
Determining the International Spread of B.1.1.523 SARS-CoV-2 Lineage with a Set of Mutations Highly Associated with Reduced Immune Neutralization

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Collection of SARS-CoV-2 Sequences and Initial Data Processing
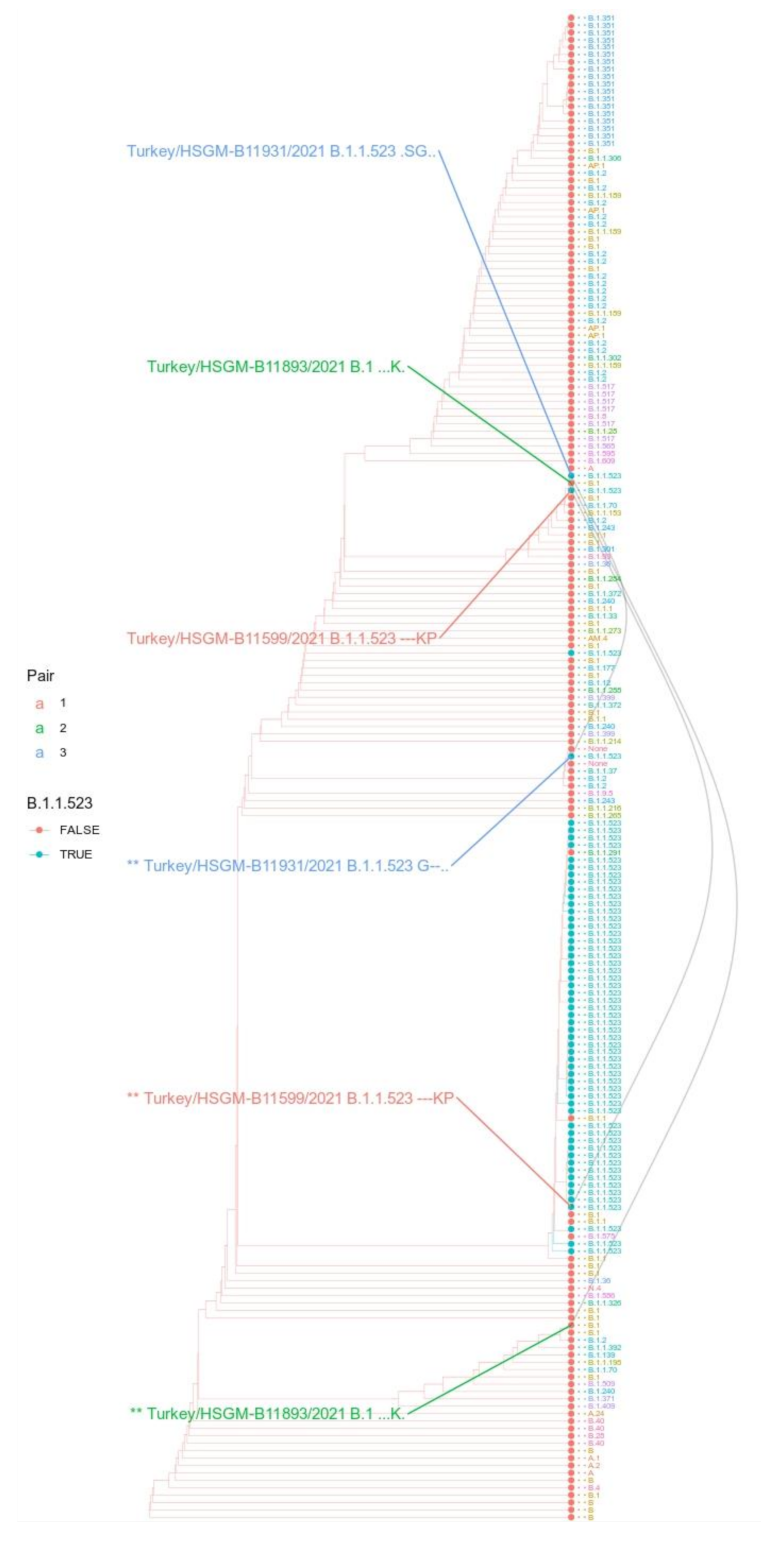
2.2. Transmission Cluster Analysis
2.3. S Protein Phylogeny Analysis
2.4. Analysis of Potential Recombination Events
2.5. Antibody Escape Effect Estimation
3. Results
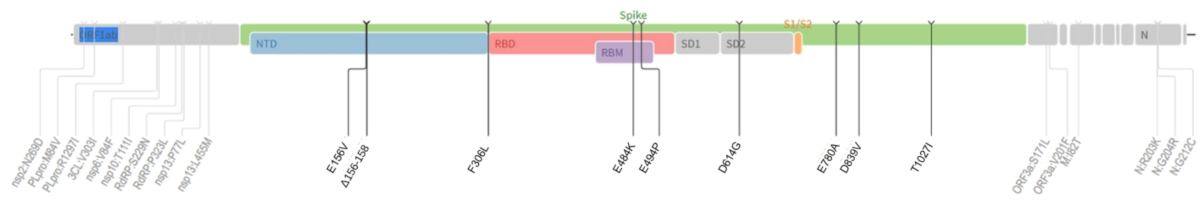
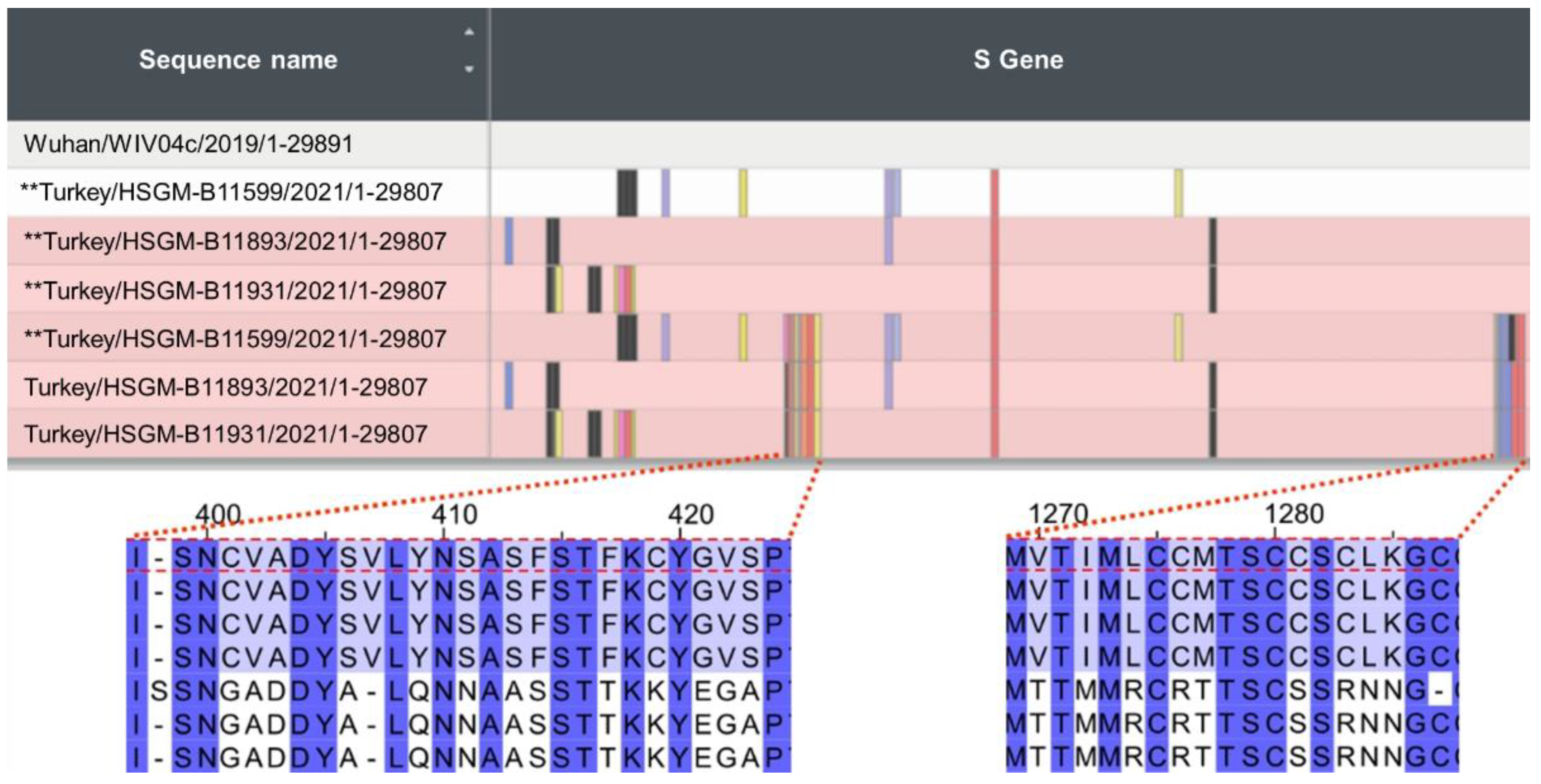
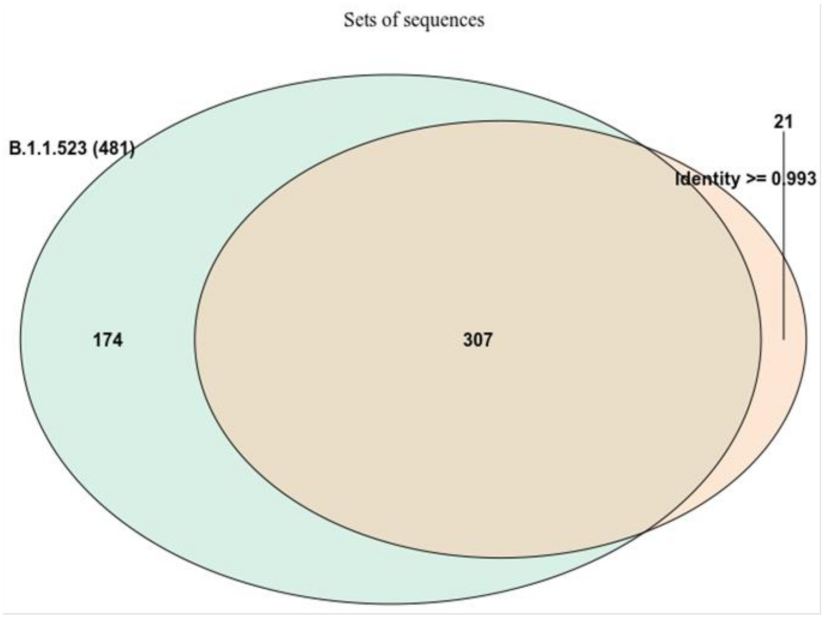
3.1. Mutation Review of B.1.1.523
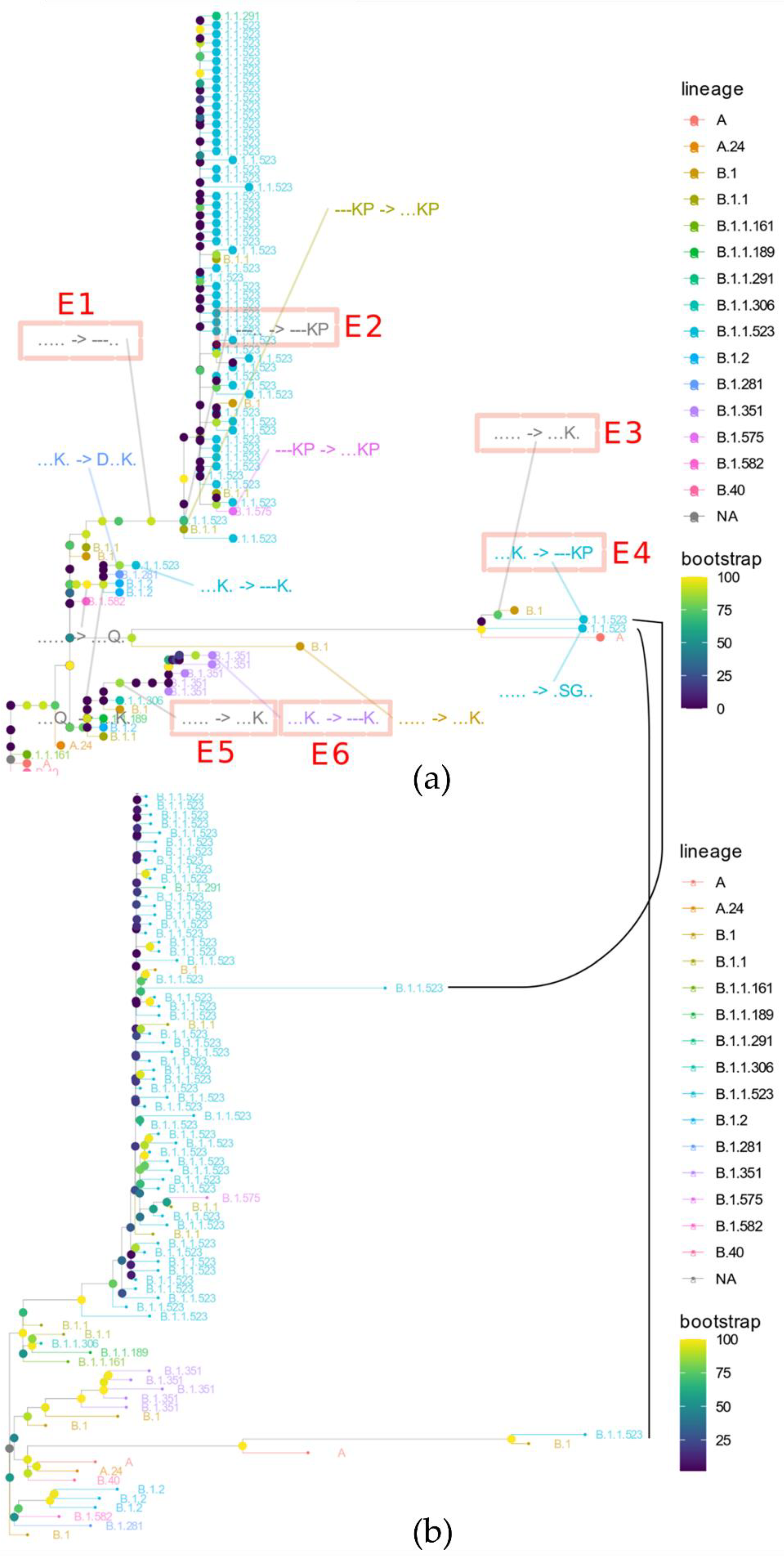
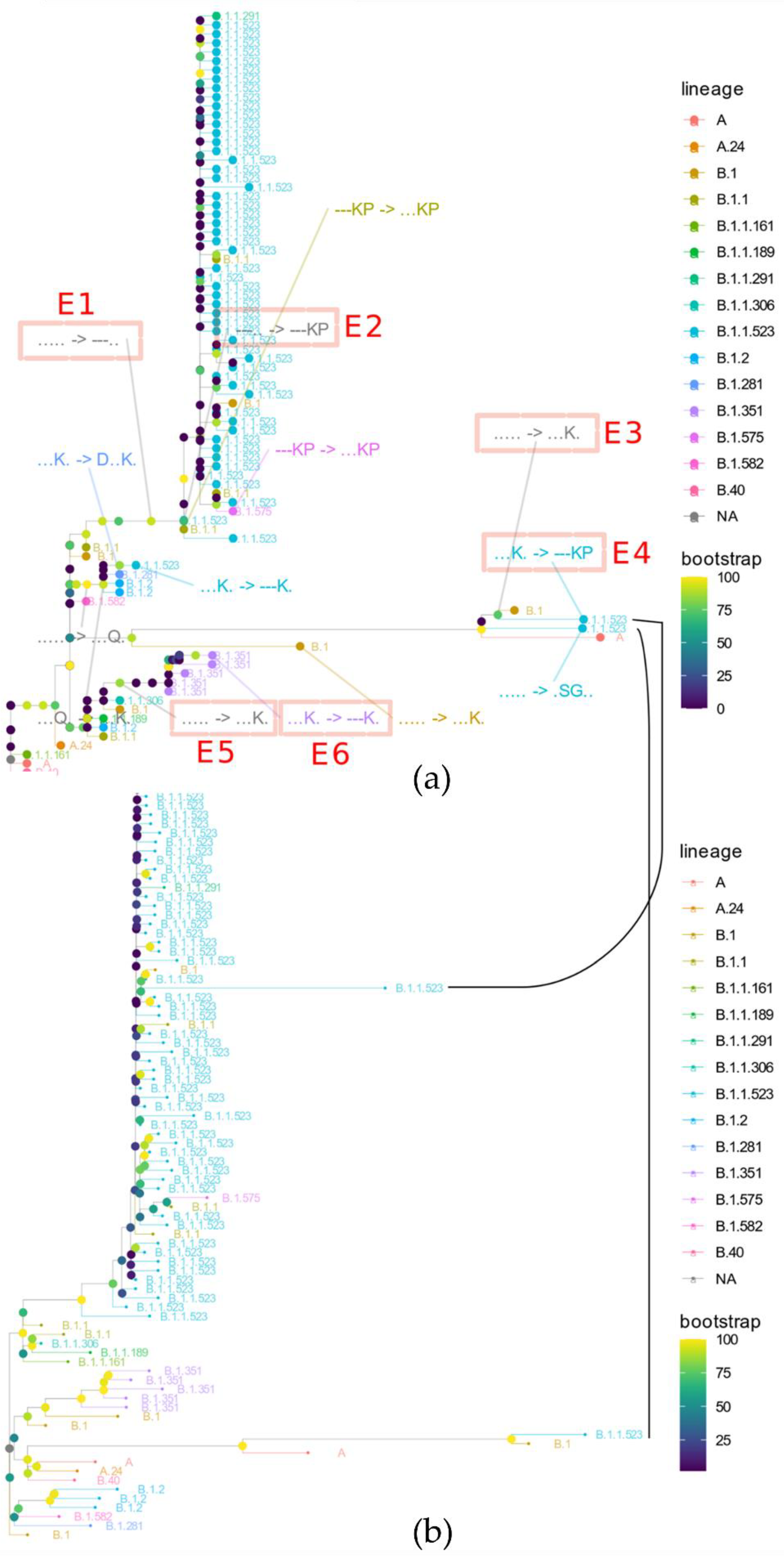
3.2. The Origin and Formation of Key S Protein Formation of the Lineage
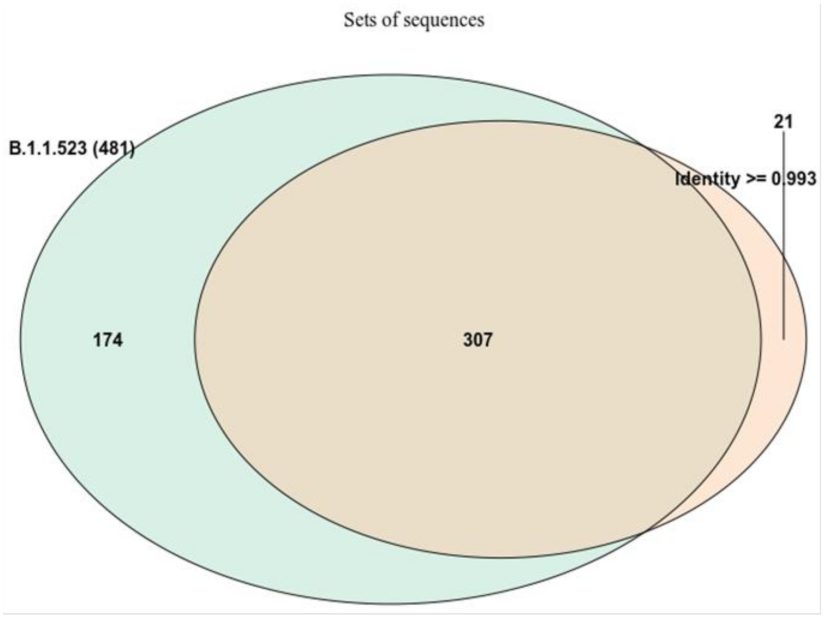
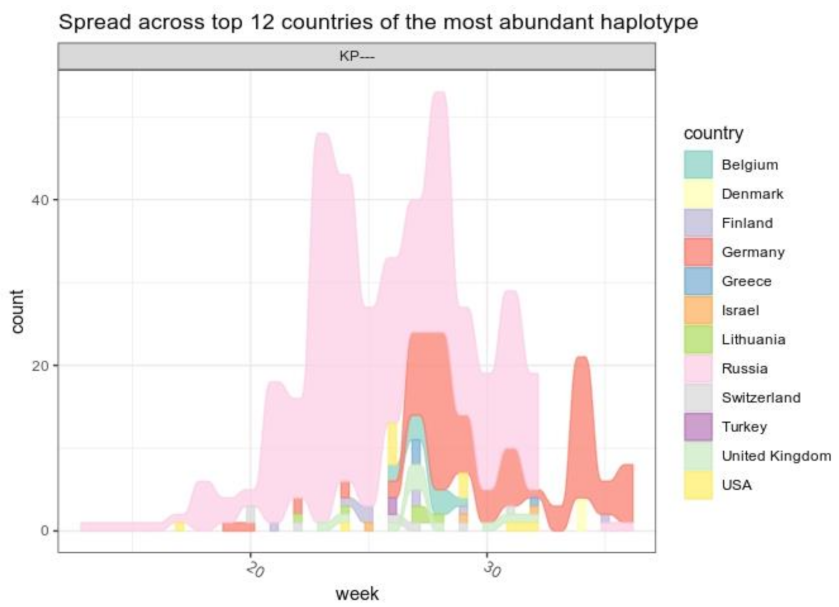
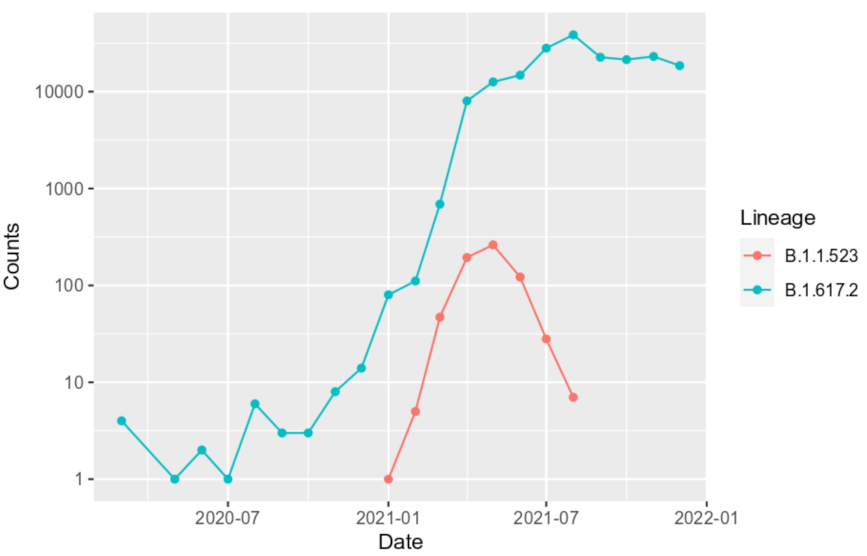
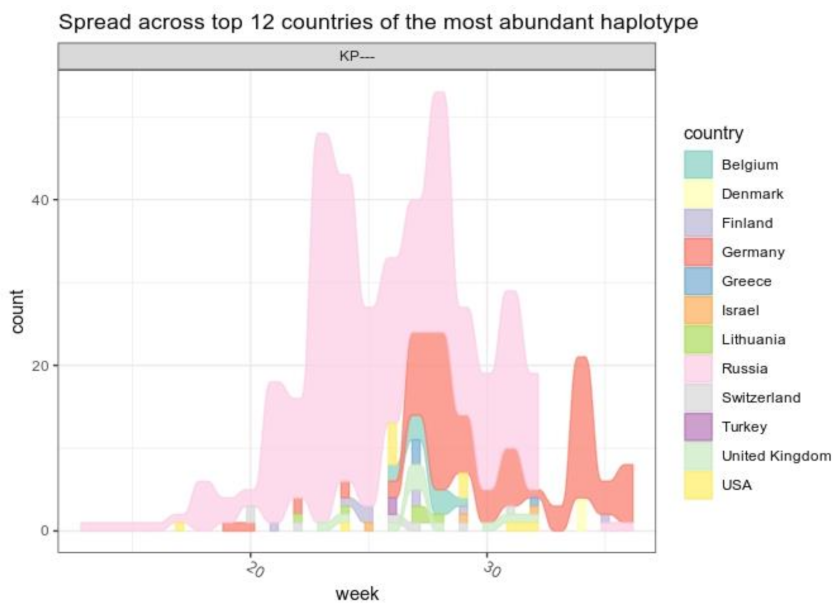
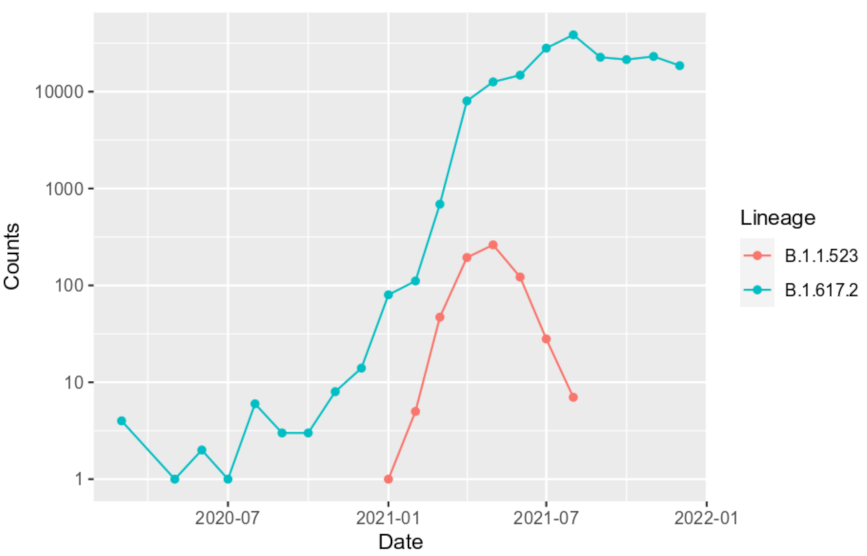
3.3. B.1.1.523 Spread Worldwide
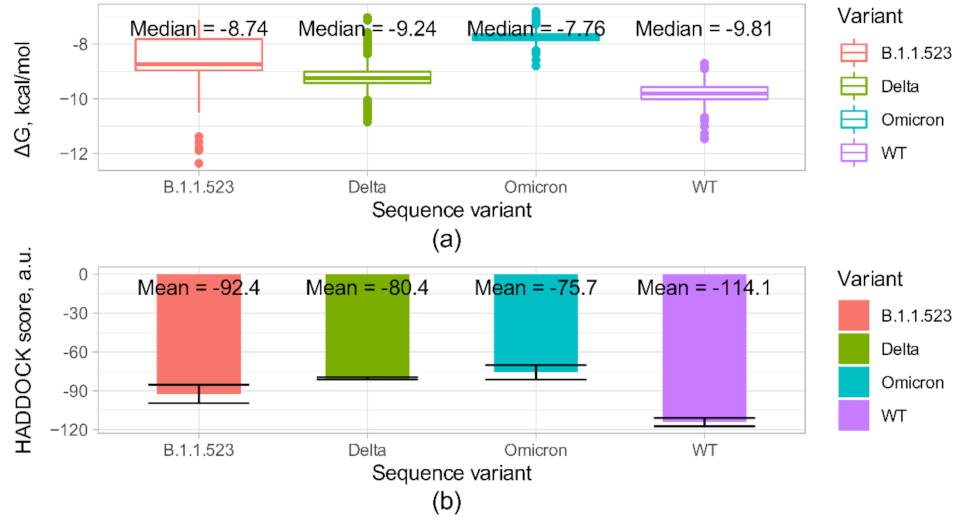
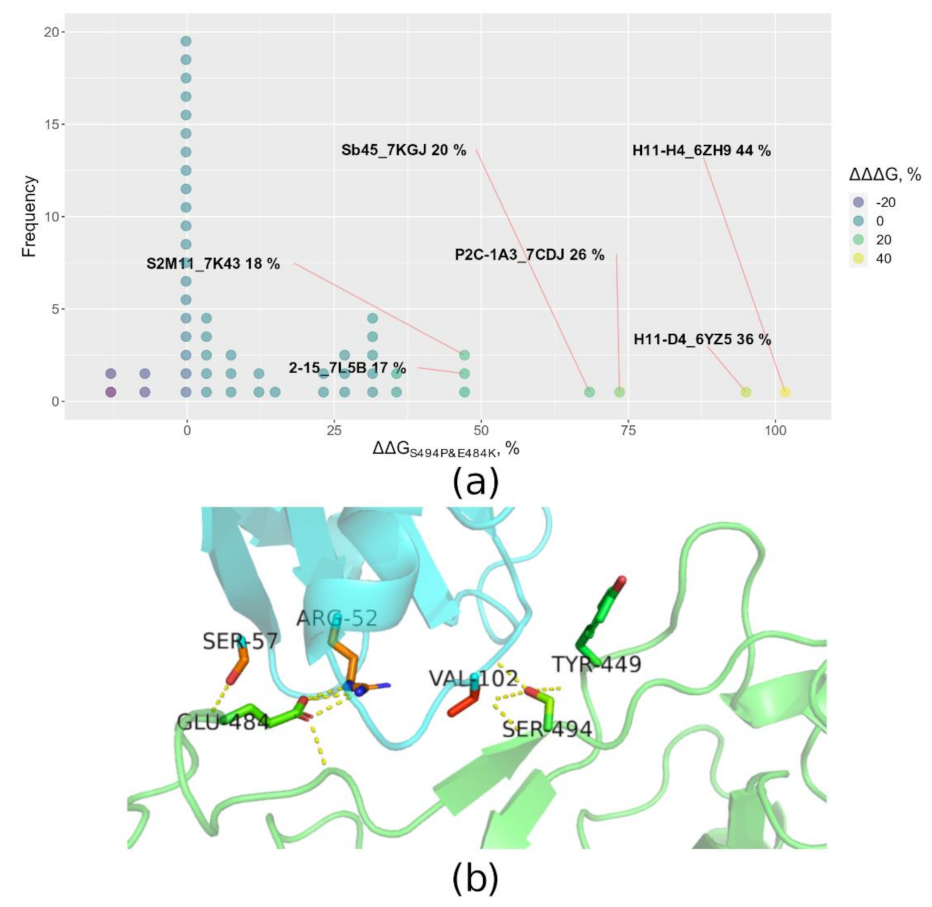
3.4. B.1.1.523 Antibody Escape
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
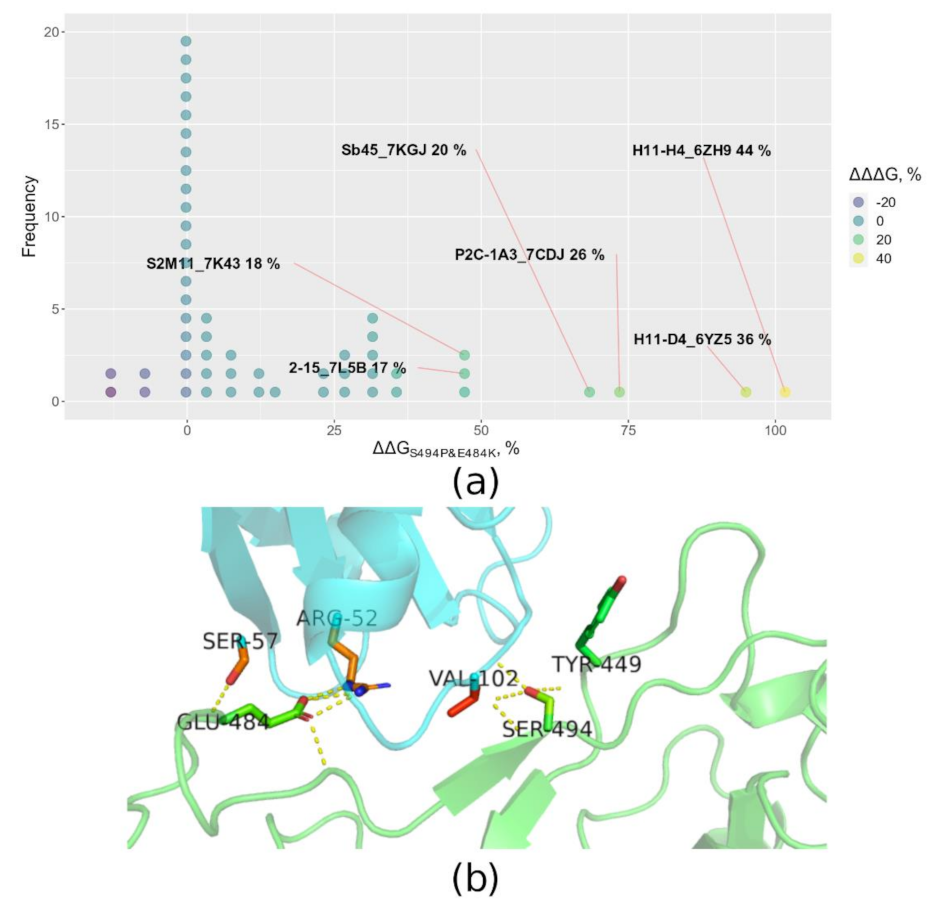{kind=link}
{kind=link}
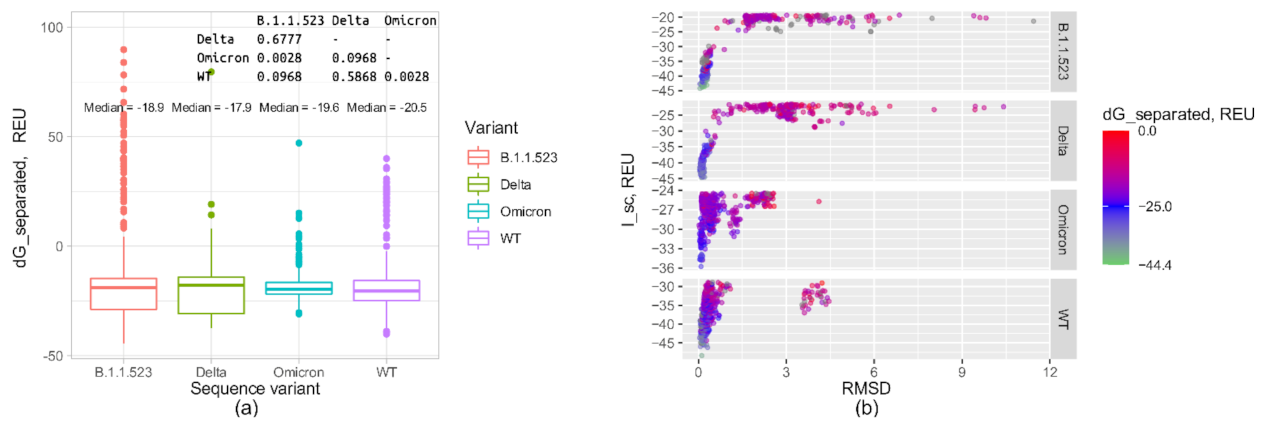{kind=link}
| Sequence Variant | |||||
|---|---|---|---|---|---|
| WT | B.1.1.523 | Delta | Omicron | ||
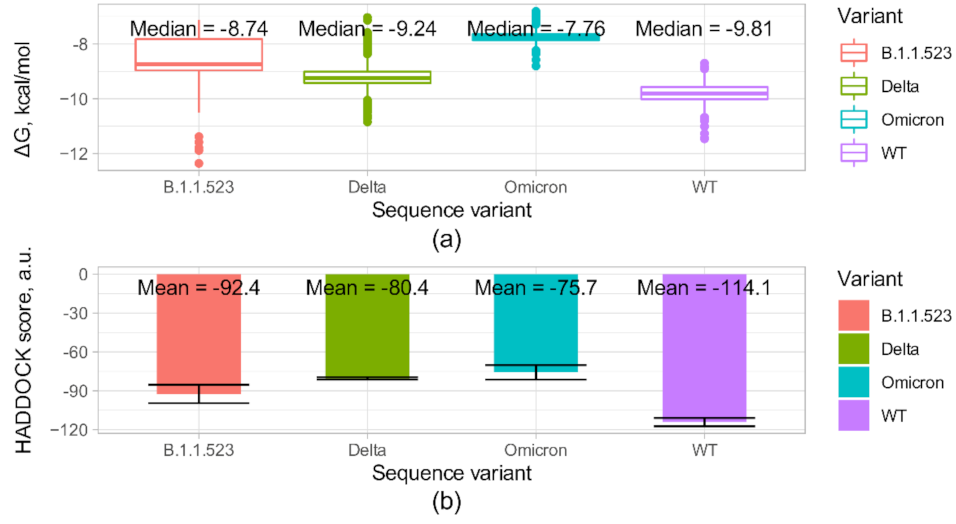
| Predicted binding ∆G, kcal/mol | −11.2 | −9 | −9.6 | −8.8 | |
| Number of Interfacial Contacts (ICs) | ICs charged-charged | 8 | 3 | 5 | 3 |
| ICs charged-polar | 9 | 7 | 9 | 8 | |
| ICs charged-apolar | 14 | 6 | 7 | 17 | |
| ICs polar-polar | 2 | 3 | 4 | 4 | |
| ICs polar-apolar | 14 | 12 | 14 | 7 | |
| ICs apolar-apolar | 14 | 14 | 17 | 9 | |
| Non-Interacting Surface (NIS) | NIS charged, | 17.95 | 17.81 | 17.86 | 18.73 |
| NIS apolar, | 38.72 | 39.95 | 39.8 | 39.49 | |
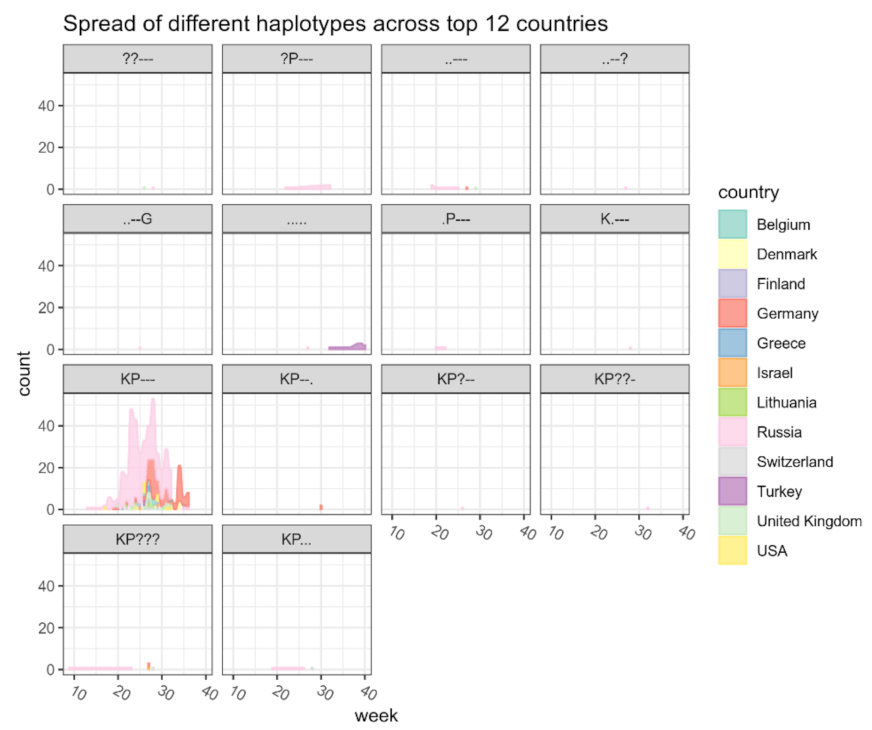
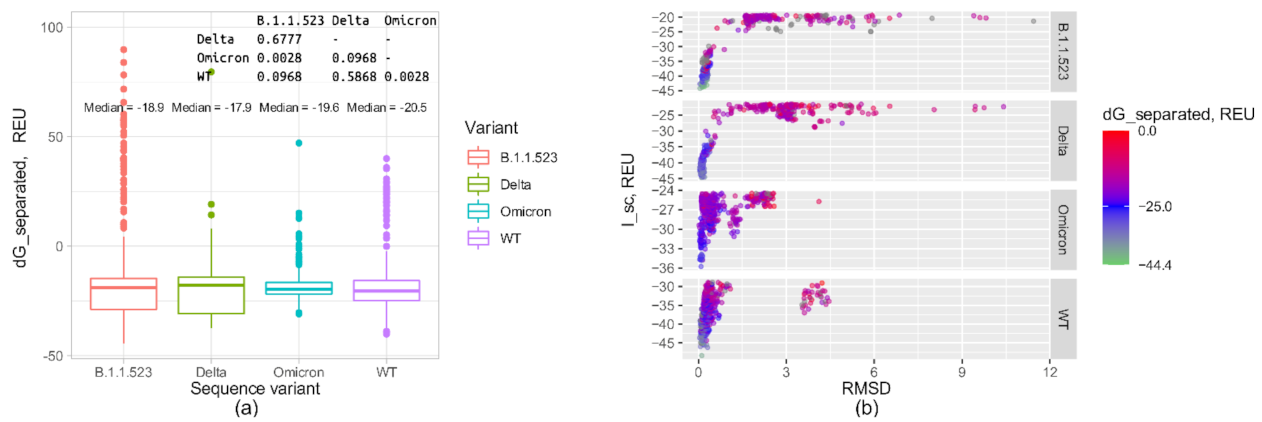
References
- World Health Organisation. WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 1 October 2021).
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Centre for Disease Prevention and Control. How ECDC Collects and Processes COVID-19 Data. Available online: https://www.ecdc.europa.eu/en/covid-19/data-collection (accessed on 1 October 2021).
- Yuelong, S.; John, M. GISAID: Global initiative on sharing all influenza data—From vision to reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [Green Version]
- World Health Organisation. Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 1 October 2021).
- European Centre for Disease Prevention and Control. SARS-CoV-2 Variants of Concern as of 30 September 2021. Available online: https://www.ecdc.europa.eu/en/covid-19/variants-concern (accessed on 1 October 2021).
- O’Toole, Á.; Hill, V.; Pybus, O.G.; Watts, A.; Bogoch, I.I.; Khan, K.; Messina, J.P.; Tegally, H.; Lessells, R.R.; Giandhari, J.; et al. Tracking the international spread of SARS-CoV-2 lineages B.1.1.7 and B.1.351/501Y-V2. Wellcome Open Res. 2021, 6, 121. [Google Scholar] [CrossRef]
- Aksamentov, I.; Roemer, C.; Hodcroft, E.B.; Neher, R.A. Nextclade: Clade assignment, mutation calling and quality control for viral genomes. J. Open Source Softw. 2021, 6, 3773. [Google Scholar] [CrossRef]
- Li, H. New strategies to improve minimap2 alignment accuracy. Bioinformatics 2021, 37, 4572–4574. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Tavaré, S. Some Probabilistic and Statistical Problems in the Analysis of DNA Sequences; Miura, R., Ed.; 1985 Revis; The American Mathematical Society: Providence, RI, USA, 1986. [Google Scholar]
- Gu, X.; Fu, Y.X.; Li, W.H. Maximum likelihood estimation of the heterogeneity of substitution rate among nucleotide sites. Mol. Biol. Evol. 1995, 12, 546–557. [Google Scholar] [CrossRef] [Green Version]
- Han, A.; Parker, E.; Maurer-Stroh, S.; Russell, C.A. Inferring putative transmission clusters with Phydelity. Virus Evol. 2019, 5, vez039. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Piñeiro, C.; Abuín, J.M.; Pichel, J.C. Very Fast Tree: Speeding up the estimation of phylogenies for large alignments through parallelization and vectorization strategies. Bioinformatics 2020, 36, 4658–4659. [Google Scholar] [CrossRef]
- Lemoine, F.; Gascuel, O. Gotree/Goalign: Toolkit and Go API to facilitate the development of phylogenetic workflows. NAR Genom. Bioinform. 2021, 3, lqab075. [Google Scholar] [CrossRef]
- Foley, G.; Mora, A.; Ross, C.M.; Bottoms, S.; Sützl, L.; Lamprecht, M.L.; Zaugg, J.; Essebier, A.; Balderson, B.; Newell, R.; et al. Identifying and engineering ancient variants of enzymes using Graphical Representation of Ancestral Sequence Predictions (GRASP). bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.Y. GGTREE: An r package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Hölzer, M.; Marz, M. PoSeiDon: A Nextflow pipeline for the detection of evolutionary recombination events and positive selection. Bioinformatics 2021, 37, 1018–1020. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D.W. GARD: A genetic algorithm for recombination detection. Bioinformatics 2006, 22, 3096–3098. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Q.; Tiedje, K.; Ruybal-Pesántez, S.; Tonkin-Hill, G.; Duffy, M.; Day, K.; Shim, H.; Chan, Y. A scalable method for identifying recombinants from unaligned sequences. Bioinformatics 2022, 38, 1823–1829. [Google Scholar] [CrossRef]
- Lam, H.M.; Ratmann, O.; Boni, M.F. Improved Algorithmic Complexity for the 3SEQ Recombination Detection Algorithm. Mol. Biol. Evol. 2018, 35, 247–251. [Google Scholar] [CrossRef] [Green Version]
- Cerutti, G.; Guo, Y.; Zhou, T.; Gorman, J.; Lee, M.; Rapp, M.; Reddem, E.R.; Yu, J.; Bahna, F.; Bimela, J.; et al. Potent SARS-CoV-2 neutralizing antibodies directed against spike N-terminal domain target a single supersite. Cell Host Microbe 2021, 29, 819–833.e7. [Google Scholar] [CrossRef]
- Leman, J.K.; Weitzner, B.D.; Lewis, S.M.; Adolf-Bryfogle, J.; Alam, N.; Alford, R.F.; Aprahamian, M.; Baker, D.; Barlow, K.A.; Barth, P.; et al. Macromolecular modeling and design in Rosetta: Recent methods and frameworks. Nat. Methods 2020, 17, 665–680. [Google Scholar] [CrossRef]
- Van Zundert, G.C.P.; Rodrigues, J.P.G.L.M.; Trellet, M.; Schmitz, C.; Kastritis, P.L.; Karaca, E.; Melquiond, A.S.J.; Van Dijk, M.; De Vries, S.J.; Bonvin, A.M.J.J. The HADDOCK2.2 Web Server: User-Friendly Integrative Modeling of Biomolecular Complexes. J. Mol. Biol. 2016, 428, 720–725. [Google Scholar] [CrossRef] [Green Version]
- Schymkowitz, J.; Borg, J.; Stricher, F.; Nys, R.; Rousseau, F.; Serrano, L. The FoldX web server: An online force field. Nucleic Acids Res. 2005, 33, W382–W388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vangone, A.; Bonvin, A.M.J.J. Contacts-based prediction of binding affinity in protein–protein complexes. eLife 2015, 4, e07454. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger PyMOL. Available online: https://pymol.org/ (accessed on 28 June 2022).
- Planas, D.; Veyer, D.; Baidaliuk, A.; Staropoli, I.; Guivel-Benhassine, F.; Rajah, M.M.; Planchais, C.; Porrot, F.; Robillard, N.; Puech, J.; et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature 2021, 596, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Collier, D.A.; Marco, A.D.; Ferreira, I.A.T.M.; Meng, B.; Datir, R.; Walls, A.C.; Kemp S, S.A.; Bassi, J.; Pinto, D.; Fregni, C.S.; et al. SARS-CoV-2 B.1.1.7 escape from mRNA vaccine-elicited neutralizing antibodies. Nature 2021, 593, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Greaney, A.J.; Loes, A.N.; Crawford, K.H.D.; Starr, T.N.; Malone, K.D.; Chu, H.Y.; Bloom, J.D. Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe 2021, 29, 463–476.e6. [Google Scholar] [CrossRef]
- Jungreis, I.; Sealfon, R.; Kellis, M. SARS-CoV-2 gene content and COVID-19 mutation impact by comparing 44 Sarbecovirus genomes. Nat. Commun. 2021, 12, 2642. [Google Scholar] [CrossRef]
- Shneider, A.D.B.; Su, M.; Hinrichs, A.; Wang, J.; Amin, H.; Bell, J.; Wadford, D.A.; O’toole, A.; Scher, E.; Perry, M.D.; et al. SARS-CoV-2 Lineage Assignment Is More Stable with UShER. Available online: https://virological.org/t/sars-cov-2-lineage-assignment-is-more-stable-with-usher/781 (accessed on 20 June 2022).
- Chen, Z.; Azman, A.S.; Chen, X.; Zou, J.; Tian, Y.; Sun, R.; Xu, X.; Wu, Y.; Lu, W.; Ge, S.; et al. Global landscape of SARS-CoV-2 genomic surveillance and data sharing. Nat. Genet. 2022, 54, 499–507. [Google Scholar] [CrossRef]
- Spiliotopoulos, D.; Kastritis, P.L.; Melquiond, A.S.J.; Bonvin, A.M.J.J.; Musco, G.; Rocchia, W.; Spitaleri, A. dMM-PBSA: A new HADDOCK scoring function for protein-peptide docking. Front. Mol. Biosci. 2016, 3, 46. [Google Scholar] [CrossRef] [Green Version]
- Gorman, J.; Rapp, M.; Kwong, P.D.; Shapiro, L. Cryo-EM Structure of NTD-Directed Neutralizing Antibody 4–8 Fab in Complex with SARS-CoV-2 S2P Spike (RCSB PDB-7LQV). Available online: https://www.rcsb.org/structure/7LQV (accessed on 29 January 2022).
- Koenig, P.A.; Das, H.; Liu, H.; Kümmerer, B.M.; Gohr, F.N.; Jenster, L.M.; Schiffelers, L.D.J.; Tesfamariam, Y.M.; Uchima, M.; Wuerth, J.D.; et al. Structure-guided multivalent nanobodies block SARS-CoV-2 infection and suppress mutational escape. Science 2021, 371, eabe6230. [Google Scholar] [CrossRef]
- Huo, J.; Le Bas, A.; Ruza, R.R.; Duyvesteyn, H.M.E.; Mikolajek, H.; Malinauskas, T.; Tan, T.K.; Rijal, P.; Dumoux, M.; Ward, P.N.; et al. Neutralizing nanobodies bind SARS-CoV-2 spike RBD and block interaction with ACE2. Nat. Struct. Mol. Biol. 2020, 279, 846–854. [Google Scholar] [CrossRef]
- Ahmad, J.; Jiang, J.; Boyd, L.F.; Zeher, A.; Huang, R.; Xia, D.; Natarajan, K.; Margulies, D.H. Structures of synthetic nanobody–SARS-CoV-2 receptor-binding domain complexes reveal distinct sites of interaction. J. Biol. Chem. 2021, 297, 101202. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.; Wang, R.; Ju, B.; Zhang, Q.; Sun, J.; Chen, P.; Zhang, S.; Tian, Y.; Shan, S.; Cheng, L.; et al. Antibody neutralization of SARS-CoV-2 through ACE2 receptor mimicry. Nat. Commun. 2021, 12, 250. [Google Scholar] [CrossRef] [PubMed]
- Tortorici, M.A.; Beltramello, M.; Lempp, F.A.; Pinto, D.; Dang, H.V.; Rosen, L.E.; McCallum, M.; Bowen, J.; Minola, A.; Jaconi, S.; et al. Ultrapotent human antibodies protect against SARS-CoV-2 challenge via multiple mechanisms. Science 2020, 370, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Latif, A.A.; Mullen, J.L.; Alkuzweny, M.; Tsueng, G.; Cano, M.; Haag, E.; Zhou, J.; Zeller, M.; Hufbauer, E.; Matteson, N.; et al. B.1.1.523 Lineage Report. Available online: https://outbreak.info/situation-reports?pango=B.1.1.523 (accessed on 31 August 2021).
- McCallum, M.; Walls, A.C.; Sprouse, K.R.; Bowen, J.E.; Rosen, L.; Dang, H.V.; deMarco, A.; Franko, N.; Tilles, S.W.; Logue, J.; et al. Molecular basis of immune evasion by the delta and kappa SARS-CoV-2 variants. Science. 2021, 374, 1621–1626. [Google Scholar] [CrossRef] [PubMed]
- Wise, J. COVID-19: The E484K mutation and the risks it poses. BMJ 2021, 372, n359. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.V.; VanBlargan, L.A.; Bloyet, L.M.; Rothlauf, P.W.; Chen, R.E.; Stumpf, S.; Zhao, H.; Errico, J.M.; Theel, E.S.; Liebeskind, M.J.; et al. Identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe 2021, 29, 477–488.e4. [Google Scholar] [CrossRef]
- Lassaunière, R.; Polacek, C.; Fonager, J.; Bennedbæk, M.; Boding, L.; Rasmussen, M.; Fomsgaard, A. Neutralisation of the SARS-CoV-2 Delta variant sub-lineages AY.4.2 and B.1.617.2 with the mutation E484K by Comirnaty (BNT162b2 mRNA) vaccine-elicited sera, Denmark, 1 to 26 November 2021. Eurosurveillance 2021, 26, 2101059. [Google Scholar] [CrossRef]
- Li, T.; Cui, Z.; Jia, Y.; Liang, Z.; Nie, J.; Zhang, L.; Wang, M.; Li, Q.; Wu, J.; Xu, N.; et al. Aggregation of high-frequency RBD mutations of SARS-CoV-2 with three VOCs did not cause significant antigenic drift. J. Med. Virol. 2022, 94, 2108–2125. [Google Scholar] [CrossRef]
- Baj, A.; Novazzi, F.; Pasciuta, R.; Genoni, A.; Ferrante, F.D.; Valli, M.; Partenope, M.; Tripiciano, R.; Ciserchia, A.; Catanoso, G.; et al. Breakthrough Infections of E484K-Harboring SARS-CoV-2 Delta Variant, Lombardy, Italy. Emerg. Infect. Dis. 2021, 27, 3180–3182. [Google Scholar] [CrossRef]
- Hu, J.; Peng, P.; Cao, X.; Wu, K.; Chen, J.; Wang, K.; Tang, N.; Huang, A. Long increased immune escape of the new SARS-CoV-2 variant of concern Omicron. Cell. Mol. Immunol. 2022, 19, 293–295. [Google Scholar] [CrossRef]
- Kisa, S.; Kisa, A. Under-reporting of COVID-19 cases in Turkey. Int. J. Health Plan. Manag. 2020, 35, 1009–1013. [Google Scholar] [CrossRef] [PubMed]
- Gowthaman, R.; Guest, J.D.; Yin, R.; Adolf-Bryfogle, J.; Schief, W.R.; Pierce, B.G. CoV3D: A database of high resolution coronavirus protein structures. Nucleic Acids Res. 2021, 49, D282–D287. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zemaitis, L.; Alzbutas, G.; Gecys, D.; Pautienius, A.; Ugenskiene, R.; Sukys, M.; Lesauskaite, V. Determining the International Spread of B.1.1.523 SARS-CoV-2 Lineage with a Set of Mutations Highly Associated with Reduced Immune Neutralization. Microorganisms 2022, 10, 1356. https://doi.org/10.3390/microorganisms10071356
Zemaitis L, Alzbutas G, Gecys D, Pautienius A, Ugenskiene R, Sukys M, Lesauskaite V. Determining the International Spread of B.1.1.523 SARS-CoV-2 Lineage with a Set of Mutations Highly Associated with Reduced Immune Neutralization. Microorganisms. 2022; 10(7):1356. https://doi.org/10.3390/microorganisms10071356
Chicago/Turabian StyleZemaitis, Lukas, Gediminas Alzbutas, Dovydas Gecys, Arnoldas Pautienius, Rasa Ugenskiene, Marius Sukys, and Vaiva Lesauskaite. 2022. "Determining the International Spread of B.1.1.523 SARS-CoV-2 Lineage with a Set of Mutations Highly Associated with Reduced Immune Neutralization" Microorganisms 10, no. 7: 1356. https://doi.org/10.3390/microorganisms10071356
APA StyleZemaitis, L., Alzbutas, G., Gecys, D., Pautienius, A., Ugenskiene, R., Sukys, M., & Lesauskaite, V. (2022). Determining the International Spread of B.1.1.523 SARS-CoV-2 Lineage with a Set of Mutations Highly Associated with Reduced Immune Neutralization. Microorganisms, 10(7), 1356. https://doi.org/10.3390/microorganisms10071356