
Unamplified, Long-Read Metagenomic Sequencing Approach to Close Endosymbiont Genomes of Low-Biomass Insect Populations



Abstract
:1. Introduction
2. Materials and Methods
2.1. Insect Colony and DNA Extraction
2.2. Phenol-Chloroform Extraction
2.3. Quantitative PCR
2.4. DNA Sequencing
2.5. Assembly and Polishing
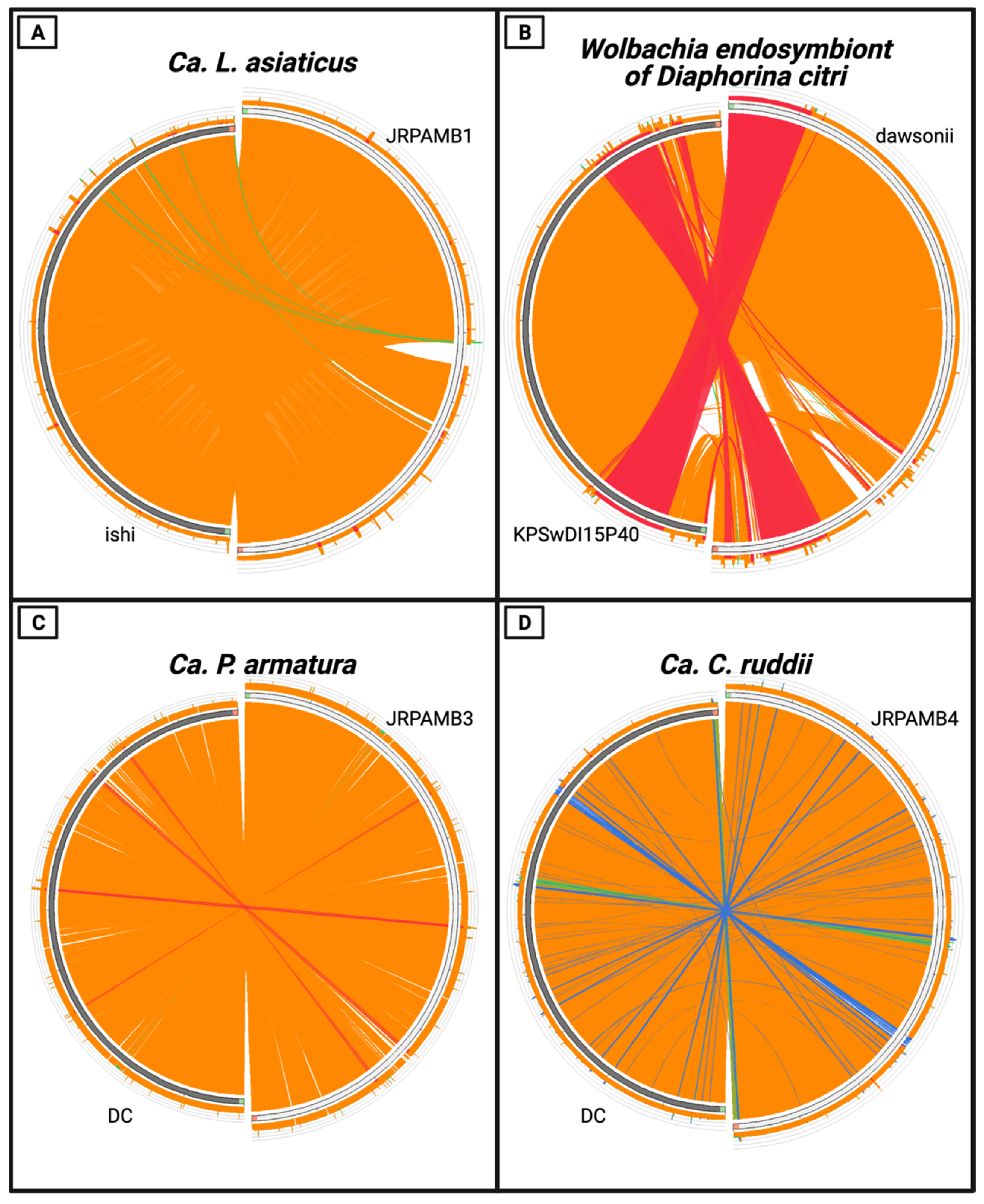
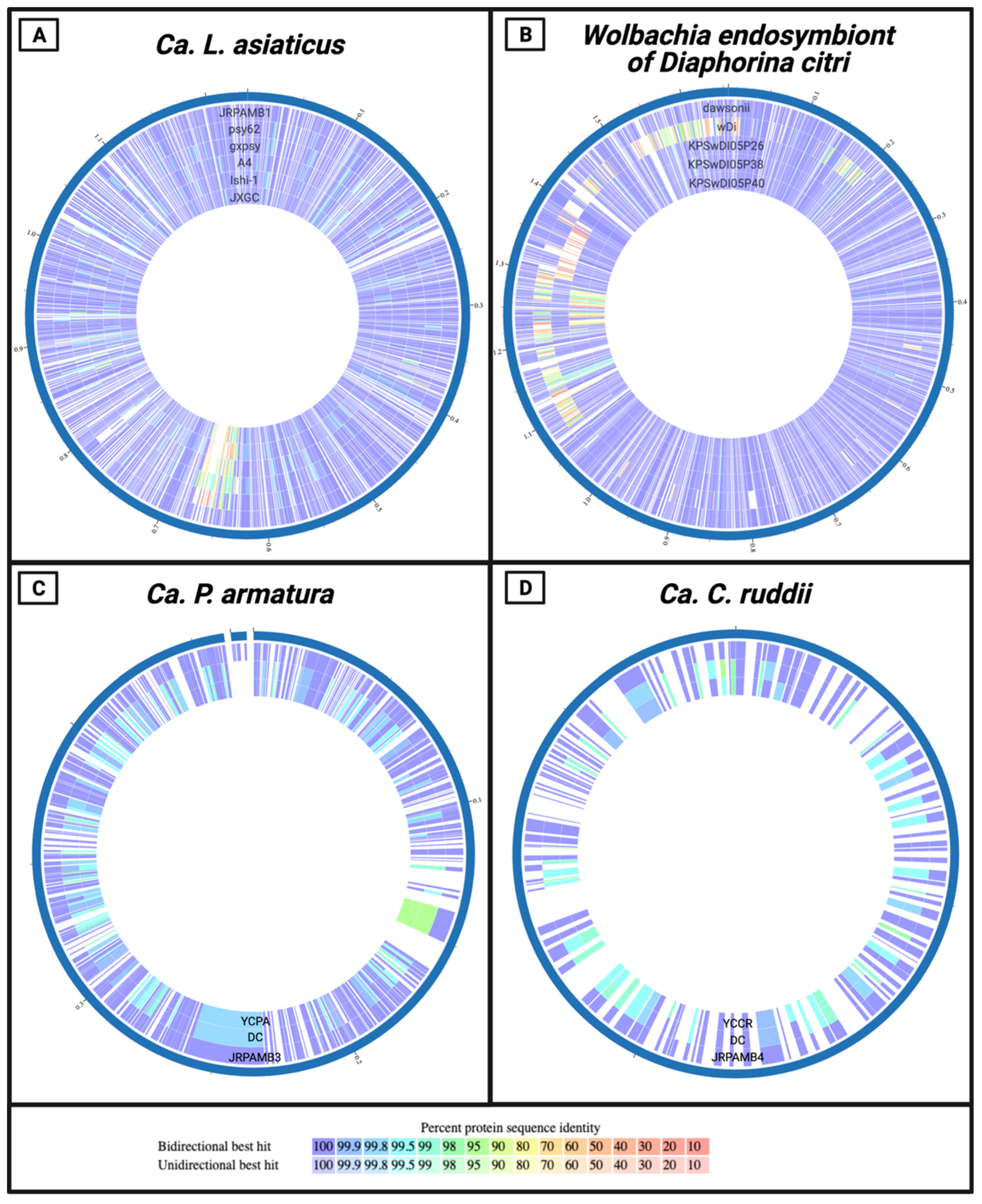
2.6. Genome Comparison
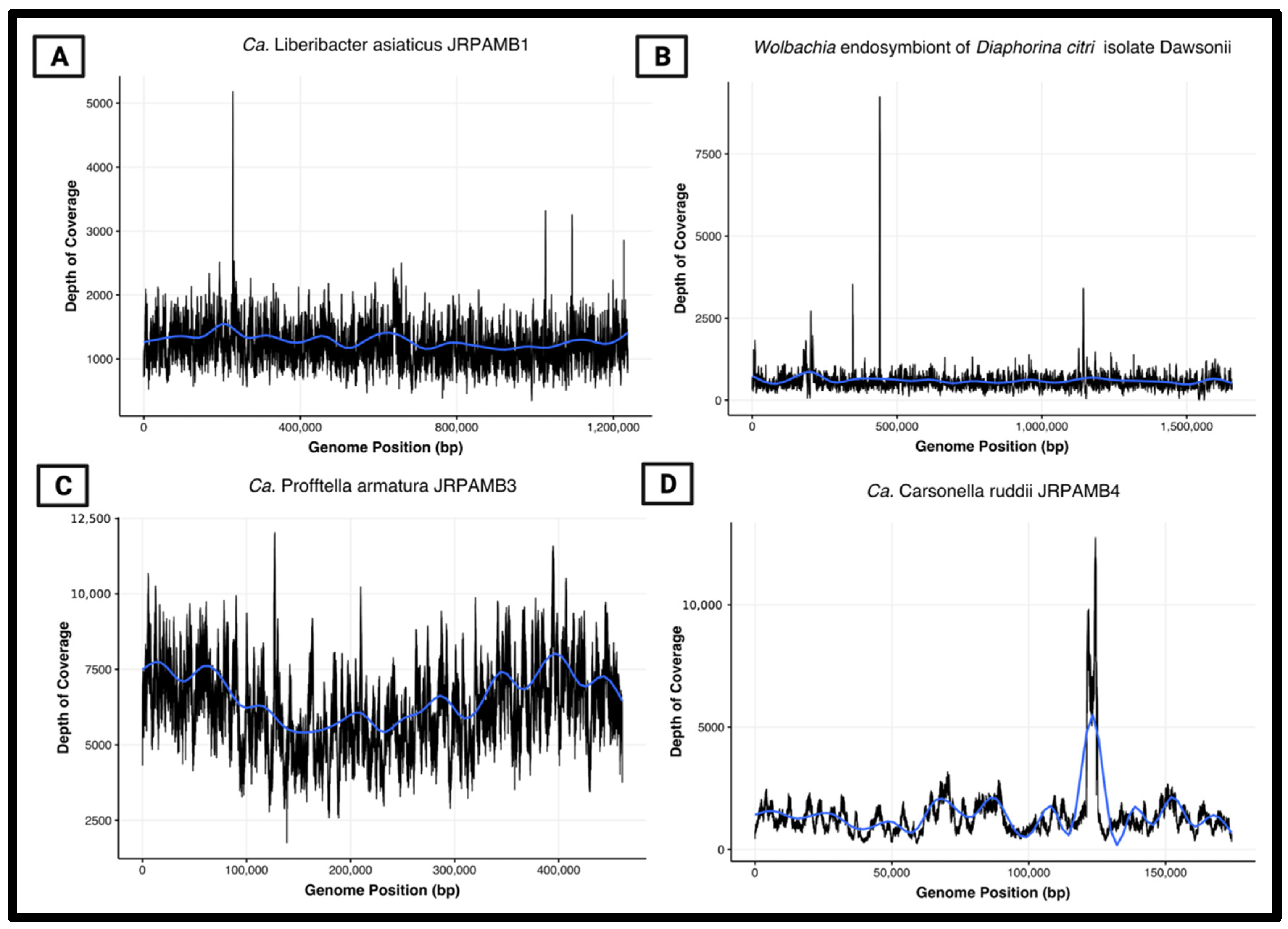
2.7. Peak-to-Trough Analysis
3. Results
3.1. Infected Psyllid DNA Pool
3.2. Initial Sequencing Results and Genome Assembly
3.3. Long-Read Sequencing Leads to the First Circularization of the Wolbachia Strain Genome
3.4. Whole-Genome Nucleotide Similarity of the Genomes
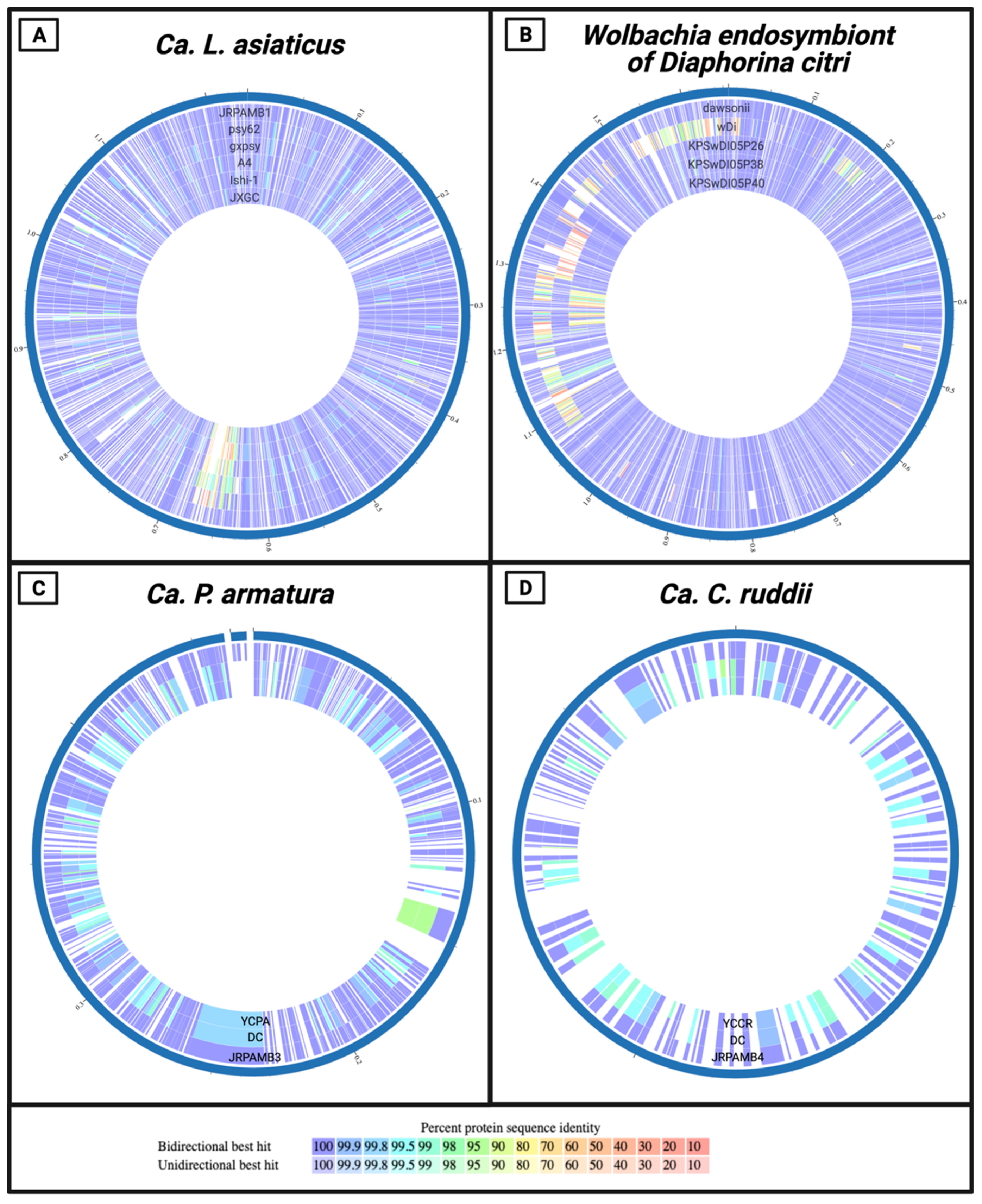
3.5. Protein-Level Comparison of the Genomes
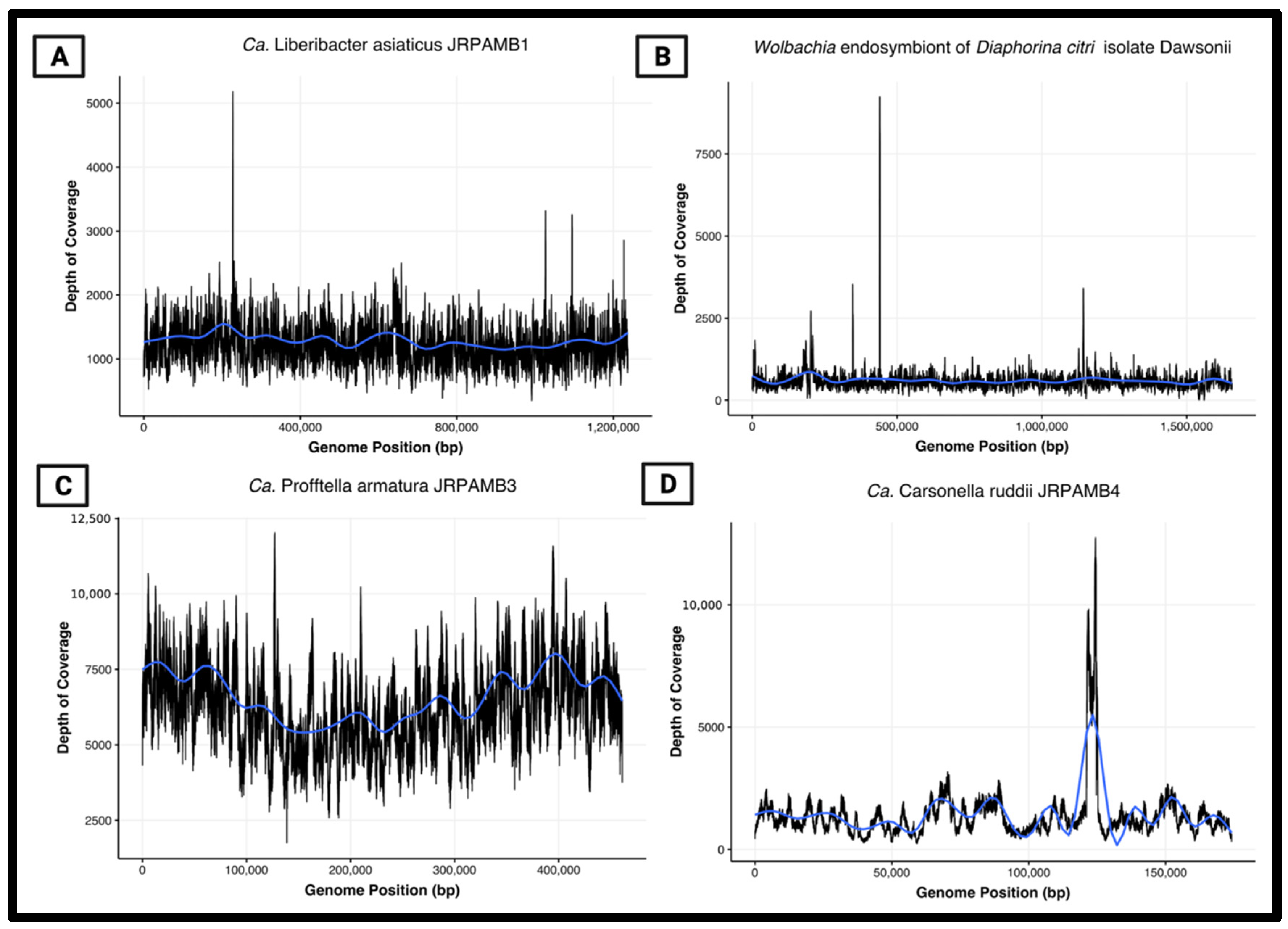
3.6. Peak-to-Trough Ratios Suggest Ca. Profftella Armatura Actively Replicating in the Population
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ballare, K.M.; Pope, N.S.; Castilla, A.R.; Cusser, S.; Metz, R.P.; Jha, S. Utilizing field collected insects for next generation sequencing: Effects of sampling, storage, and DNA extraction methods. Ecol. Evol. 2019, 9, 13690–13705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagisz, M.; Port, G.; Wolff, K. A cost-effective, simple and high-throughput method for DNA extraction from insects. Insect Sci. 2010, 17, 465–470. [Google Scholar] [CrossRef]
- Wenninger, E.J.; Hall, D.G. Daily and Seasonal Patterns in Abdominal Color in Diaphorina citri (Hemiptera: Psyllidae). Ann. Entomol. Soc. Am. 2008, 101, 585–592. [Google Scholar] [CrossRef]
- Wu, F.; Deng, X.; Liang, G.; Huang, J.; Cen, Y.; Chen, J. Whole-Genome Sequence of “Candidatus Profftella armatura” from Diaphorina citri in Guangdong, China. Genome Announc. 2015, 3, e01282-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Z.; Deng, X.; Chen, J. Whole-Genome Sequence of “Candidatus Liberibacter asiaticus” from Guangdong, China. Genome Announc. 2014, 2, e00273-14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Z.; Bao, M.; Wu, F.; Van Horn, C.; Chen, J.; Deng, X. A Type 3 Prophage of “Candidatus Liberibacter asiaticus” Carrying a Restriction-Modification System. Phytopathology 2018, 108, 454–461. [Google Scholar] [CrossRef] [Green Version]
- Katoh, H.; Miyata, S.-I.; Inoue, H.; Iwanami, T. Unique features of a Japanese “Candidatus Liberibacter asiaticus” strain revealed by whole genome sequencing. PLoS ONE 2014, 9, e106109. [Google Scholar] [CrossRef] [Green Version]
- Tay, W.T.; Elfekih, S.; Polaszek, A.; Court, L.N.; Evans, G.A.; Gordon, K.H.J.; De Barro, P.J. Novel molecular approach to define pest species status and tritrophic interactions from historical Bemisia specimens. Sci. Rep. 2017, 7, 429. [Google Scholar] [CrossRef]
- Ye, Z.; Vollhardt, I.M.G.; Girtler, S.; Wallinger, C.; Tomanovic, Z.; Traugott, M. An effective molecular approach for assessing cereal aphid-parasitoid-endosymbiont networks. Sci. Rep. 2017, 7, 3138. [Google Scholar] [CrossRef]
- Wilding, C.S.; Weetman, D.; Steen, K.; Donnelly, M.J. Accurate determination of DNA yield from individual mosquitoes for population genomic applications. Insect Sci. 2009, 16, 361–363. [Google Scholar] [CrossRef]
- Hong, S.; Bunge, J.; Leslin, C.; Jeon, S.; Epstein, S.S. Polymerase chain reaction primers miss half of rRNA microbial diversity. ISME J. 2009, 3, 1365–1373. [Google Scholar] [CrossRef] [PubMed]
- Thoendel, M.; Jeraldo, P.; Greenwood-Quaintance, K.E.; Yao, J.; Chia, N.; Hanssen, A.D.; Abdel, M.P.; Patel, R. Impact of Contaminating DNA in Whole-Genome Amplification Kits Used for Metagenomic Shotgun Sequencing for Infection Diagnosis. J. Clin. Microbiol. 2017, 55, 1789–1801. [Google Scholar] [CrossRef] [Green Version]
- Roux, S.; Trubl, G.; Goudeau, D.; Nath, N.; Couradeau, E.; Ahlgren, N.A.; Zhan, Y.; Marsan, D.; Chen, F.; Fuhrman, J.A.; et al. Optimizing de novo genome assembly from PCR-amplified metagenomes. PeerJ 2019, 7, e6902. [Google Scholar] [CrossRef] [Green Version]
- Kozarewa, I.; Ning, Z.; Quail, M.A.; Sanders, M.J.; Berriman, M.; Turner, D.J. Amplification-free Illumina sequencing-library preparation facilitates improved mapping and assembly of (G+C)-biased genomes. Nat. Methods 2009, 6, 291–295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chu, C.-C.; Gill, T.A.; Hoffmann, M.; Pelz-Stelinski, K.S. Inter-Population Variability of Endosymbiont Densities in the Asian Citrus Psyllid (Diaphorina citri Kuwayama). Microb. Ecol. 2016, 71, 999–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, Y.; Zhou, L.; Hall, D.G.; Li, W.; Doddapaneni, H.; Lin, H.; Liu, L.; Vahling, C.M.; Gabriel, D.W.; Williams, K.P.; et al. Complete Genome Sequence of Citrus Huanglongbing Bacterium, ‘Candidatus Liberibacter asiaticus’ Obtained through Metagenomics. Mol. Plant Microbe Interact. 2009, 22, 1011–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Thaochan, N.; Huang, J.; Chen, J.; Deng, X.; Zheng, Z. Genome Sequence Resource of ‘Candidatus Liberibacter asiaticus’ from Thailand. Plant Dis. 2019, 104, 624–626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Z.; Sun, X.; Deng, X.; Chen, J. Whole-Genome Sequence of “Candidatus Liberibacter asiaticus” from a Huanglongbing-Affected Citrus Tree in Central Florida. Genome Announc. 2015, 3, e00169-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.; Han, C.S.; Liu, B.; Lou, B.; Bai, X.; Deng, C.; Civerolo, E.L.; Gupta, G. Complete Genome Sequence of a Chinese Strain of “Candidatus Liberibacter asiaticus”. Genome Announc. 2013, 1, e00184-13. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Hunter, W.B.; Reese, J.; Morgan, J.K.; Marutani-Hert, M.; Huang, H.; Lindeberg, M. Survey of endosymbionts in the Diaphorina citri metagenome and assembly of a Wolbachia wDi draft genome. PLoS ONE 2012, 7, e50067. [Google Scholar] [CrossRef] [Green Version]
- Quick, J. Ultra-long read sequencing protocol for RAD004. Protocols 2018, io10, 17504. [Google Scholar] [CrossRef]
- Tyson, J. Bead-free long fragment LSK109 library preparation. eLife 2020, 10, e66405. [Google Scholar] [CrossRef]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [Green Version]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple Genome Alignment with Gene Gain, Loss and Rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, M.; Rosselló-Móra, R.; Oliver Glöckner, F.; Peplies, J. JSpeciesWS: A web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 2016, 32, 929–931. [Google Scholar] [CrossRef]
- Marçais, G.; Delcher, A.L.; Phillippy, A.M.; Coston, R.; Salzberg, S.L.; Zimin, A. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef] [Green Version]
- Darzentas, N. Circoletto: Visualizing sequence similarity with Circos. Bioinformatics 2010, 26, 2620–2621. [Google Scholar] [CrossRef]
- Davis, J.J.; Wattam, A.R.; Aziz, R.K.; Brettin, T.; Butler, R.; Butler, R.M.; Chlenski, P.; Conrad, N.; Dickerman, A.; Dietrich, E.M.; et al. The PATRIC Bioinformatics Resource Center: Expanding data and analysis capabilities. Nucleic Acids Res. 2020, 48, D606–D612. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- RStudio Team. RStudio: Integrated Development for R; RStudio, PBC: Boston, MA, USA, 2020. [Google Scholar]
- Chin, C.-S.; Alexander, D.H.; Marks, P.; Klammer, A.A.; Drake, J.; Heiner, C.; Clum, A.; Copeland, A.; Huddleston, J.; Eichler, E.E.; et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 2013, 10, 563–569. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunt, M.; Silva, N.D.; Otto, T.D.; Parkhill, J.; Keane, J.A.; Harris, S.R. Circlator: Automated circularization of genome assemblies using long sequencing reads. Genome Biol. 2015, 16, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaser, R.; Sović, I.; Nagarajan, N.; Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 2017, 27, 737–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolmogorov, M.; Bickhart, D.M.; Behsaz, B.; Gurevich, A.; Rayko, M.; Shin, S.B.; Kuhn, K.; Yuan, J.; Polevikov, E.; Smith, T.P.L.; et al. metaFlye: Scalable long-read metagenome assembly using repeat graphs. Nat. Methods 2020, 17, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wu, J.; Yang, J.; Sun, S.; Xiao, J.; Yu, J. PGAP: Pan-genomes analysis pipeline. Bioinformatics 2012, 28, 416–418. [Google Scholar] [CrossRef] [Green Version]
- Saha, S.; Hosmani, P.; Flores, M.; Hunter, W.; Brown, S.; Mueller, L.A. Using long reads, optical maps and long-range scaffolding to improve the Diaphorina citri genome. figshare 2017. [Google Scholar] [CrossRef]
- Neupane, S.; Bonilla, S.I.; Manalo, A.M.; Pelz-Stelinski, K.S. Near-Complete Genome Sequences of a Wolbachia Strain Isolated from Diaphorina citri Kuwayama (Hemiptera: Liviidae). Microbiol. Resour. Announc. 2020, 9, e00560-20. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.-X.; Anantharaman, K.; Shaiber, A.; Eren, A.M.; Banfield, J.F. Accurate and complete genomes from metagenomes. Genome Res. 2020, 30, 315–333. [Google Scholar] [CrossRef] [Green Version]
- Lui, L.M.; Nielsen, T.N.; Arkin, A.P. A method for achieving complete microbial genomes and improving bins from metagenomics data. PLoS Comput. Biol. 2021, 17, e1008972. [Google Scholar] [CrossRef] [PubMed]
- Korem, T.; Zeevi, D.; Suez, J.; Weinberger, A.; Avnit-Sagi, T.; Pompan-Lotan, M.; Matot, E.; Jona, G.; Harmelin, A.; Cohen, N.; et al. Growth dynamics of gut microbiota in health and disease inferred from single metagenomic samples. Science 2015, 349, 1101–1106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Worning, P.; Jensen, L.J.; Hallin, P.F.; Stærfeldt, H.-H.; Ussery, D.W. Origin of replication in circular prokaryotic chromosomes. Environ. Microbiol. 2006, 8, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Grigoriev, A. Analyzing genomes with cumulative skew diagrams. Nucleic Acids Res. 1998, 26, 2286–2290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrone, J.R.; University of Florida Department of Microbiology and Cell Sciences, Gainesville, FL, USA. GenSkew Program Run on Genomes. 2021. [Google Scholar]
- Brown, C.T.; Olm, M.R.; Thomas, B.C.; Banfield, J.F. Measurement of bacterial replication rates in microbial communities. Nat. Biotechnol. 2016, 34, 1256–1263. [Google Scholar] [CrossRef] [Green Version]
- Watson, M.; Warr, A. Errors in long-read assemblies can critically affect protein prediction. Nat. Biotechnol. 2019, 37, 124–126. [Google Scholar] [CrossRef] [Green Version]
- Kerkhof, L.J. Is Oxford Nanopore sequencing ready for analyzing complex microbiomes? FEMS Microbiol. Ecol. 2021, 97, fiab001. [Google Scholar] [CrossRef]
- Goldstein, S.; Beka, L.; Graf, J.; Klassen, J.L. Evaluation of strategies for the assembly of diverse bacterial genomes using MinION long-read sequencing. BMC Genom. 2019, 20, 23. [Google Scholar] [CrossRef] [Green Version]
- Hommelsheim, C.M.; Frantzeskakis, L.; Huang, M.; Ülker, B. PCR amplification of repetitive DNA: A limitation to genome editing technologies and many other applications. Sci. Rep. 2014, 4, 5052. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ca. L. asiaticus JRPAMB1/ ishi | Wolbachia Dawsonii/ KPSwDI05P40 | Ca. P. armatura JRPAMB3/ DC | Ca. C. rudii JRPAMB4/ DC | |
|---|---|---|---|---|
| Genes (total) | 1111/1057 | 1561/1409 | 405/401 | 226/221 |
| Protein CDSs | 1032/980 | 1332/1234 | 360/356 | 198/196 |
| tRNAs | 44/44 | 34/34 | 34/34 | 22/22 |
| Pseudo Genes | 23/21 | 188/134 | 5/5 | 3/0 |
| Size (bp) | 1,237,165/1,190,853 | 1,656,288/1,538,623 | 461,109/459,399 (5459/5458) | 174,118/174,014 |
| GC Content (%) | 36.4/36.3 | 34.0/33.9 | 24.2/24.2 | 17.8/17.6 |
| Assembler | HGAP v4 | Flye v2.8.1-meta | Canu v1.8 | Canu v1.8 |
| Polisher | Arrow Resequencing SMRT Linkv7.0.0 | (4×) Minimap2v2.16 + Racon v1.4.3 | (4×) Minimap2v2.16 + Racon v1.4.3 | Arrow Resequencing SMRT Linkv7.0.0 |
| Contigs | 1/1 | 1/1 | 1/1 | 1/1 |
| CheckM(%) Completeness Contamination | 97.30/97.30 0.00/0.00 | 100/100 0.43/0.43 | 33.47/33.47 0.00/0.00 | 16.60/16.60 0.00/0.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petrone, J.R.; Muñoz-Beristain, A.; Glusberger, P.R.; Russell, J.T.; Triplett, E.W. Unamplified, Long-Read Metagenomic Sequencing Approach to Close Endosymbiont Genomes of Low-Biomass Insect Populations. Microorganisms 2022, 10, 513. https://doi.org/10.3390/microorganisms10030513
Petrone JR, Muñoz-Beristain A, Glusberger PR, Russell JT, Triplett EW. Unamplified, Long-Read Metagenomic Sequencing Approach to Close Endosymbiont Genomes of Low-Biomass Insect Populations. Microorganisms. 2022; 10(3):513. https://doi.org/10.3390/microorganisms10030513
Chicago/Turabian StylePetrone, Joseph R., Alam Muñoz-Beristain, Paula Rios Glusberger, Jordan T. Russell, and Eric W. Triplett. 2022. "Unamplified, Long-Read Metagenomic Sequencing Approach to Close Endosymbiont Genomes of Low-Biomass Insect Populations" Microorganisms 10, no. 3: 513. https://doi.org/10.3390/microorganisms10030513
APA StylePetrone, J. R., Muñoz-Beristain, A., Glusberger, P. R., Russell, J. T., & Triplett, E. W. (2022). Unamplified, Long-Read Metagenomic Sequencing Approach to Close Endosymbiont Genomes of Low-Biomass Insect Populations. Microorganisms, 10(3), 513. https://doi.org/10.3390/microorganisms10030513