
An Age Effect of Rumen Microbiome in Dairy Buffaloes Revealed by Metagenomics

 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Animals and Sample Collection
2.3. DNA Extraction and High-Throughput Sequencing
2.4. Bioinformatics Analysis
2.5. Taxonomic and Functional Annotation from Rumen Metagenomes
2.6. Statistical Analysis
2.7. Nucleotide Sequence Accession Numbers
3. Results
3.1. Profiling of the Rumen Metagenome
3.2. Comparison of Buffalo Rumen Microbiome across Different Age Groups
3.3. Changing Pattern of Rumen Microbial Functions with Increasing Age
3.3.1. CAZyme Functional Annotation
3.3.2. COG Functional Annotation
3.3.3. KEGG Functional Annotation
3.4. Functional Contribution Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xue, M.Y.; Sun, H.Z.; Wu, X.H.; Liu, J.X.; Guan, L.L. Multi-omics reveals that the rumen microbiome and its metabolome together with the host metabolome contribute to individualized dairy cow performance. Microbiome 2020, 8, 64. [Google Scholar] [CrossRef] [PubMed]
- Furman, O.; Shenhav, L.; Sasson, G.; Kokou, F.; Honig, H.; Jacoby, S.; Hertz, T.; Cordero, O.X.; Halperin, E.; Mizrahi, I. Stochasticity constrained by deterministic effects of diet and age drive rumen microbiome assembly dynamics. Nat. Commun. 2020, 11, 1904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, C.Y.; Ji, S.K.; Yan, H.; Wang, Y.J.; Liu, J.J.; Cao, Z.J.; Yang, H.J.; Zhang, W.J.; Li, S.L. Dynamic change of the gastrointestinal bacterial ecology in cows from birth to adulthood. MicrobiologyOpen 2020, 9, e1119. [Google Scholar] [CrossRef] [PubMed]
- Jami, E.; Israel, A.; Kotser, A.; Mizrahi, I. Exploring the bovine rumen bacterial community from birth to adulthood. ISME J. 2013, 7, 1069–1079. [Google Scholar] [CrossRef] [Green Version]
- Dill-McFarland, K.A.; Breaker, J.D.; Suen, G. Microbial succession in the gastrointestinal tract of dairy cows from 2 weeks to first lactation. Sci. Rep. 2017, 7, 40864. [Google Scholar] [CrossRef] [Green Version]
- Dias, J.; Marcondes, M.I.; Motta, D.S.S.; Cardoso, D.M.E.S.; Fontes, N.M.; Tassinari, R.R.; Machado, F.S.; Cuquetto, M.H.; Dill-McFarland, K.A.; Suen, G. Bacterial Community Dynamics across the Gastrointestinal Tracts of Dairy Calves during Preweaning Development. Appl. Environ. Microbiol. 2018, 84, e02675-17. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Zhang, K.; Li, C.; Wang, X.; Chen, Y.; Yang, Y. Characterization and comparison of microbiota in the gastrointestinal tracts of the goat (Capra hircus) during preweaning development. Front. Microbiol. 2019, 10, 2125. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.H.; Wang, Y.C.; Luo, H.P.; Qiu, W.Q.; Zhang, H.L.; Hu, L.R.; Wang, Y.J.; Dong, G.H.; Guo, G. The association between inflammaging and Age-Related changes in the ruminal and fecal microbiota among lactating holstein cows. Front. Microbiol. 2019, 10, 1803. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Meng, Q.H.; Chen, Y.H.; Xu, M.S.; Shen, M.; Gao, R.; Gan, S.Q. Role of Age-Related shifts in rumen bacteria and methanogens in methane production in cattle. Front. Microbiol. 2017, 8, 1563. [Google Scholar] [CrossRef] [Green Version]
- Alipour, M.J.; Jalanka, J.; Pessa-Morikawa, T.; Kokkonen, T.; Satokari, R.; Hynonen, U.; Iivanainen, A.; Niku, M. The composition of the perinatal intestinal microbiota in cattle. Sci. Rep. 2018, 8, 10437. [Google Scholar] [CrossRef]
- Mayer, M.; Abenthum, A.; Matthes, J.M.; Kleeberger, D.; Ege, M.J.; Holzel, C.; Bauer, J.; Schwaiger, K. Development and genetic influence of the rectal bacterial flora of newborn calves. Vet. Microbiol. 2012, 161, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Pineda, P.S.; Flores, E.B.; Herrera, J.R.V.; Low, W.Y. Opportunities and challenges for improving the productivity of swamp buffaloes in southeastern asia. Front. Genet. 2021, 12, 629861. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.U.; Hassan, F.U.; Luo, X.; Li, Z.P.; Liu, Q.Y. Whole-Genome sequencing and characterization of buffalo genetic resources: Recent advances and future challenges. Animals 2021, 11, 904. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Colli, L.; Barker, J. Asian water buffalo: Domestication, history and genetics. Anim. Genet. 2020, 51, 177–191. [Google Scholar] [CrossRef]
- Chanthakhoun, V.; Wanapat, M.; Kongmun, P.; Cherdthong, A. Comparison of ruminal fermentation characteristics and microbial population in swamp buffalo and cattle. Livest. Sci. 2012, 2-3, 172–176. [Google Scholar] [CrossRef]
- Wanapat, M. Rumen manipulation to increase the efficient use of local feed resources and productivity of ruminants in the tropics. Asian-Aust. J. Anim. Sci. 2000, 13, 59–67. [Google Scholar]
- Nguyen, V.T.; Wanapat, M.; Khejornsart, P.; Kongmun, P. Nutrient digestibility and ruminal fermentation characteristic in swamp buffaloes fed on chemically treated rice straw and urea. Trop Anim Health Prod. 2012, 44, 629–636. [Google Scholar] [CrossRef]
- Wanapat, M.; Cherdthong, A. Use of real-time PCR technique in studying rumen cellulolytic bacteria population as affected by level of roughage in Swamp buffalo. Curr. Microbiol. 2009, 58, 294–299. [Google Scholar] [CrossRef]
- Dehority, B.A. Rumen Microbiology; Nottingham University Press: Thrumpton, UK, 2003; p. 372. [Google Scholar]
- Malakar, D.; Walli, T.K. Relative fibre degradation (in vitro) by bacteria and fungi using inoculum from cow and buffalo rumen. Indian J. Dairy Sci. 1995, 48, 295–301. [Google Scholar]
- Tong, F.; Wang, T.; Gao, N.L.; Liu, Z.Y.; Cui, K.Q.; Duan, Y.Q.; Wu, S.C.; Luo, Y.H.; Li, Z.P.; Yang, C.J. The microbiome of the buffalo digestive tract. Nat. Commun. 2022, 13, 823. [Google Scholar] [CrossRef]
- Singh, K.M.; Reddy, B.; Patel, D.; Patel, A.K.; Parmar, N.; Patel, A.; Patel, J.B.; Joshi, C.G. High potential source for biomass degradation enzyme discovery and environmental aspects revealed through metagenomics of Indian buffalo rumen. Biomed. Res. Int. 2014, 2014, 267189. [Google Scholar] [CrossRef] [PubMed]
- Parmar, N.R.; Solanki, J.V.; Patel, A.B.; Shah, T.M.; Patel, A.K.; Parnerkar, S.; Kumar, J.I.; Joshi, C.G. Metagenome of Mehsani buffalo rumen microbiota: An assessment of variation in feed-dependent phylogenetic and functional classification. J. Mol. Microbiol. Biotechnol. 2014, 24, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.D.; Patel, A.K.; Parmar, N.R.; Shah, T.M.; Patel, J.B.; Pandya, P.R.; Joshi, C.G. Microbial and Carbohydrate Active Enzyme profile of buffalo rumen metagenome and their alteration in response to variation in the diet. Gene 2014, 545, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.M.; Ahir, V.B.; Tripathi, A.K.; Ramani, U.V.; Sajnani, M.; Koringa, P.G.; Jakhesara, S.; Pandya, P.R.; Rank, D.N.; Murty, D.S. Metagenomic analysis of Surti buffalo (Bubalus bubalis) rumen: A preliminary study. Mol. Biol. Rep. 2012, 39, 4841–4848. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.M.; Tripathi, A.K.; Pandya, P.R.; Parnerkar, S.; Rank, D.N.; Kothari, R.K.; Joshi, C.G. Methanogen diversity in the rumen of Indian Surti buffalo (Bubalus bubalis), assessed by 16S rDNA analysis. Res. Vet. Sci. 2012, 92, 451–455. [Google Scholar] [CrossRef]
- Pandya, P.R.; Singh, K.M.; Parnerkar, S.; Tripathi, A.K.; Mehta, H.H.; Rank, D.N.; Kothari, R.K.; Joshi, C.G. Bacterial diversity in the rumen of Indian Surti buffalo (Bubalus bubalis), assessed by 16S rDNA analysis. J. Appl. Genet. 2010, 51, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.Z.; Peng, K.L.; Xue, M.Y.; Liu, J.X. Metagenomics analysis revealed the distinctive ruminal microbiome and resistive profiles in dairy buffaloes. Anim. Microbiome 2021, 3, 44. [Google Scholar] [CrossRef]
- Shen, J.S.; Chai, Z.; Song, L.J.; Liu, J.X.; Wu, Y.M. Insertion depth of oral stomach tubes may affect the fermentation parameters of ruminal fluid collected in dairy cows. J. Dairy Sci. 2012, 95, 5978–5984. [Google Scholar] [CrossRef] [Green Version]
- Wilson, K. Preparation of genomic DNA from bacteria. Curr. Protoc. Mol. Biol. 2001. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, H.; Park, J.; Takagi, T. MetaGene: Prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res. 2006, 34, 5623–5630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, L.; Niu, B.; Zhu, Z.W.; Wu, S.; Li, W.Z. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Li, R.Q.; Yu, C.; Li, Y.R.; Lam, T.W.; Yiu, S.M.; Kristiansen, K.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.Z.; Huang, J.J.; Ding, Y.; Wu, J.M.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L.P. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [Green Version]
- Jensen, L.J.; Julien, P.; Kuhn, M.; von Mering, C.; Muller, J.; Doerks, T.; Bork, P. EggNOG: Automated construction and annotation of orthologous groups of genes. Nucleic Acids Res. 2008, 36, D250–D254. [Google Scholar] [CrossRef]
- Lombard, V.; Golaconda, R.H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [Green Version]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [Green Version]
- Clarke, K.R. Non-parametric multivariate analyses of changes in community structure. Aust. J. Ecol. 1993, 18, 117–143. [Google Scholar] [CrossRef]
- Hammer, Ø.; Harper, D.A.T.; Ryan, P.D. PAST-Palaeontological Statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar]
- Zhang, Y.; Xu, J.; Riera, N.; Jin, T.; Li, J.Y.; Wang, N. Huanglongbing impairs the rhizosphere-torhizoplane enrichment process of the citrus root-associated microbiome. Microbiome 2017, 5, 97. [Google Scholar] [CrossRef]
- Ofek-Lalzar, M.; Sela, N.; Goldman-Voronov, M.; Green, S.J.; Hadar, Y.; Minz, D. Niche and host-associated functional signatures of the root surface microbiome. Nat. Commun. 2014, 5, 4950. [Google Scholar] [CrossRef] [PubMed]
- Khedkar, C.D.; Kalyankar, S.D.; Deosarkar, S.S. Buffalo milk. In Encyclopedia of Food and Health; Academic Press: Cambridge, MA, USA, 2016; pp. 522–528. [Google Scholar]
- Koringa, P.G.; Thakkar, J.R.; Pandit, R.J.; Hinsu, A.T.; Parekh, M.J.; Shah, R.K.; Jakhesara, S.J.; Joshi, C.G. Metagenomic characterisation of ruminal bacterial diversity in buffaloes from birth to adulthood using 16S rRNA gene amplicon sequencing. Funct. Integr. Genom. 2019, 19, 237–247. [Google Scholar] [CrossRef]
- McCann, J.C.; Wickersham, T.A.; Loor, J.J. High-throughput methods redefine the rumen microbiome and its relationship with nutrition and metabolism. Bioinform. Biol. Insights 2014, 8, 109–125. [Google Scholar] [CrossRef] [Green Version]
- Lin, B.; Henderson, G.; Zou, C.; Cox, F.; Liang, X.; Janssen, P.H.; Attwood, G.T. Characterization of the rumen microbial community composition of buffalo breeds consuming diets typical of dairy production systems in Southern China. Anim. Feed Sci. Technol. 2015, 207, 75–84. [Google Scholar] [CrossRef]
- Lapébie, P.; Lombard, V.; Drula, E.; Terrapon, N.; Henrissat, B. Bacteroidetes use thousands of enzyme combinations to break down glycans. Nat. Commun. 2019, 10, 2043. [Google Scholar] [CrossRef]
- Jumpertz, R.; Le, D.S.; Turnbaugh, P.J.; Trinidad, C.; Bogardus, C.; Gordon, J.I.; Krakoff, J. Energy-balance studies reveal associations between gut microbes, caloric load, and nutrient absorption in humans. Am. J. Clin. Nutr. 2011, 94, 58–65. [Google Scholar] [CrossRef] [Green Version]
- Rahayu, E.S.; Mariyatun, M.; Manurung, N.E.P.; Hasan, P.N.; Therdtatha, P.; Mishima, R.; Komalasari, H.; Mahfuzah, N.A.; Pamungkaningtyas, F.H.; Yoga, W.K.; et al. Effect of probiotic Lactobacillus plantarum Dad-13 powder consumption on the gut microbiota and intestinal health of overweight adults. World J. Gastroenterol. 2021, 27, 107–128. [Google Scholar] [CrossRef]
- Amat, S.; Alexander, T.W.; Holman, D.B.; Schwinghamer, T.; Timsit, E. Intranasal bacterial therapeutics reduce colonization by the respiratory pathogen mannheimia haemolytica in dairy calves. mSystems 2020, 5, e00629-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Omar, J.M.; Chan, Y.; Jones, M.L.; Prakash, S.; Jones, P.J.H. Lactobacillus fermentum and Lactobacillus amylovorus as probiotics alter body adiposity and gut microflora in healthy persons. J. Funct. Foods 2013, 1, 116–123. [Google Scholar] [CrossRef]
- Adetoye, A.; Pinloche, E.; Adeniyi, B.A.; Ayeni, F.A. Characterization and anti-salmonella activities of lactic acid bacteria isolated from cattle faeces. BMC Microbiol. 2018, 18, 96. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, S.; Fraga, M.; Silveyra, E.; Trombert, A.N.; Rabaza, A.; Pla, M.; Zunino, P. Probiotic properties of native Lactobacillus spp. Strains for dairy calves. Benef. Microbes 2018, 9, 613–624. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Indugu, N.; Vecchiarelli, B.; Pitta, D.W. Associative patterns among anaerobic fungi, methanogenic archaea, and bacterial communities in response to changes in diet and age in the rumen of dairy cows. Front. Microbiol. 2015, 6, 781. [Google Scholar] [CrossRef] [Green Version]
- Janssen, P.H.; Kirs, M. Structure of the archaeal community of the rumen. Appl. Environ. Microbiol. 2008, 74, 3619–3625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, A.D.; Ma, X.; Obispo, N.E. Methanobrevibacter phylotypes are the dominant methanogens in sheep from Venezuela. Microb. Ecol. 2008, 56, 390–394. [Google Scholar] [CrossRef]
- Singh, K.M.; Patel, A.K.; Shah, R.K.; Reddy, B.; Joshi, C.G. Potential functional gene diversity involved in methanogenesis and methanogenic community structure in Indian buffalo (Bubalus bubalis) rumen. J. Appl. Genet. 2015, 56, 411–426. [Google Scholar] [CrossRef]
- Chaudhary, P.P.; Sirohi, S.K. Dominance of Methanomicrobium phylotype in methanogen population present in Murrah buffaloes (Bubalus bubalis). Lett. Appl. Microbiol. 2009, 49, 274–277. [Google Scholar] [CrossRef]
- Hook, S.E.; Wright, A.D.; McBride, B.W. Methanogens: Methane producers of the rumen and mitigation strategies. Archaea 2010, 2010, 945785. [Google Scholar] [CrossRef] [Green Version]
- Jeyanathan, J.; Kirs, M.; Ronimus, R.S.; Hoskin, S.O.; Janssen, P.H. Methanogen community structure in the rumens of farmed sheep, cattle and red deer fed different diets. FEMS Microbiol. Ecol. 2011, 76, 311–326. [Google Scholar] [CrossRef]
- Jami, E.; Mizrahi, I. Composition and similarity of bovine rumen microbiota across individual animals. PLoS ONE 2012, 7, e33306. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Bian, G.; Zhu, Z.G.; Smidt, H.; Zhu, W.Y. Early methanogenic colonisation in the faeces of Meishan and Yorkshire piglets as determined by pyrosequencing analysis. Archaea 2014, 2014, 547908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.W.; Connor, E.E.; Li, C.; Baldwin, V.R.; Sparks, M.E. Characterization of the rumen microbiota of pre-ruminant calves using metagenomic tools. Environ. Microbiol. 2012, 14, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, J.K.; Waters, J.L.; Poole, A.C.; Sutter, J.L.; Koren, O.; Blekhman, R.; Beaumont, M.; Van Treuren, W.; Knight, R.; Bell, J.T. Human genetics shape the gut microbiome. Cell 2014, 159, 789–799. [Google Scholar] [CrossRef] [Green Version]
- Pitta, D.W.; Indugu, N.; Kumar, S.; Vecchiarelli, B.; Sinha, R.; Baker, L.D.; Bhukya, B.; Ferguson, J.D. Metagenomic assessment of the functional potential of the rumen microbiome in Holstein dairy cows. Anaerobe 2016, 38, 50–60. [Google Scholar] [CrossRef] [PubMed]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef]
- Prüß, B.M. Involvement of Two-Component Signaling on Bacterial Motility and Biofilm Development. J. Bacteriol. 2017, 199, e00259-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niedernhofer, L.J.; Gurkar, A.U.; Wang, Y.S.; Vijg, J.; Hoeijmakers, J.H.J.; Robbins, P.D. Nuclear genomic instability and aging. Annu. Rev. Biochem. 2018, 87, 295–322. [Google Scholar] [CrossRef]
- Ferreira, L.S.; Bittar, C.M.M. Performance and plasma metabolites of dairy calves fed starter containing sodium butyrate, calcium propionate or sodium monensin. Animal 2011, 5, 239–245. [Google Scholar] [CrossRef] [Green Version]
- Sheperd, A.C.; Combs, D.K. Long-term effects of acetate and propionate on voluntary feed intake by midlactation cows. J. Dairy Sci. 1998, 81, 2240–2250. [Google Scholar] [CrossRef]
- Ogunade, I.M.; Lay, J.; Andries, K.; McManus, C.J.; Bebe, F. Effects of live yeast on differential genetic and functional attributes of rumen microbiota in beef cattle. J. Anim. Sci. Biotechnol. 2019, 10, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brüssow, H. Microbiota and healthy aging: Observational and nutritional intervention studies. Microb. Biotechnol. 2013, 6, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Zhang, k.; Meng, M.; Gao, L.; Tu, Y.; Bai, Y. Rumen-derived lipopolysaccharide induced ruminal epithelium barrier damage in goats fed a high-concentrate diet. Microb. Pathog. 2019, 131, 81–86. [Google Scholar] [CrossRef]
- Giger-Reverdin, S.; Domange, C.; Broudiscou, L.P.; Sauvant, D.; Berthelot, V. Rumen function in goats, an example of adaptive capacity. J. Dairy Res. 2020, 87, 45–51. [Google Scholar] [CrossRef] [Green Version]
- Stewart, R.D.; Auffret, M.D.; Warr, A.; Wiser, A.H.; Press, M.O.; Langford, K.W.; Liachko, I.; Snelling, T.J.; Dewhurst, R.J.; Walker, A.W. Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen. Nat. Commun. 2018, 9, 870. [Google Scholar] [CrossRef] [Green Version]
- Gharechahi, J.; Salekdeh, G.H. A metagenomic analysis of the camel rumen’s microbiome identifies the major microbes responsible for lignocellulose degradation and fermentation. Biotechnol. Biofuels 2018, 11, 216. [Google Scholar] [CrossRef] [Green Version]
- Nascimento, A.S.; Muniz, J.R.C.; Aparicio, R.; Golubev, A.M.; Polikarpov, I. Insights into the structure and function of fungal beta-mannosidases from glycoside hydrolase family 2 based on multiple crystal structures of the Trichoderma harzianum enzyme. FEBS J. 2014, 281, 4165–4178. [Google Scholar] [CrossRef] [Green Version]
- Krause, D.O.; Denman, S.E.; Mackie, R.I.; Morrison, M.; Rae, A.L.; Attwood, G.T.; McSweeney, C.S. Opportunities to improve fiber degradation in the rumen: Microbiology, ecology, and genomics. FEMS Microbiol. Rev. 2003, 27, 663–693. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.M.; Li, J.F.; Wang, J.Q.; Yang, Y.J.; Wu, M.C. Determinants for the improved thermostability of a mesophilic family 11 xylanase predicted by computational methods. Biotechnol. Biofuels 2014, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Kasmaei, K.M.; Sundh, J. Identification of novel putative bacterial feruloyl esterases from anaerobic ecosystems by use of whole-Genome shotgun metagenomics and genome binning. Front. Microbiol. 2019, 10, 2673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, M.; Peng, Y.J.; Chen, Y.; Klinger, C.M.; Oba, M.; Liu, J.X.; Guan, L.L. Assessment of microbiome changes after rumen transfaunation: Implications on improving feed efficiency in beef cattle. Microbiome 2018, 6, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.S.; Whon, T.W.; Sung, H.; Jeong, Y.S.; Jung, E.S.; Shin, N.R.; Hyun, D.W.; Kim, P.S.; Lee, J.W.; Lee, C.H.; et al. Longitudinal evaluation of fecal microbiota transplantation for ameliorating calf diarrhea and improving growth performance. Nat. Commun. 2021, 12, 161. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Ji, S.; Duan, C.; Ju, S.; Zhang, Y.; Yan, H.; Liu, H. Rumen fluid transplantation affects growth performance of weaned lambs by altering gastrointestinal microbiota, immune function and feed digestibility. Animal 2021, 15, 100076. [Google Scholar] [CrossRef]
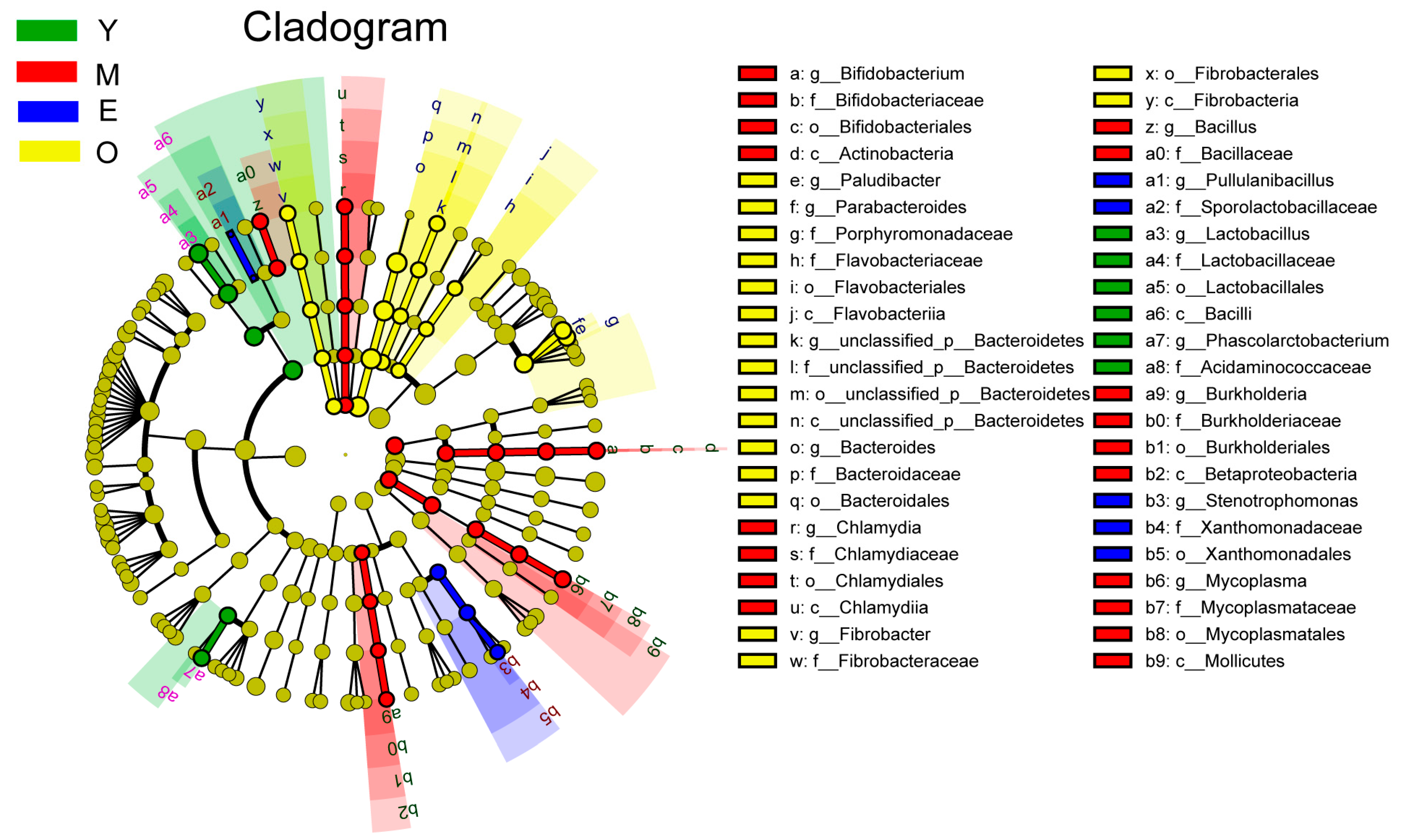
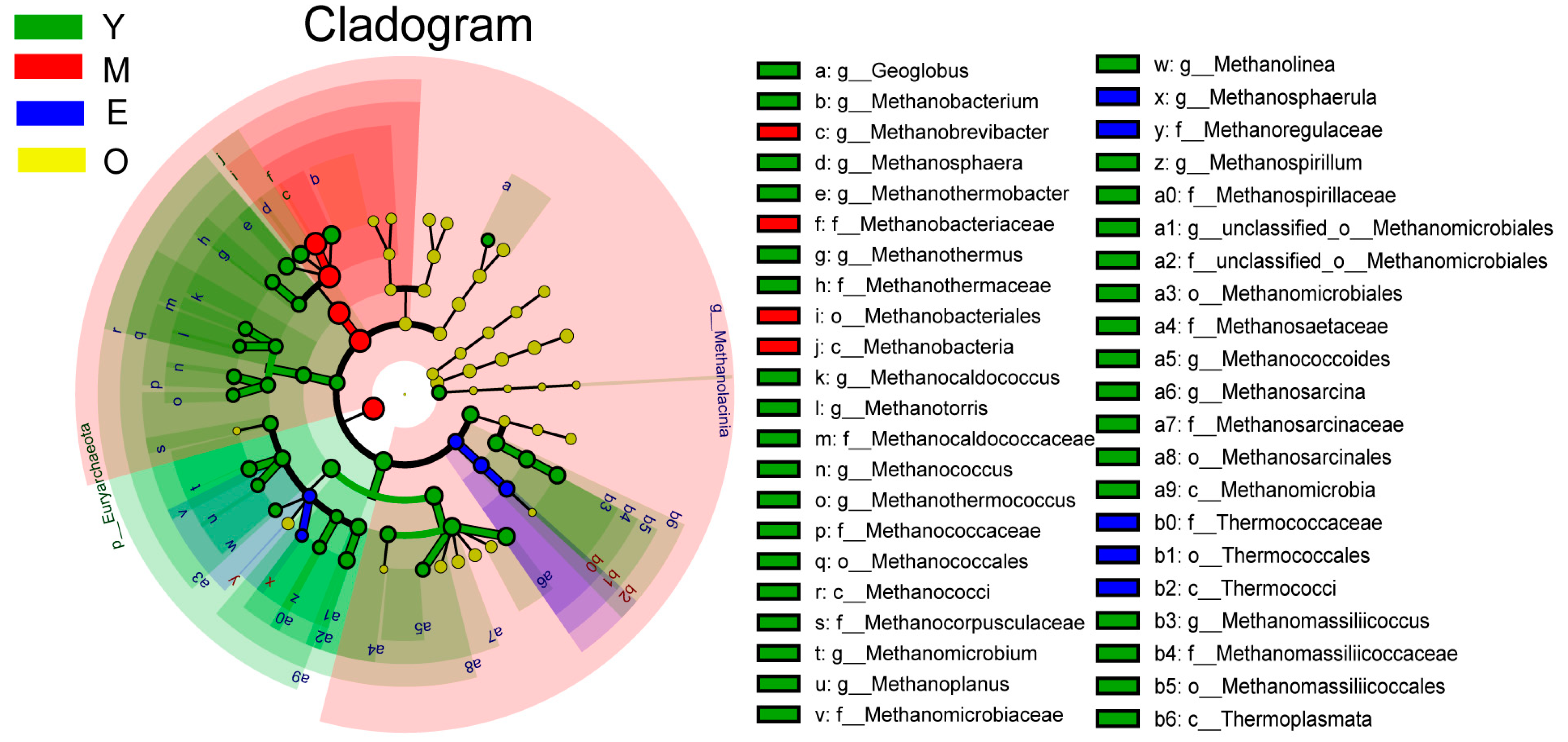
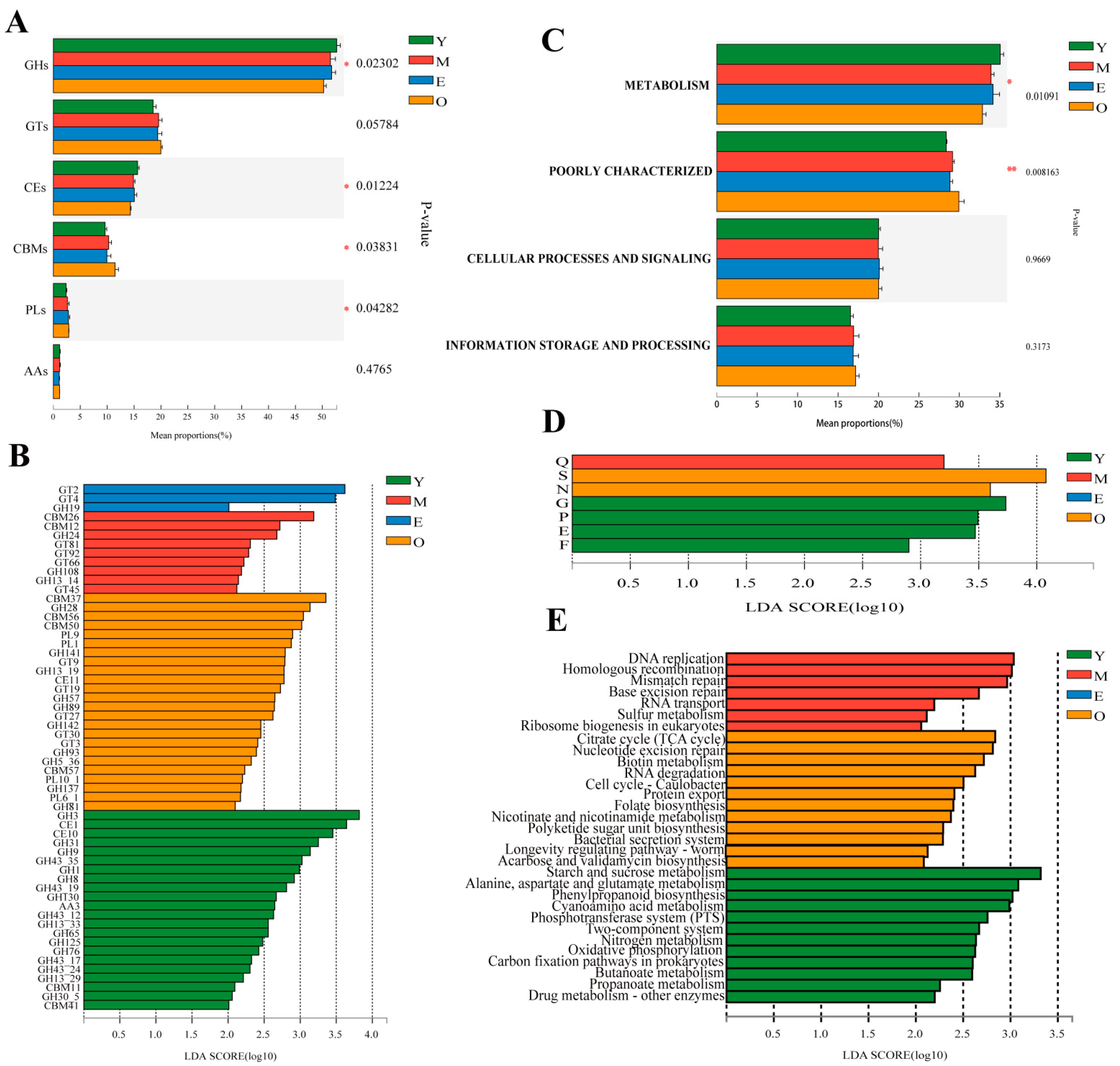
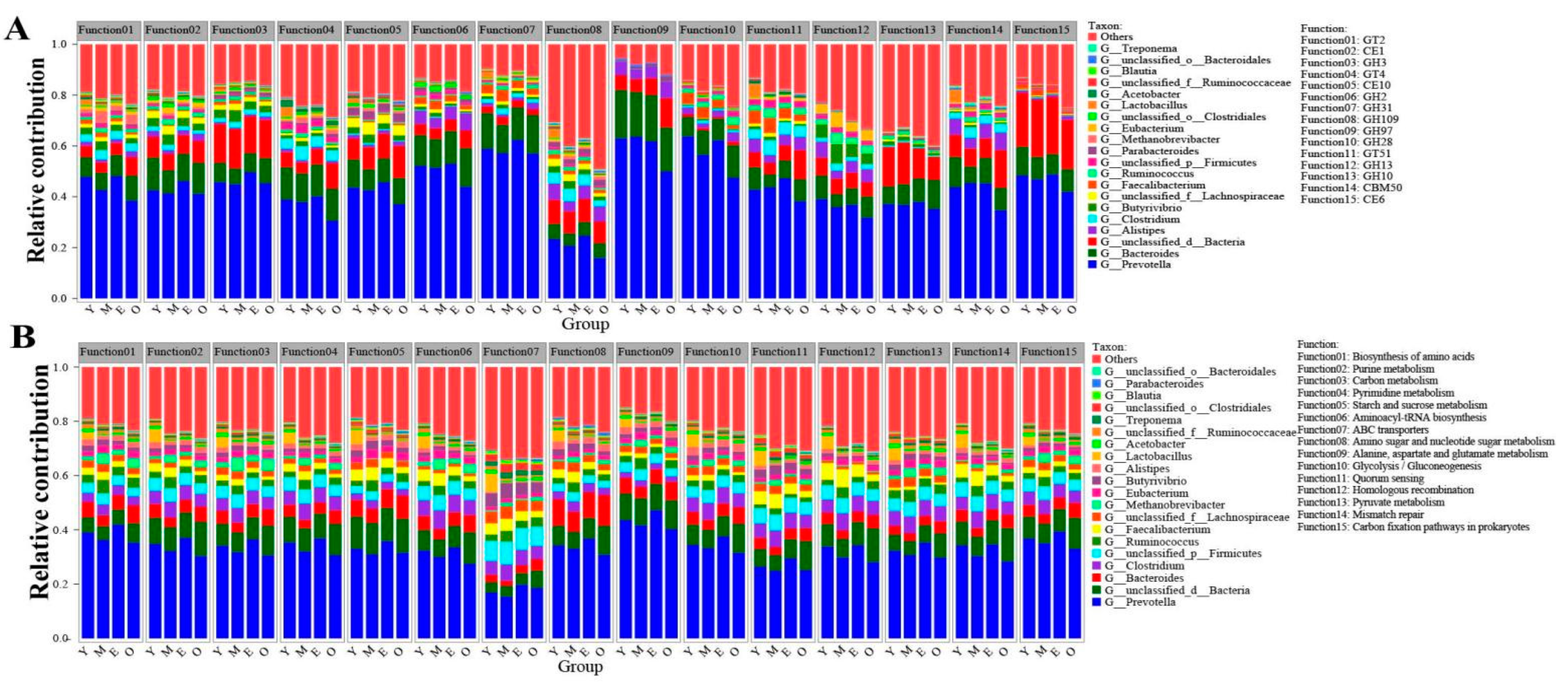

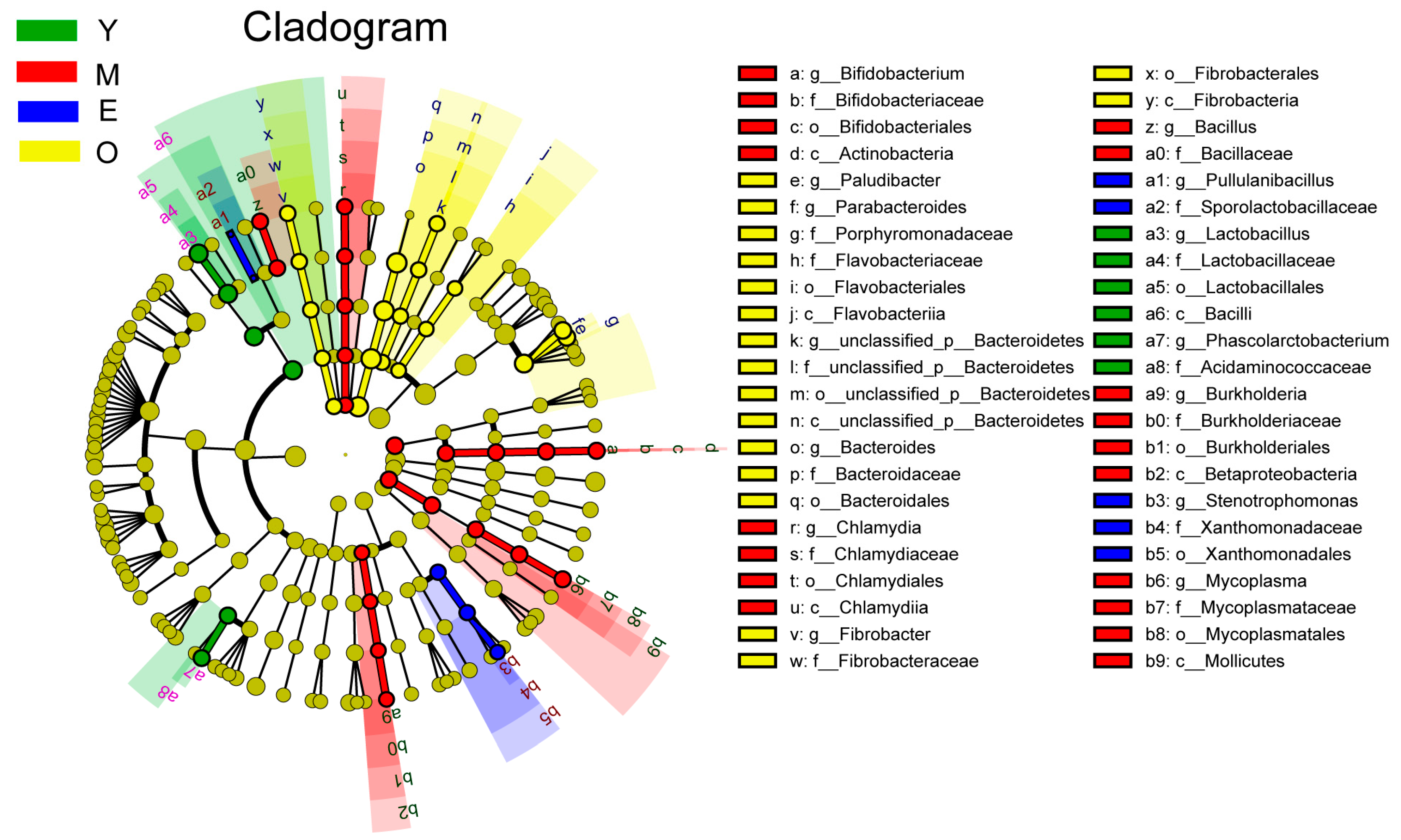{kind=link}
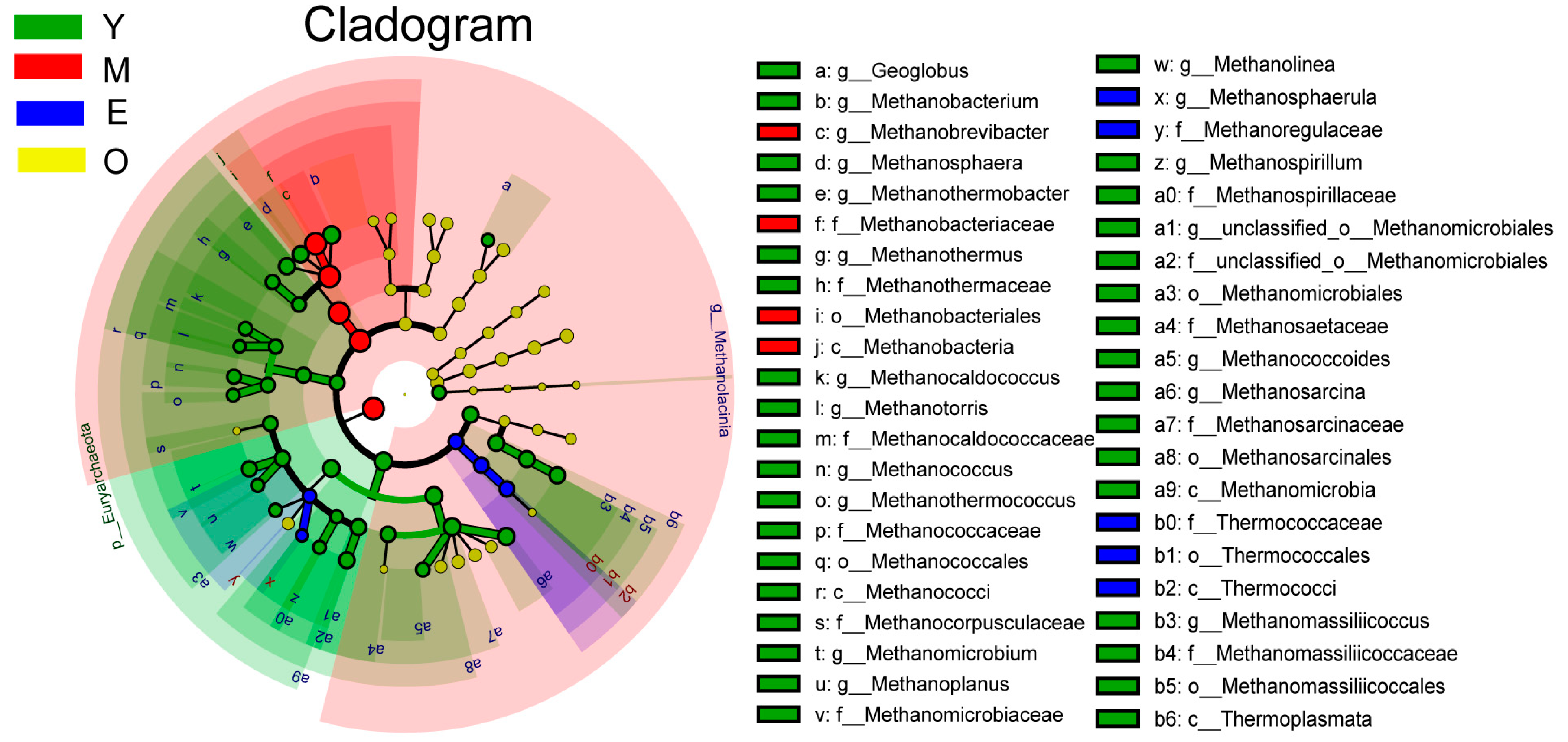{kind=link}
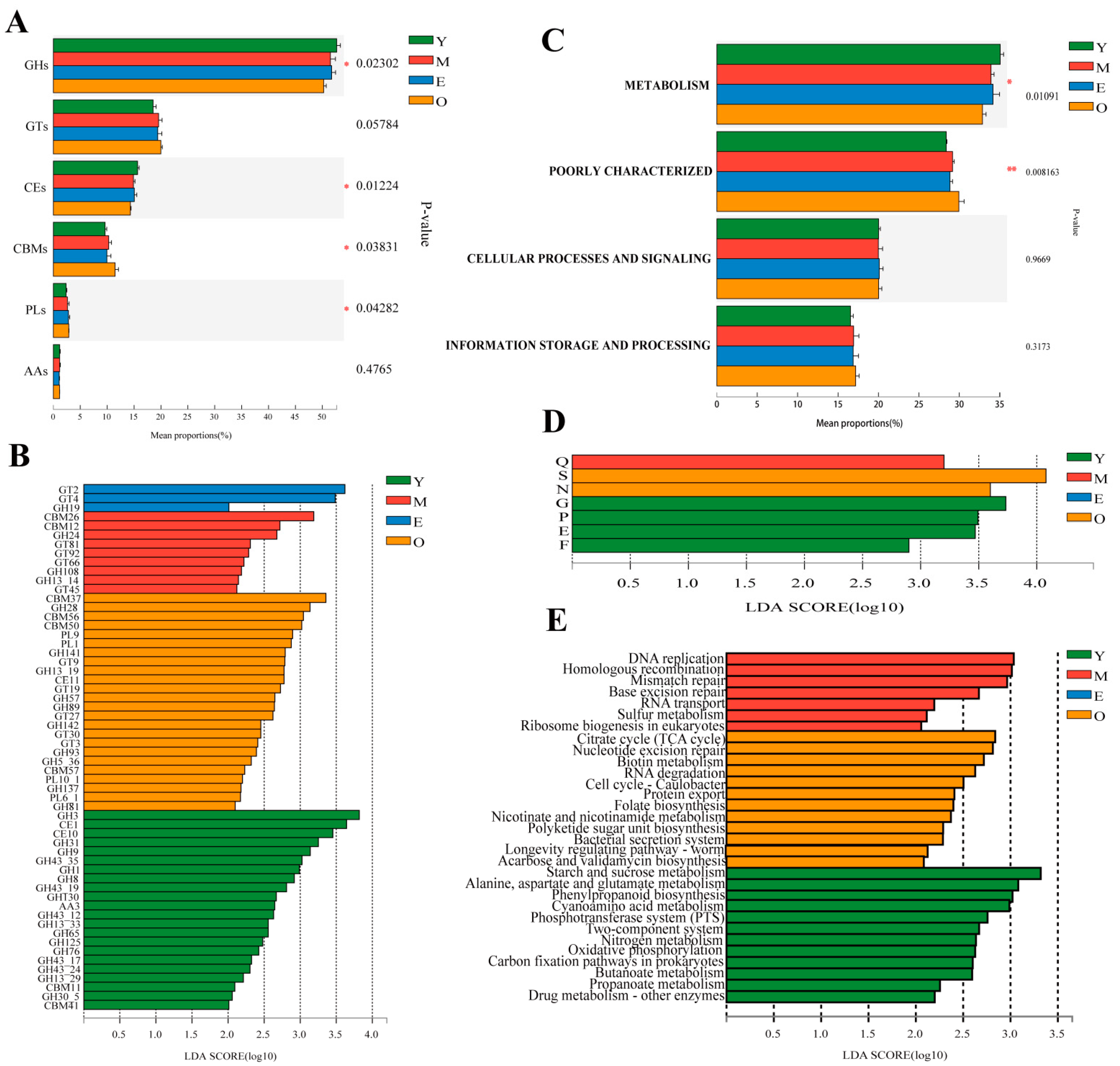{kind=link}
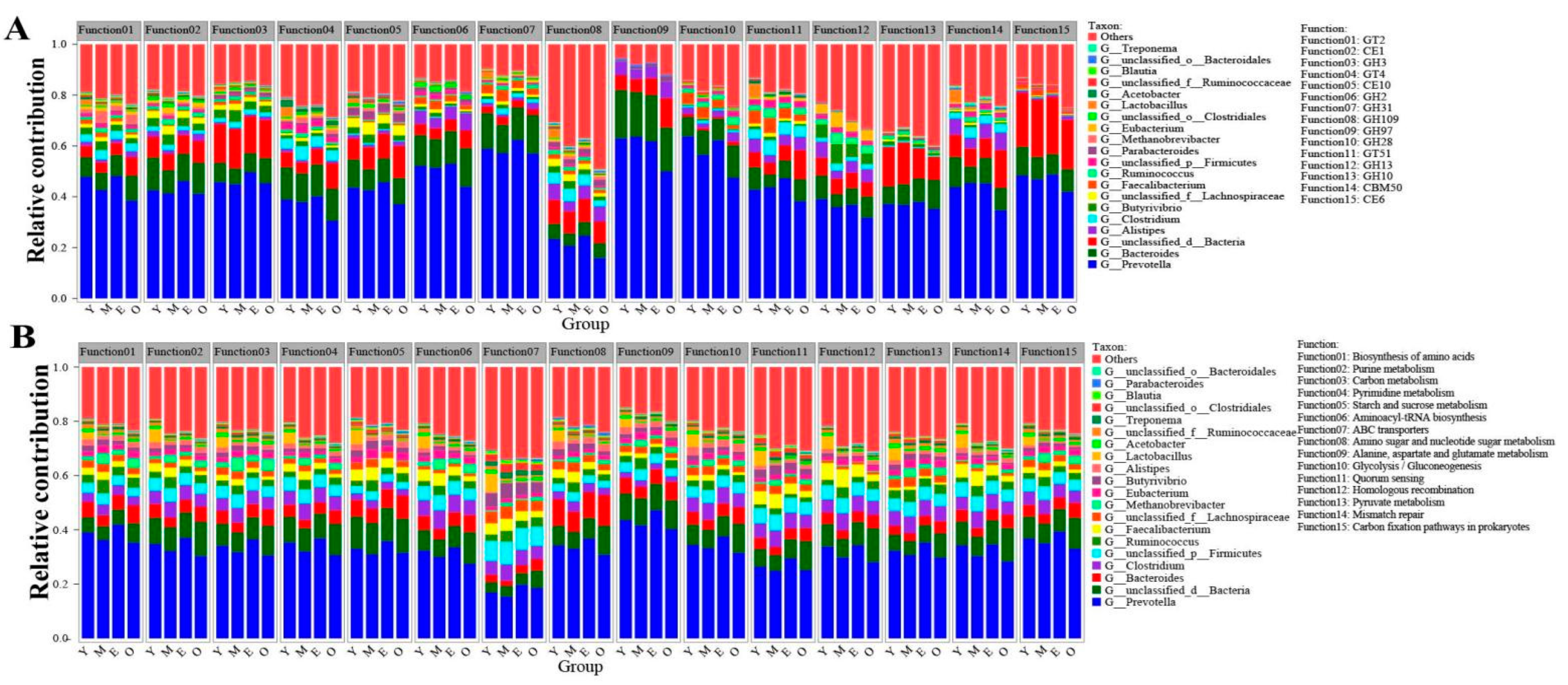{kind=link}
| Domain | Groups 1 | SEM | p-Value | |||
|---|---|---|---|---|---|---|
| Y | M | E | O | |||
| Bacteroidetes | 97.80 a | 95.38 b | 97.54 a | 95.97 b | 0.302 | 0.013 |
| Archaea | 1.18 a | 3.13 b | 1.46 a | 2.81 b | 0.262 | 0.010 |
| Eukaryota | 0.63 | 0.84 | 0.42 | 0.64 | 0.107 | 0.783 |
| Viruses | 0.13 a | 0.41 b | 0.32 b | 0.33 b | 0.034 | 0.009 |
| unclassified | 0.26 | 0.24 | 0.26 | 0.25 | 0.004 | 0.306 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, L.-P.; Peng, K.-L.; Xue, M.-Y.; Zhu, S.-L.; Liu, J.-X.; Sun, H.-Z. An Age Effect of Rumen Microbiome in Dairy Buffaloes Revealed by Metagenomics. Microorganisms 2022, 10, 1491. https://doi.org/10.3390/microorganisms10081491
Li L-P, Peng K-L, Xue M-Y, Zhu S-L, Liu J-X, Sun H-Z. An Age Effect of Rumen Microbiome in Dairy Buffaloes Revealed by Metagenomics. Microorganisms. 2022; 10(8):1491. https://doi.org/10.3390/microorganisms10081491
Chicago/Turabian StyleLi, Long-Ping, Ke-Lan Peng, Ming-Yuan Xue, Sen-Lin Zhu, Jian-Xin Liu, and Hui-Zeng Sun. 2022. "An Age Effect of Rumen Microbiome in Dairy Buffaloes Revealed by Metagenomics" Microorganisms 10, no. 8: 1491. https://doi.org/10.3390/microorganisms10081491
APA StyleLi, L.-P., Peng, K.-L., Xue, M.-Y., Zhu, S.-L., Liu, J.-X., & Sun, H.-Z. (2022). An Age Effect of Rumen Microbiome in Dairy Buffaloes Revealed by Metagenomics. Microorganisms, 10(8), 1491. https://doi.org/10.3390/microorganisms10081491