
Typing of the Gut Microbiota Community in Japanese Subjects

 , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Subjects and Data Collection
2.2. Sample Collection and DNA Extraction
2.3. Sequencing of the 16S rRNA Gene
2.4. Microbiome Analysis and Community Typing
2.5. Statistical Analysis
2.6. Ethics Statements
3. Results
3.1. Enrolled Study Participants
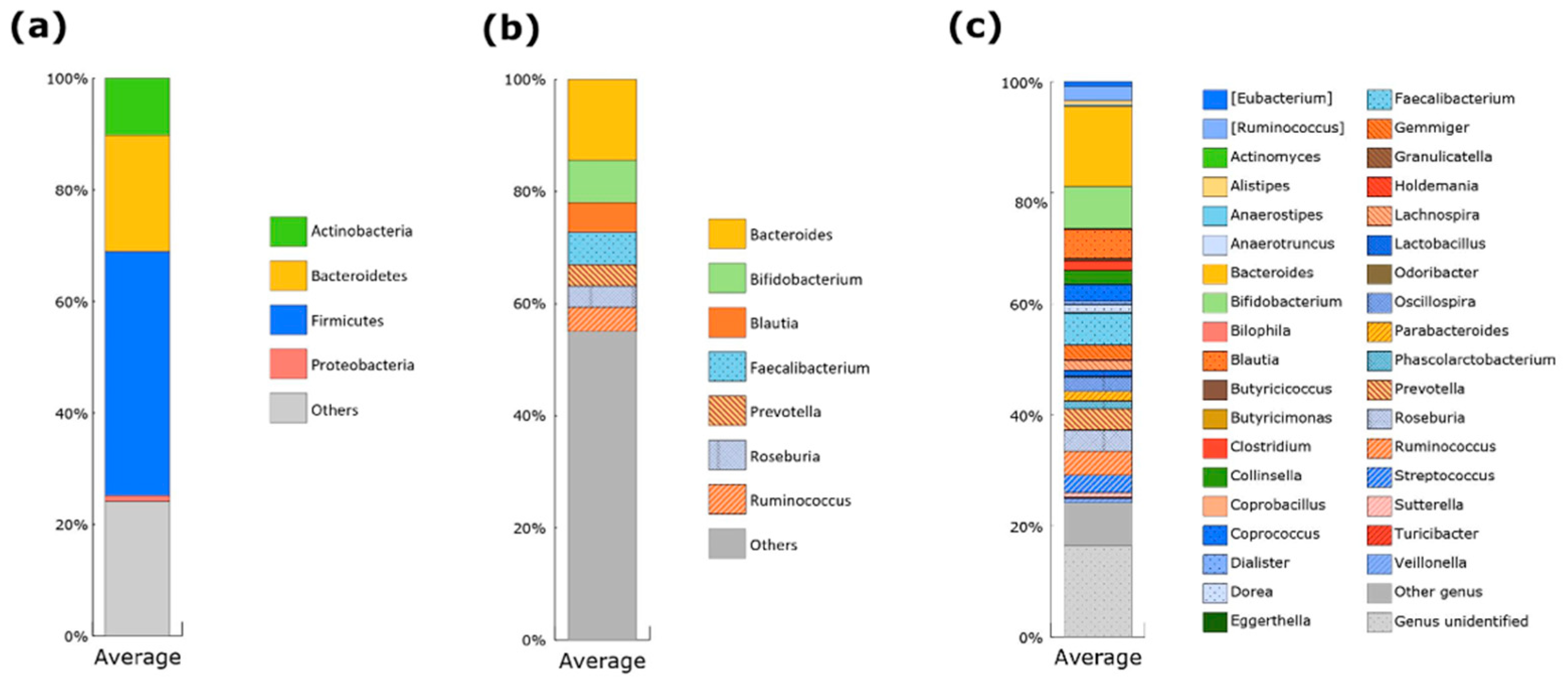
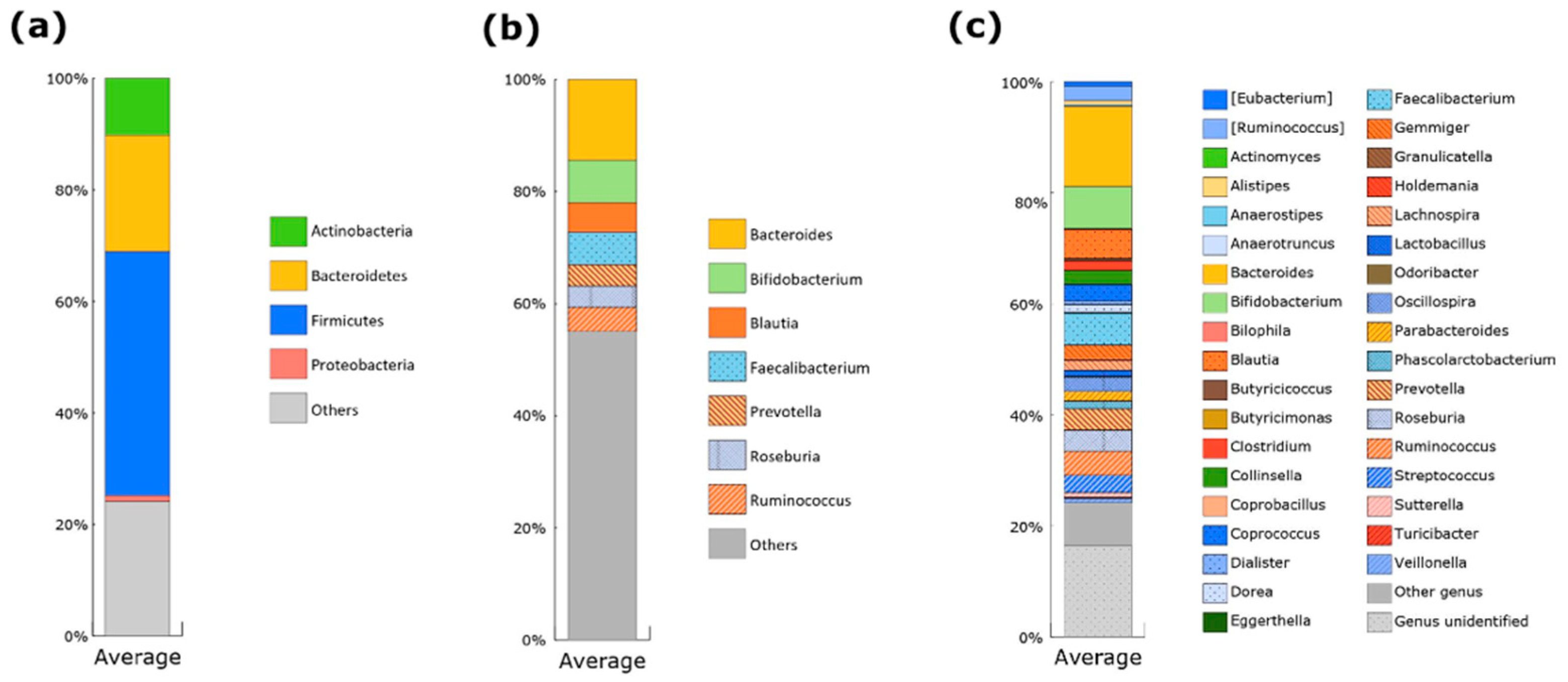
3.2. Gut Microbiota of Japanese Participants
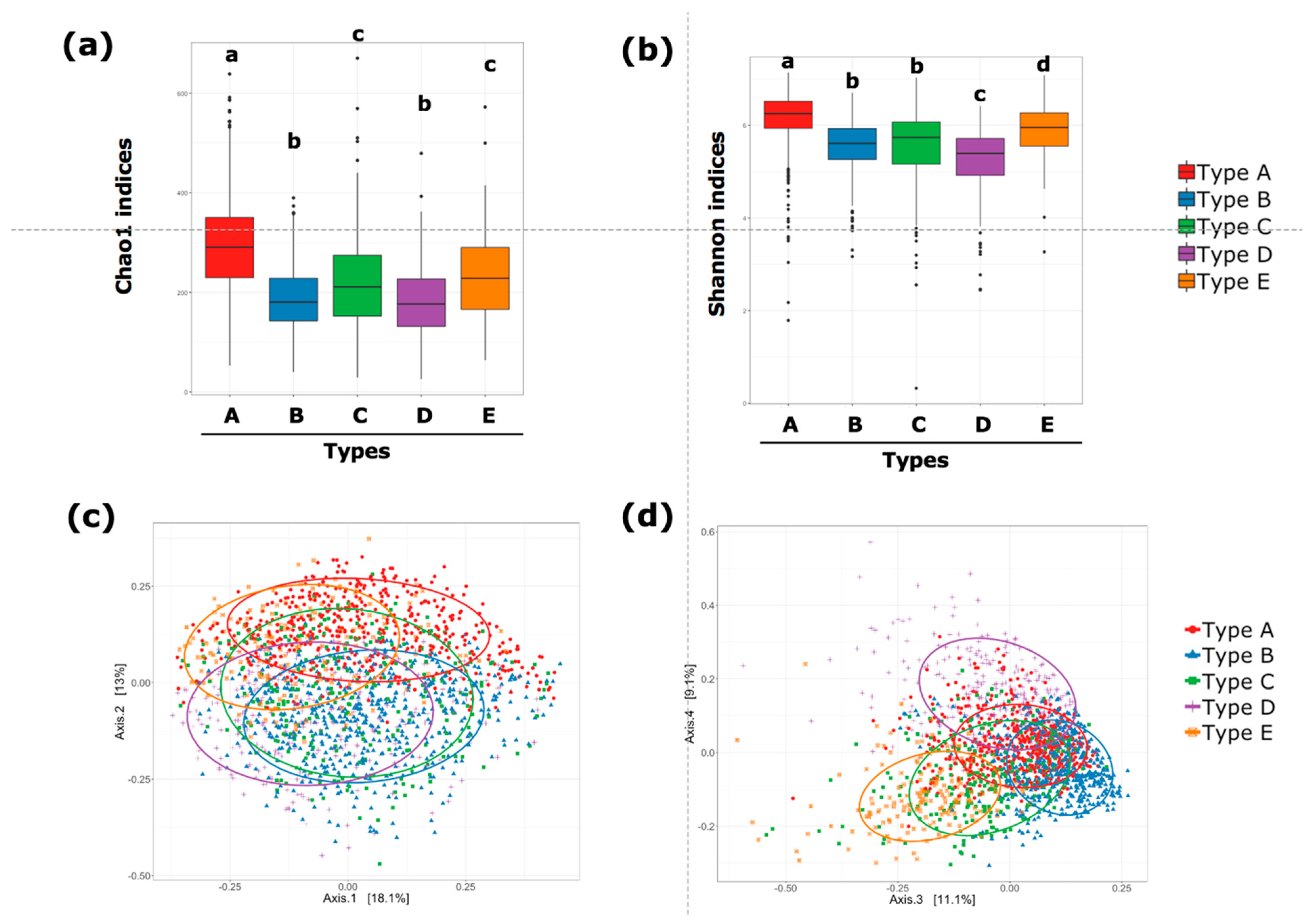
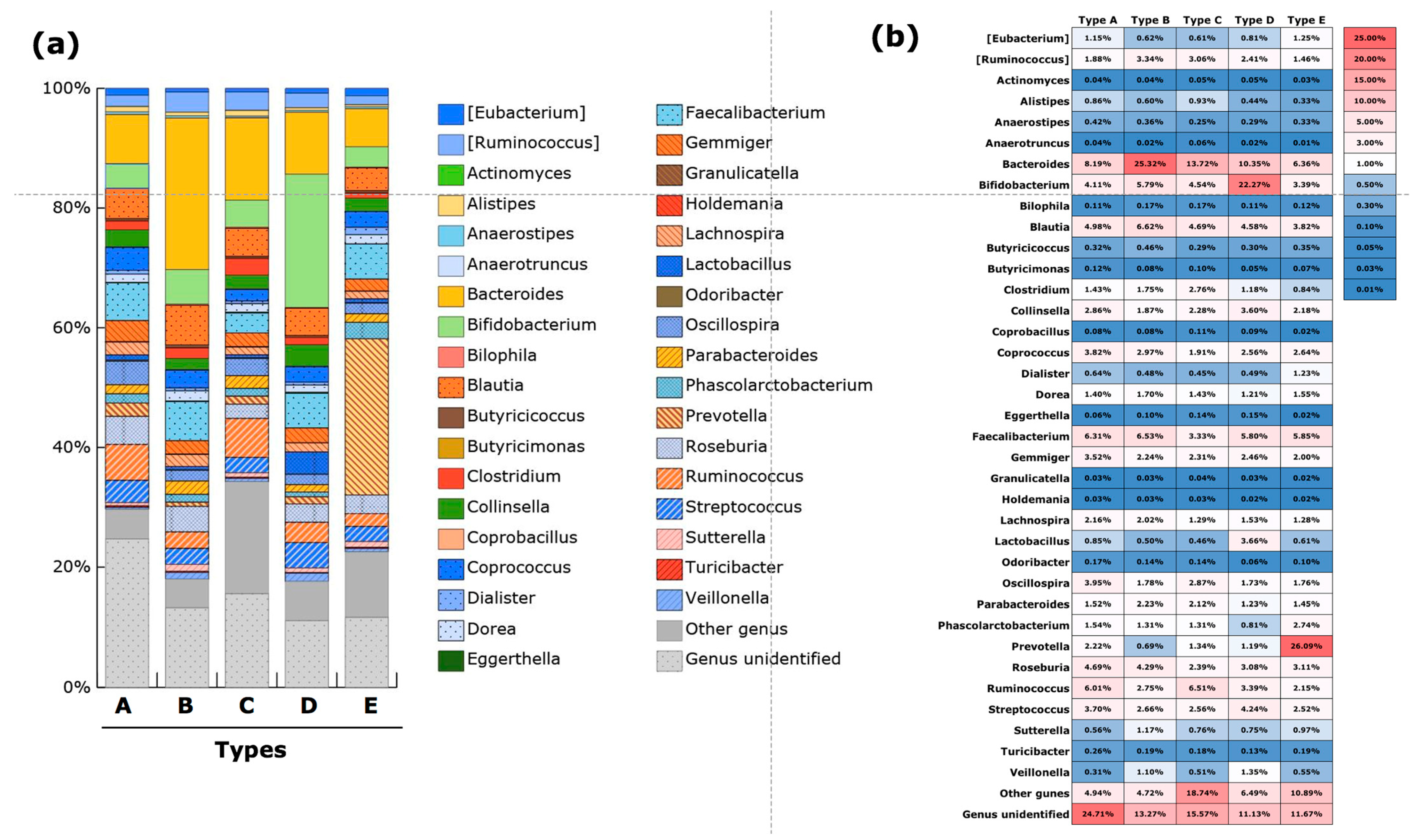
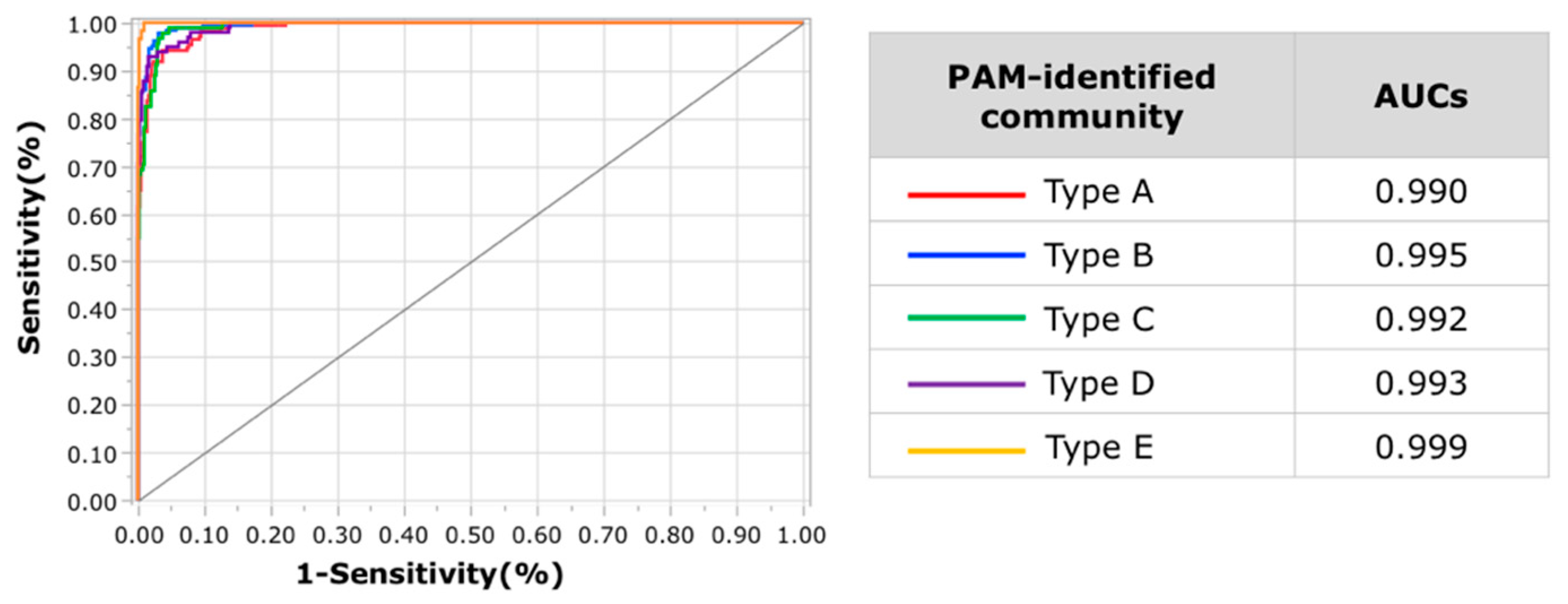
3.3. Gut Microbiota Community Typing in Japanese Participants Enrolled in the Study
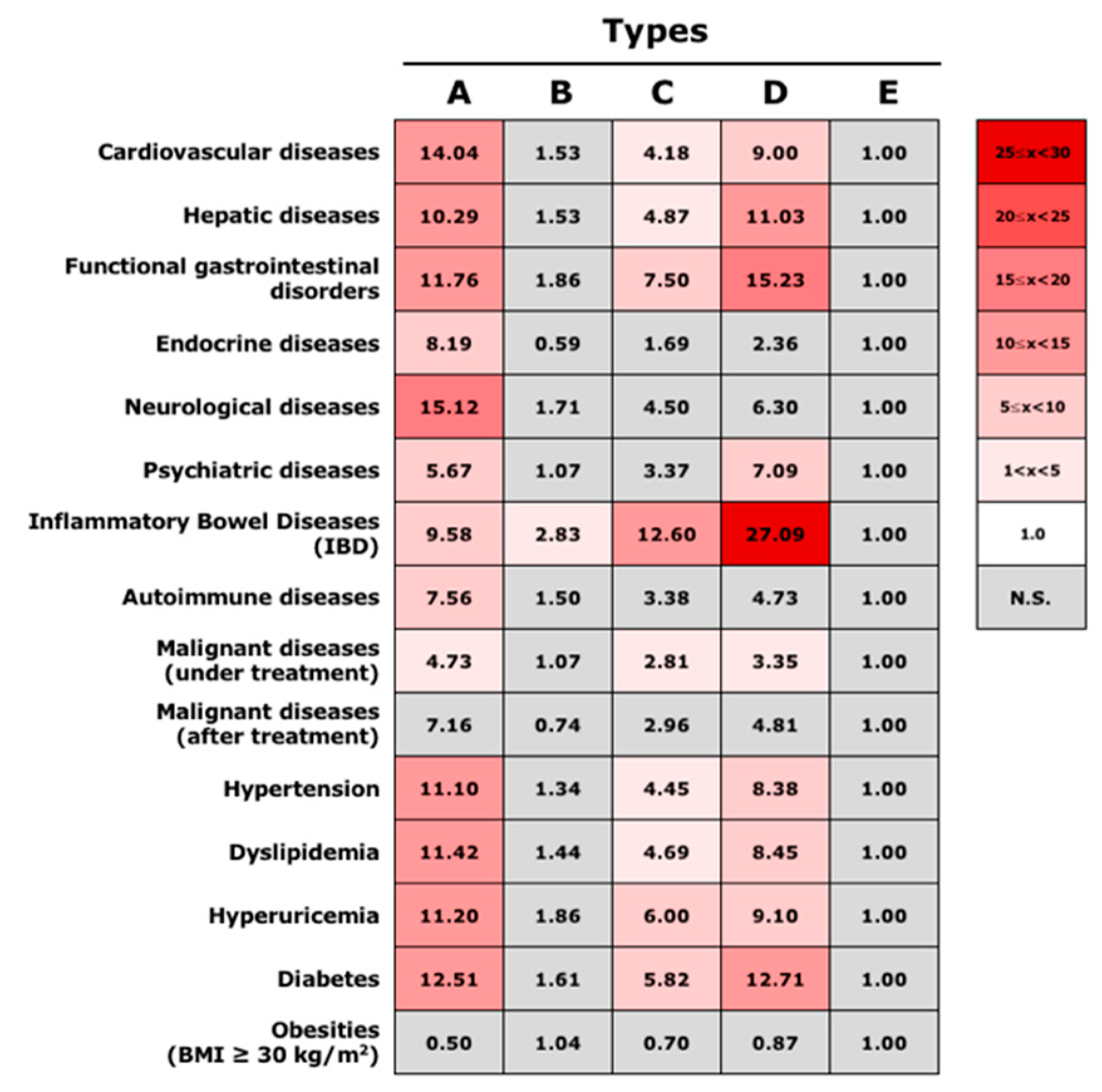
3.4. Association between Gut Microbiota Community Types and Disease Status
3.5. Gut Microbiota Communities Identified via DMM-Based Clustering
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Marchesi, J.R.; Adams, D.H.; Fava, F.; Hermes, G.D.; Hirschfield, G.M.; Hold, G.; Quraishi, M.N.; Kinross, J.; Smidt, H.; Tuohy, K.M.; et al. The gut microbiota and host health: A new clinical frontier. Gut 2016, 65, 330–339. [Google Scholar] [CrossRef] [Green Version]
- Lynch, S.V.; Pedersen, O. The Human Intestinal Microbiome in Health and Disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Fu, L.; Niu, B.; Wu, S.; Wooley, J. Ultrafast clustering algorithms for metagenomic sequence analysis. Brief. Bioinform. 2012, 13, 656–668. [Google Scholar] [CrossRef]
- Adenote, A.; Dumic, I.; Madrid, C.; Barusya, C.; Nordstrom, C.W.; Rueda Prada, L. NAFLD and Infection, a Nuanced Relationship. Can. J. Gastroenterol. Hepatol. 2021, 2021, 5556354. [Google Scholar] [CrossRef]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef] [Green Version]
- Vandeputte, D.; Kathagen, G.; D’Hoe, K.; Vieira-Silva, S.; Valles-Colomer, M.; Sabino, J.; Wang, J.; Tito, R.Y.; De Commer, L.; Darzi, Y.; et al. Quantitative microbiome profiling links gut community variation to microbial load. Nature 2017, 551, 507–511. [Google Scholar] [CrossRef]
- Holmes, I.; Harris, K.; Quince, C. Dirichlet multinomial mixtures: Generative models for microbial metagenomics. PLoS ONE 2012, 7, e30126. [Google Scholar] [CrossRef] [Green Version]
- Vandeputte, D.; Falony, G.; Vieira-Silva, S.; Tito, R.Y.; Joossens, M.; Raes, J. Stool consistency is strongly associated with gut microbiota richness and composition, enterotypes and bacterial growth rates. Gut 2016, 65, 57–62. [Google Scholar] [CrossRef] [Green Version]
- Vieira-Silva, S.; Sabino, J.; Valles-Colomer, M.; Falony, G.; Kathagen, G.; Caenepeel, C.; Cleynen, I.; van der Merwe, S.; Vermeire, S.; Raes, J. Quantitative microbiome profiling disentangles inflammation- and bile duct obstruction-associated microbiota alterations across PSC/IBD diagnoses. Nat. Microbiol. 2019, 4, 1826–1831. [Google Scholar] [CrossRef]
- Vieira-Silva, S.; Falony, G.; Belda, E.; Nielsen, T.; Aron-Wisnewsky, J.; Chakaroun, R.; Forslund, S.K.; Assmann, K.; Valles-Colomer, M.; Nguyen, T.T.D.; et al. Statin therapy is associated with lower prevalence of gut microbiota dysbiosis. Nature 2020, 581, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Nishijima, S.; Suda, W.; Oshima, K.; Kim, S.W.; Hirose, Y.; Morita, H.; Hattori, M. The gut microbiome of healthy Japanese and its microbial and functional uniqueness. DNA Res. 2016, 23, 125–133. [Google Scholar] [CrossRef] [Green Version]
- Hosoda, S.; Nishijima, S.; Fukunaga, T.; Hattori, M.; Hamada, M. Revealing the microbial assemblage structure in the human gut microbiome using latent Dirichlet allocation. Microbiome 2020, 8, 95. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, J.; Watanabe, K.; Jiang, J.; Matsuda, K.; Chao, S.H.; Haryono, P.; La-Ongkham, O.; Sarwoko, M.A.; Sujaya, I.N.; Zhao, L.; et al. Diversity in gut bacterial community of school-age children in Asia. Sci. Rep. 2015, 5, 8397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, T.; Naito, Y.; Kashiwagi, S.; Uchiyama, K.; Mizushima, K.; Kamada, K.; Ishikawa, T.; Inoue, R.; Okuda, K.; Tsujimoto, Y.; et al. Changes in the Gut Microbiota are Associated with Hypertension, Hyperlipidemia, and Type 2 Diabetes Mellitus in Japanese Subjects. Nutrients 2020, 12, 2996. [Google Scholar] [CrossRef] [PubMed]
- Takagi, T.; Naito, Y.; Inoue, R.; Kashiwagi, S.; Uchiyama, K.; Mizushima, K.; Tsuchiya, S.; Okayama, T.; Dohi, O.; Yoshida, N.; et al. The influence of long-term use of proton pump inhibitors on the gut microbiota: An age-sex-matched case-control study. J. Clin. Biochem. Nutr. 2018, 62, 100–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takagi, T.; Naito, Y.; Inoue, R.; Kashiwagi, S.; Uchiyama, K.; Mizushima, K.; Tsuchiya, S.; Dohi, O.; Yoshida, N.; Kamada, K.; et al. Differences in gut microbiota associated with age, sex, and stool consistency in healthy Japanese subjects. J. Gastroenterol. 2019, 54, 53–63. [Google Scholar] [CrossRef]
- Park, J.; Kato, K.; Murakami, H.; Hosomi, K.; Tanisawa, K.; Nakagata, T.; Ohno, H.; Konishi, K.; Kawashima, H.; Chen, Y.A.; et al. Comprehensive analysis of gut microbiota of a healthy population and covariates affecting microbial variation in two large Japanese cohorts. BMC Microbiol. 2021, 21, 151. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo-Cantabrana, C.; Delgado, S.; Ruiz, L.; Ruas-Madiedo, P.; Sánchez, B.; Margolles, A. Bifidobacteria and Their Health-Promoting Effects. Microbiol. Spectr. 2017, 5, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Fuyuki, A.; Higurashi, T.; Kessoku, T.; Ashikari, K.; Yoshihara, T.; Misawa, N.; Iwaki, M.; Kobayashi, T.; Ohkubo, H.; Yoneda, M.; et al. Efficacy of Bifidobacterium bifidum G9-1 in improving quality of life in patients with chronic constipation: A prospective intervention study. Biosci. Microbiota Food Health 2021, 40, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Yamashita, T.; Watanabe, H.; Kami, K.; Yoshida, N.; Tabata, T.; Emoto, T.; Sasaki, N.; Mizoguchi, T.; Irino, Y.; et al. Gut Microbiome and Plasma Microbiome-Related Metabolites in Patients With Decompensated and Compensated Heart Failure. Circ. J. 2018, 83, 182–192. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, Y.; Hamaguchi, M.; Kaji, A.; Sakai, R.; Osaka, T.; Inoue, R.; Kashiwagi, S.; Mizushima, K.; Uchiyama, K.; Takagi, T.; et al. Intake of sucrose affects gut dysbiosis in patients with type 2 diabetes. J. Diabetes Investig. 2020, 11, 1623–1634. [Google Scholar] [CrossRef]
- Kondo, Y.; Hashimoto, Y.; Hamaguchi, M.; Ando, S.; Kaji, A.; Sakai, R.; Inoue, R.; Kashiwagi, S.; Mizushima, K.; Uchiyama, K.; et al. Unique Habitual Food Intakes in the Gut Microbiota Cluster Associated with Type 2 Diabetes Mellitus. Nutrients 2021, 13, 3816. [Google Scholar] [CrossRef]
- Wang, S.P.; Rubio, L.A.; Duncan, S.H.; Donachie, G.E.; Holtrop, G.; Lo, G.; Farquharson, F.M.; Wagner, J.; Parkhill, J.; Louis, P.; et al. Pivotal Roles for pH, Lactate, and Lactate-Utilizing Bacteria in the Stability of a Human Colonic Microbial Ecosystem. mSystems 2020, 5, e00645-20. [Google Scholar] [CrossRef]
- Marquet, P.; Duncan, S.H.; Chassard, C.; Bernalier-Donadille, A.; Flint, H.J. Lactate has the potential to promote hydrogen sulphide formation in the human colon. FEMS Microbiol. Lett. 2009, 299, 128–134. [Google Scholar] [CrossRef] [Green Version]
- Vernia, P.; Caprilli, R.; Latella, G.; Barbetti, F.; Magliocca, F.M.; Cittadini, M. Fecal lactate and ulcerative colitis. Gastroenterology 1988, 95, 1564–1568. [Google Scholar] [CrossRef]
- Hove, H.; Nordgaard-Andersen, I.; Mortensen, P.B. Faecal DL-lactate concentration in 100 gastrointestinal patients. Scand. J. Gastroenterol. 1994, 29, 255–259. [Google Scholar] [CrossRef]
- Chen, L.; Li, R.; Wang, Z.; Zhang, Z.; Wang, J.; Qiao, Y.; Huang, Y.; Liu, W. Lactate-utilizing bacteria ameliorates DSS-induced colitis in mice. Life Sci. 2022, 288, 120179. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| N | Male (Age ± SD) | Female (Age ± SD) | |
|---|---|---|---|
| Total | 1803 | 983 (63.2 ± 16.2) | 820 (65.5 ± 13.4) |
| Healthy subjescts | 283 | 177 (43.4 ± 11.1) | 106 (49.2 ± 12.3) |
| Cardiovascular diseases | 104 | 71 (74.6 ± 8.2) | 33 (73.5 ± 6.9) |
| Hepatic diseases | 168 | 89 (64.4 ± 12.7) | 79 (69.0 ± 10.8) |
| Functional gastrointestinal disorders | 109 | 61 (68.5 ± 18.0) | 48 (67.8 ± 13.5) |
| Endocrine diseases | 57 | 26 (68.9 ± 8.3) | 31 (68.8 ± 9.1) |
| Neurological diseases | 15 | 7 (66.7 ± 15.1) | 8 (65.3 ± 15.5) |
| Psychiatric diseases | 38 | 19 (65.5 ± 13.7) | 19 (71.3 ± 13.5) |
| Inflammatory Bowel Diseases (IBD) | 128 | 76 (48.4 ± 18.5) | 52 (52.3 ± 15.6) |
| Autoimmune diseases | 21 | 7 (72.1 ± 8.7) | 14 (66.9 ± 12.3) |
| Malignant diseases (under treatment) | 123 | 81 (69.2 ± 9.6) | 42 (69.7 ± 8.8) |
| Malignant diseases (after treatment) | 160 | 99 (71.1 ± 9.4) | 61 (68.4 ± 9.5) |
| Hypertension | 619 | 313 (70.2 ± 9.8) | 306 (70.2 ± 9.1) |
| Dyslipidemia | 819 | 422 (68.3 ± 11.4) | 397 (69.1 ± 9.9) |
| Hyperuricemia | 138 | 99 (68.5 ± 12.4) | 39 (72.5 ± 7.9) |
| Diabetes | 474 | 268 (67.4 ± 11.3) | 206 (66.3 ± 10.7) |
| Obesities (BMI ≥ 30 kg/m2) | 96 | 40 (51.2 ± 17.2) | 56 (55.2 ± 15.3) |
| The Number of Enrolled Subjects | The Number of Healthy Subjects | The Rate of Healthy Subjects (%) | Male (Age ± SD) | Female (Age ± SD) | |
|---|---|---|---|---|---|
| Type A | 512 | 25 | 4.9 | 264 (69.8 ± 13.0) | 248 (69.9 ± 9.7) |
| Type B | 552 | 147 | 26.6 | 299 (58.4 ± 17.1) | 253 (62.9 ± 14.6) |
| Type C | 271 | 28 | 10.3 | 151 (64.4 ± 16.4) | 120 (66.6 ± 11.9) |
| Type D | 292 | 20 | 6.8 | 133 (65.5 ± 14.9) | 159 (62.4 ± 14.5) |
| Type E | 176 | 63 | 35.8 | 136 (57.3 ± 15.7) | 40 (62.4 ± 16.7) |
| Characteristic Feature | Other Features | |
|---|---|---|
| Type A | family Ruminococcaceae | genera Coprococcus, Gemminger and Roseburia |
| Type B | genus Bacteroides | genera Blautia and Faecalibacterium |
| Type C | genus Bacteroides | genera Megamonus, Fusobacterium and Proteus |
| Type D | genus Bifidobacterium | genera Lactobacillus and Streptococcus |
| Type E | genus Prevotella |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takagi, T.; Inoue, R.; Oshima, A.; Sakazume, H.; Ogawa, K.; Tominaga, T.; Mihara, Y.; Sugaya, T.; Mizushima, K.; Uchiyama, K.; et al. Typing of the Gut Microbiota Community in Japanese Subjects. Microorganisms 2022, 10, 664. https://doi.org/10.3390/microorganisms10030664
Takagi T, Inoue R, Oshima A, Sakazume H, Ogawa K, Tominaga T, Mihara Y, Sugaya T, Mizushima K, Uchiyama K, et al. Typing of the Gut Microbiota Community in Japanese Subjects. Microorganisms. 2022; 10(3):664. https://doi.org/10.3390/microorganisms10030664
Chicago/Turabian StyleTakagi, Tomohisa, Ryo Inoue, Akira Oshima, Hiroshi Sakazume, Kenta Ogawa, Tomo Tominaga, Yoichi Mihara, Takeshi Sugaya, Katsura Mizushima, Kazuhiko Uchiyama, and et al. 2022. "Typing of the Gut Microbiota Community in Japanese Subjects" Microorganisms 10, no. 3: 664. https://doi.org/10.3390/microorganisms10030664
APA StyleTakagi, T., Inoue, R., Oshima, A., Sakazume, H., Ogawa, K., Tominaga, T., Mihara, Y., Sugaya, T., Mizushima, K., Uchiyama, K., Itoh, Y., & Naito, Y. (2022). Typing of the Gut Microbiota Community in Japanese Subjects. Microorganisms, 10(3), 664. https://doi.org/10.3390/microorganisms10030664