bacterium and virus proteins. BMC Microbiol. 2013, 13, 48. [Google Scholar] [CrossRef] [PubMed]
- Merdanovic, M.; Clausen, T.; Kaiser, M.; Huber, R.; Ehrmann, M. Protein quality control in the bacterial periplasm. Annu. Rev. Microbiol. 2011, 65, 149–168. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, X.; Lai, Q.; Du, Y.; Sun, F.; Shao, Z. Muricauda indica sp. nov., isolated from deep sea water. Int. J. Syst. Evol. Microbiol. 2018, 68, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Q.; Sun, Q.L.; Sun, Y.Y.; Yu, C.; Sun, L. Muricauda iocasae sp. Nov., isolated from deep sea sediment of the south China sea. Int. J. Syst. Evol. Microbiol. 2018, 68, 2538–2544. [Google Scholar] [CrossRef]
- Lee, S.Y.; Park, S.; Oh, T.K.; Yoon, J.H. Muricauda beolgyonensis sp. nov., isolated from a tidal flat. Int. J. Syst. Evol. Microbiol. 2012, 62, 1134–1139. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Li, Y.; Guo, Q.; Lai, Q.; Wei, J.; Zheng, T.; Tian, Y. Muricauda zhangzhouensis sp. nov., isolated from mangrove sediment. Int. J. Syst. Evol. Microbiol. 2013, 63, 2320–2325. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yu, M.; Zhou, S.; Fu, T.; Sun, W.; Wang, L.; Zhang, X.H. Muricauda alvinocaridis sp. nov., isolated from shrimp gill from the Okinawa Trough. Int. J. Syst. Evol. Microbiol. 2020, 70, 1666–1671. [Google Scholar] [CrossRef]
- Qunjian, Y.; Zhang, W.; Li, X.; Lihong, Z.; Xiaoqing, Q.I.; Chan, Z.; Long-Fei, W.U.; Yin, Q.; Zhang, W.; Li, X.; et al. Contribution of trimethylamine N-oxide on the growth and pressure tolerance of deep-sea bacteria. J. Oceanol. Limnol. 2019, 37, 210–222. [Google Scholar] [CrossRef]
- Nedashkovskaya, O.I.; Kim, S.B.; Lysenko, A.M.; Park, M.S.; Mikhailov, V.V.; Bae, K.S.; Park, H.Y. Roseivirga echinicomitans sp. nov., a novel marine bacterium isolated from the sea urchin Strongylocentrotus intermedius, and emended description of the genus Roseivirga. Int. J. Syst. Evol. Microbiol. 2005, 55, 1797–1800. [Google Scholar] [CrossRef]
- Shao, R.; Lai, Q.; Liu, X.; Sun, F.; Du, Y.; Li, G.; Shao, Z. Zunongwangia atlantica sp. nov., isolated from deep-sea water. Int. J. Syst. Evol. Microbiol. 2014, 64, 16–20. [Google Scholar] [CrossRef]
- Pinhassi, J.; Bowman, J.P.; Nedashkovskaya, O.I.; Lekunberri, I.; Gomez-Consarnau, L.; Pedrós-Alió, C. Leeuwenhoekiella blandensis sp. nov., a genome-sequenced marine member of the family Flavobacteriaceae. Int. J. Syst. Evol. Microbiol. 2006, 56, 1489–1493. [Google Scholar] [CrossRef]
- Johri, S.; Solanki, J.; Cantu, V.A.; Fellows, S.R.; Edwards, R.A.; Moreno, I.; Vyas, A.; Dinsdale, E.A. ‘Genome skimming’ with the MinION hand-held sequencer identifies CITES-listed shark species in India’s exports market. Sci. Rep. 2019, 9, 4476. [Google Scholar] [CrossRef]







| Sample | Sex | Base Pair Count | Sequence Count |
|---|---|---|---|
| Captive 1 | Male | 13,254,680 | 36,040 |
| Captive 2 | Female | 165,572,548 | 563,299 |
| Captive 3 | Female | 129,410,990 | 454,887 |
| Captive 4 | Female | 140,813,416 | 512,408 |
| S.C. 1 | Female | 194,456,844 | 683,590 |
| S.C. 2 | Female | 114,129,299 | 364,329 |
| S.C. 3 | Female | 106,800,378 | 351,397 |
| S.C. 4 | Female | 147,769,235 | 491,188 |
| Wild 1 | Female | 263,295,773 | 1,298,868 |
| Wild 2 | Female | 262,597,687 | 1,176,351 |
| Wild 3 | Female | 258,939,703 | 1,226,960 |
| Wild 4 | Female | 653,532,367 | 2,531,410 |
| Wild 5 | Female | 640,031,260 | 2,434,754 |
| Wild 6 | Female | 673,699,889 | 2,549,181 |
| Wild 7 | Female | 397,391,784 | 1,368,629 |
| Wild 8 | Female | 273,016,668 | 1,039,793 |
| Wild 9 | Female | 291,042,619 | 1,045,530 |
| Wild 10 | Female | 291,228,603 | 1,001,807 |
| Wild 11 | Female | 369,123,866 | 1,254,908 |
| Wild 12 | Female | 211,033,215 | 710,743 |
| Wild 13 | Female | 279,527,632 | 914,034 |
| Wild 14 | Female | 195,358,159 | 648,568 |
| Wild 15 | Female | 109,131,187 | 328,208 |
| Wild 16 | Female | 94,795,257 | 316,833 |
| Wild 17 | Female | 186,632,228 | 623,111 |
| Wild 18 | Female | 342,901,940 | 1,246,300 |
| Wild 19 | Female | 217,511,438 | 709,664 |
| Host Environment | Margalef’s (d) Index ± S.E.M. * | Pielou’s (J’) Index ± S.E.M. | Inverse Simpson (1/λ) Index ± S.E.M. |
|---|---|---|---|
| Captive | 41.21 ± 4.15 | 0.59 ± 4.3 × 10−2 | 70.50 ± 3.54 |
| Semi-captive | 41.53 ± 3.54 | 0.624 ± 3.37 × 10−2 | 73.30 ± 2.27 |
| Wild | 40.07 ± 1.44 | 0.581 ± 2.88 × 10−2 | 68.29 ± 2.29 |
| Scheme | ||
|---|---|---|
| Family Level | % Dissimilarity | Contributing Microbes |
| All vs. Water | 19.05 | Moraxellaceae, Pseudomonadaceae, Rhodobacteraceae, Planctomycetaceae, Halomnocadaceae, Shewanellaceae, Enterobacteriaceae, Rickettsiales, Parachlamydiaceae, Cyanobacteria |
| Wild vs. Captive | 17.4 | Alteromondales, Pseudoalteromonadaceae, Rhodobacterales, Alcanivoraceae, Flavobacteriaceae, Caulobacteraceae, Erythrobacteraceae, Comamonodaceae, Alteromonoadaceae, Pseudomonadaceae, Rickettsiales, Halomonodaceae, Rhodobacteraceae |
| Wild vs. Semi-Captive | 15.4 | Moraxellaceae, Pseudomonodaceae, Rickettsiales, Pseudoalteromonadaceae, Rickettsiales, Alcanivoraceae, Erythrobacteraceae, Alteromonadales, Flavobacteriaceae, Xanthomonadaceae |
| Captive vs. Semi-Captive | 13.4 | Alteromonadales, Pseudomonadaceae, Pseudoalteromonadaceae, Halomonadaceae, Rhodobacterales, Flavobacteriaceae, Caulobacteraceae, Moraxellaceae, Bradyrhizobiaceae, Rickettsiales |
| PERMANOVA | PERMDISP | ||||||
|---|---|---|---|---|---|---|---|
| Family Level | d.f. | Sum Sq | Mean Sq | Pseudo-F | p-(Perm) | F-Value | p-Value |
| Wild vs. Captive vs. Semi-Captive | 2 | 446 | 223 | 1.84 | 0.054 | 1.29 | 0.598 |
| Residual | 25 | 2913.7 | 121.4 | ||||
| Total | 27 | 3359.7 | |||||
| Genera Level | |||||||
| Wild vs. Captive vs. Semi-Captive | 2 | 1045.7 | 261.41 | 1.68 | 0.085 | 1.85 | 0.665 |
| Residual | 25 | 3886.2 | 155.45 | ||||
| Total | 27 | 4931.9 | |||||
| Gene Function: Subsystem Level 2 | |||||||
| Wild vs. Captive vs. Semi-Captive | 2 | 774.15 | 129 | 1.69 | 0.082 | 1.58 | 0.472 |
| Residual | 25 | 3048.2 | 76.21 | ||||
| Total | 27 | 3822.3 | |||||
| Gene Function: Subsystem Level 3 | |||||||
| Wild vs. Captive vs. Semi-Captive | 2 | 3712 | 618.7 | 1.79 | 0.052 | 4.02 | 0.068 |
| Residual | 25 | 13,848 | 346.2 | ||||
| Total | 27 | 17,560 | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goodman, A.Z.; Papudeshi, B.; Doane, M.P.; Mora, M.; Kerr, E.; Torres, M.; Nero Moffatt, J.; Lima, L.; Nosal, A.P.; Dinsdale, E. Epidermal Microbiomes of Leopard Sharks (Triakis semifasciata) Are Consistent across Captive and Wild Environments. Microorganisms 2022, 10, 2081. https://doi.org/10.3390/microorganisms10102081
Goodman AZ, Papudeshi B, Doane MP, Mora M, Kerr E, Torres M, Nero Moffatt J, Lima L, Nosal AP, Dinsdale E. Epidermal Microbiomes of Leopard Sharks (Triakis semifasciata) Are Consistent across Captive and Wild Environments. Microorganisms. 2022; 10(10):2081. https://doi.org/10.3390/microorganisms10102081
Chicago/Turabian StyleGoodman, Asha Z., Bhavya Papudeshi, Michael P. Doane, Maria Mora, Emma Kerr, Melissa Torres, Jennifer Nero Moffatt, Lais Lima, Andrew P. Nosal, and Elizabeth Dinsdale. 2022. "Epidermal Microbiomes of Leopard Sharks (Triakis semifasciata) Are Consistent across Captive and Wild Environments" Microorganisms 10, no. 10: 2081. https://doi.org/10.3390/microorganisms10102081
APA StyleGoodman, A. Z., Papudeshi, B., Doane, M. P., Mora, M., Kerr, E., Torres, M., Nero Moffatt, J., Lima, L., Nosal, A. P., & Dinsdale, E. (2022). Epidermal Microbiomes of Leopard Sharks (Triakis semifasciata) Are Consistent across Captive and Wild Environments. Microorganisms, 10(10), 2081. https://doi.org/10.3390/microorganisms10102081