Molecular Characterization of African Swine Fever Virus Isolates in Estonia in 2014–2019

 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. Selection of Isolates for Molecular Characterization
2.3. Detection of the Genome of ASFV
2.4. B602L Gene Amplification and Sequencing
2.5. Phylogenetic Analysis
2.6. Mapping Software and Spatial Autocorrelation
3. Results
3.1. B602L Gene Amplification and Sequencing
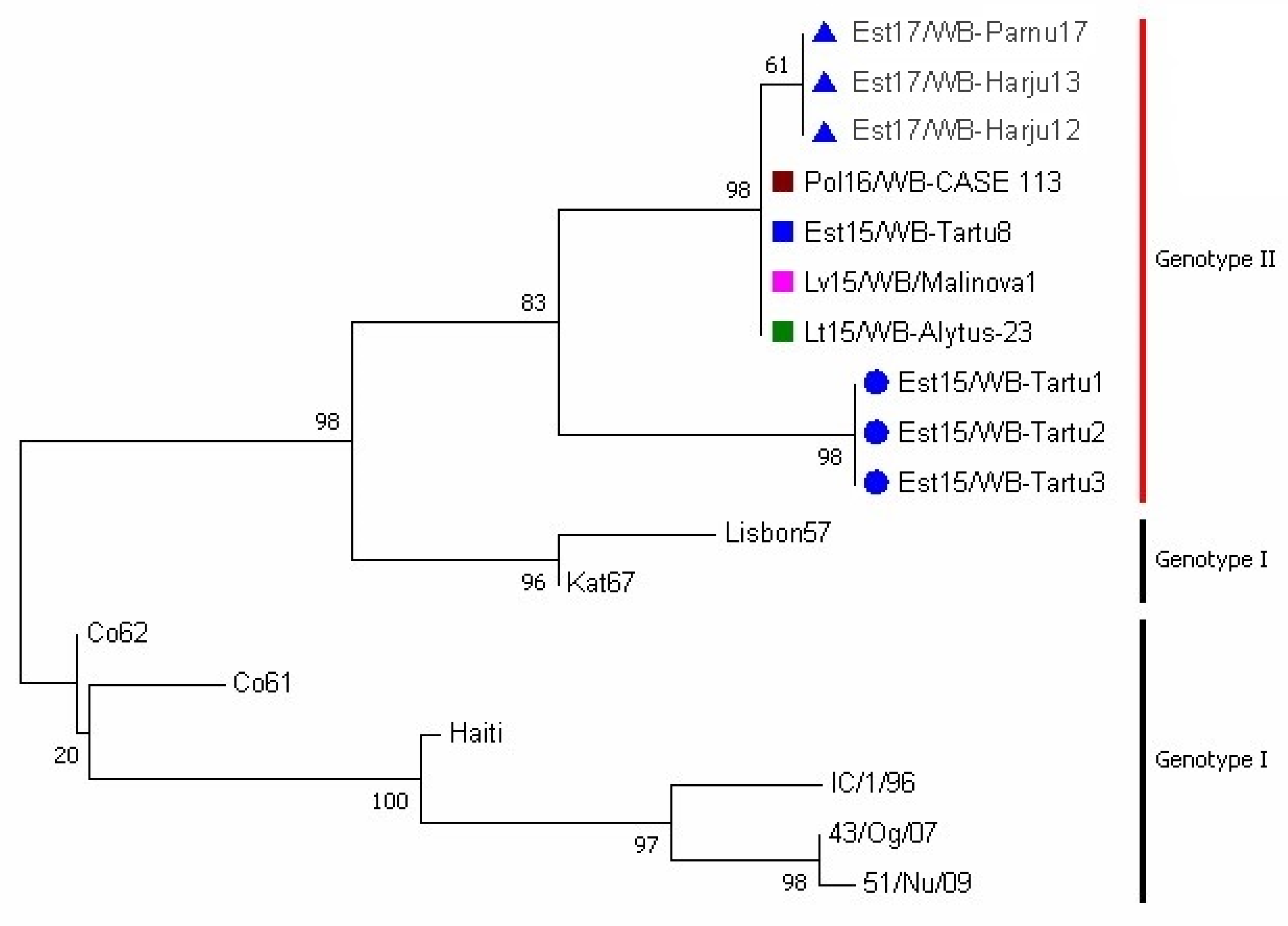
3.2. Phylogenetic Analysis
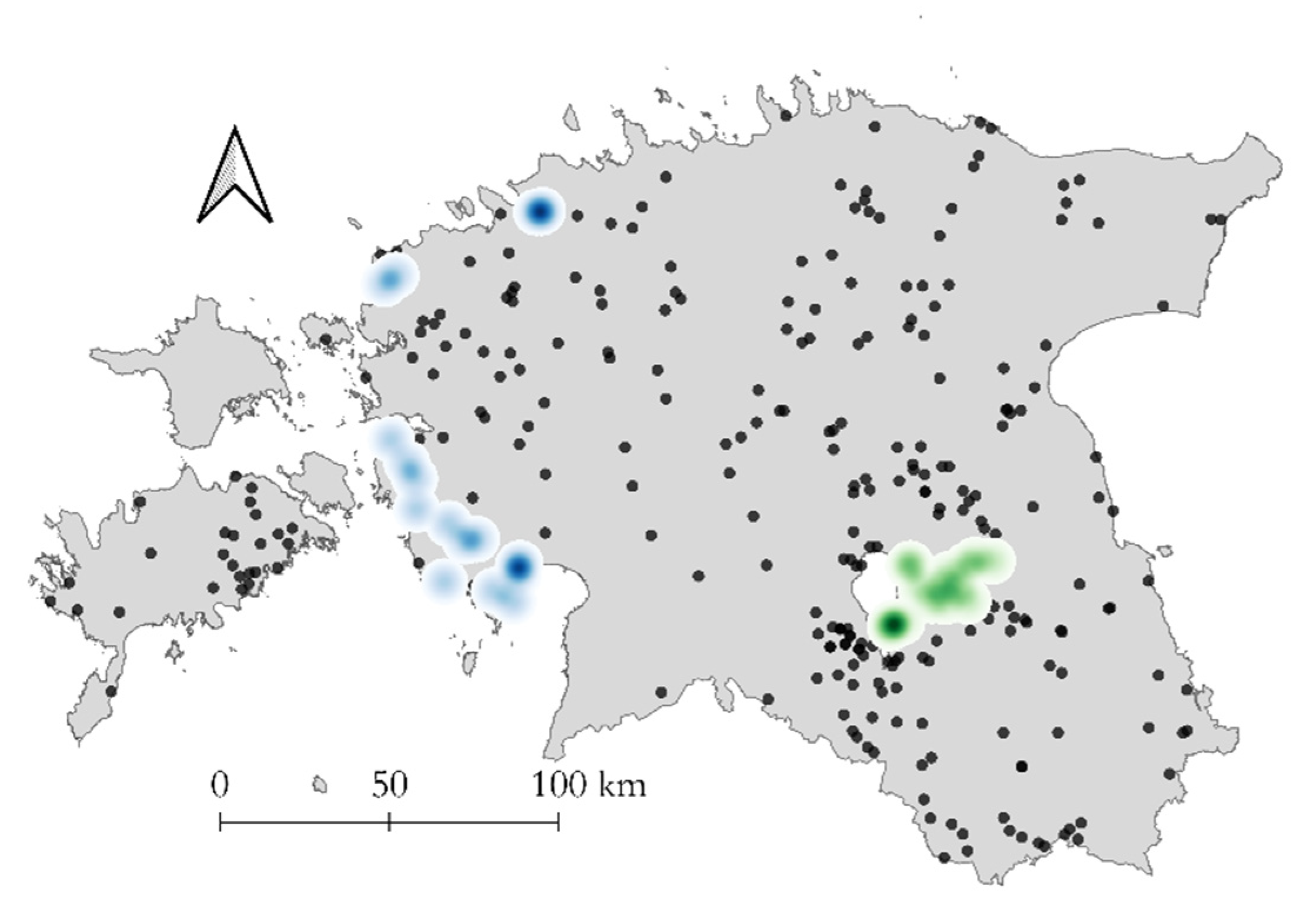
3.3. Geographic Distribution of CVR Variants
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Galindo, I.; Alonso, C. African swine fever virus: A review. Viruses 2017, 9, 103. [Google Scholar] [CrossRef] [PubMed]
- Barasona, J.A.; Gallardo, C.; Cadenas-Fernández, E.; Jurado, C.; Rivera, B.; Rodríguez-Bertos, A.; Arias, M.; Sánchez-Vizcaíno, J.M. First oral vaccination of eurasian wild boar against African swine fever virus genotype II. Front. Vet. Sci. 2019, 6, 137. [Google Scholar] [CrossRef] [PubMed]
- Malogolovkin, A.; Yelsukova, A.; Gallardo, C.; Tsybanov, S.; Kolbasov, D. Molecular characterization of African swine fever virus isolates originating from outbreaks in the Russian Federation between 2007 and 2011. Vet. Microbiol. 2012, 158, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Yáñez, R.J.; Rodriguez, J.M.; Nogal, M.L.; Yuste, L.; Enriquez, C.; Rodriguez, J.F.; Vinuela, E. Analysis of the complete nucleotide sequence of african swine fever virus. Virology 1995, 208, 249–278. [Google Scholar] [CrossRef]
- Dixon, L.K.; Chapman, D.A.G.; Netherton, C.L.; Upton, C. African swine fever virus replication and genomics. Virus Res. 2013, 173, 3–14. [Google Scholar] [CrossRef]
- Portugal, R.; Coelho, J.; Höper, D.; Little, N.S.; Smithson, C.; Upton, C.; Martins, C.; Leitão, A.; Keil, G.M. Related strains of African swine fever virus with different virulence: Genome comparison and analysis. J. Gen. Virol. 2015, 96, 408–419. [Google Scholar] [CrossRef]
- Alonso, C.; Borca, M.; Dixon, L.; Revilla, Y.; Rodriguez, F.; Escribano, J.M. ICTV report consortium ICTV virus taxonomy profile: Asfarviridae. J. Gen. Virol. 2018, 99, 613–614. [Google Scholar] [CrossRef]
- Gallardo, C.; Soler, A.; Nieto, R.; Cano, C.; Pelayo, V.; Sánchez, M.A.; Pridotkas, G.; Fernandez-Pinero, J.; Briones, V.; Arias, M. Experimental Infection of domestic pigs with African swine fever virus Lithuania 2014 genotype II field isolate. Transbound. Emerg. Dis. 2017, 64, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Arias, M.; Jurado, C.; Gallardo, C.; Fernández-Pinero, J.; Sánchez-Vizcaíno, J.M. Gaps in African swine fever: Analysis and priorities. Transbound. Emerg. Dis. 2018, 65, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Mulumba-Mfumu, L.K.; Saegerman, C.; Dixon, L.K.; Madimba, K.C.; Kazadi, E.; Mukalakata, N.T.; Oura, C.A.L.; Chenais, E.; Masembe, C.; Ståhl, K.; et al. African swine fever: Update on Eastern, Central and Southern Africa. Transbound. Emerg. Dis. 2019, tbed.13187. [Google Scholar] [CrossRef] [PubMed]
- Jurado, C.; Fernández-Carrión, E.; Mur, L.; Rolesu, S.; Laddomada, A.; Sánchez-Vizcaíno, J.M. Why is African swine fever still present in Sardinia? Transbound. Emerg. Dis. 2018, 65, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Rowlands, R.J.; Michaud, V.; Heath, L.; Hutchings, G.; Oura, C.; Vosloo, W.; Dwarka, R.; Onashvili, T.; Albina, E.; Dixon, L.K. African swine fever virus isolate, Georgia, 2007. Emerg. Infect. Dis. 2008, 14, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- World Organization for Animal Health. Current Situation of ASF, Report no 44, May 2020. Available online: https://www.oie.int/fileadmin/Home/eng/Animal_Health_in_the_World/docs/pdf/Disease_cards/ASF/Report_44_Current_situation_of_ASF.pdf (accessed on 10 June 2020).
- Word Organization for Animal Health. Immediate Notification Report, China 2018. Available online: https://www.oie.int/wahis_2/temp/reports/en_imm_0000027741_20180831_161124.pdf (accessed on 10 June 2020).
- World Organization for Animal Health. Immediate Notification Report. Papua New Guinea. 2020. Available online: https://www.oie.int/wahis_2/public/wahid.php/Reviewreport/Review?page_refer=MapFullEventReport&reportid=33803 (accessed on 10 June 2020).
- Bastos, A.D.S.; Penrith, M.-L.; Crucière, C.; Edrich, J.L.; Hutchings, G.; Roger, F.; Couacy-Hymann, E.; R.Thomson, G. Genotyping field strains of African swine fever virus by partial p72 gene characterisation. Arch. Virol. 2003, 148, 693–706. [Google Scholar] [CrossRef]
- Boshoff, C.I.; Bastos, A.D.S.; Gerber, L.J.; Vosloo, W. Genetic characterisation of African swine fever viruses from outbreaks in southern Africa (1973–1999). Vet. Microbiol. 2007, 121, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Quembo, C.J.; Jori, F.; Vosloo, W.; Heath, L. Genetic characterization of African swine fever virus isolates from soft ticks at the wildlife/domestic interface in Mozambique and identification of a novel genotype. Transbound. Emerg. Dis. 2018, 65, 420–431. [Google Scholar] [CrossRef]
- Achenbach, J.E.; Gallardo, C.; Nieto-Pelegrín, E.; Rivera-Arroyo, B.; Degefa-Negi, T.; Arias, M.; Jenberie, S.; Mulisa, D.D.; Gizaw, D.; Gelaye, E.; et al. Identification of a new genotype of African swine fever virus in domestic pigs from Ethiopia. Transbound. Emerg. Dis. 2017, 64, 1393–1404. [Google Scholar] [CrossRef]
- Arias, M.; de la Torre, A.; Dixon, L.; Gallardo, C.; Jori, F.; Laddomada, A.; Martins, C.; Parkhouse, R.M.; Revilla, Y.; Rodriguez, F.J.-M.; et al. Approaches and perspectives for development of African swine fever virus vaccines. Vaccines 2017, 5, 35. [Google Scholar] [CrossRef]
- Gallardo, C.; Mwaengo, D.M.; Macharia, J.M.; Arias, M.; Taracha, E.A.; Soler, A.; Okoth, E.; Martín, E.; Kasiti, J.; Bishop, R.P. Enhanced discrimination of African swine fever virus isolates through nucleotide sequencing of the p54, p72, and pB602L (CVR) genes. Virus Genes 2009, 38, 85–95. [Google Scholar] [CrossRef]
- Gallardo, C.; Nurmoja, I.; Soler, A.; Delicado, V.; Simón, A.; Martin, E.M.; Perez, C.P.; Nieto, R.; Arias, M. Evolution in Europe of African swine fever genotype II viruses from highly to moderately virulent. Vet. Microbiol. 2018, 219, 70–79. [Google Scholar] [CrossRef]
- Gallardo, C.; Arias, M. EURL-African Swine Fever (ASF) activities. In Proceedings of the ASF/CSF Annual Meeting, Madrid, Spain, 17 June 2019. [Google Scholar]
- Forth, J.H.; Tignon, M.; Cay, A.B.; Forth, L.F.; Höper, D.; Blome, S.; Beer, M. Comparative analysis of whole-genome sequence of African swine fever virus Belgium 2018/1. Emerg. Infect. Dis. 2019, 25, 1249–1252. [Google Scholar] [CrossRef]
- Ge, S.; Li, J.; Fan, X.; Liu, F.; Li, L.; Wang, Q.; Ren, W.; Bao, J.; Liu, C.; Wang, H.; et al. Molecular characterization of African swine fever virus, China, 2018. Emerg. Infect. Dis. 2018, 24, 2131–2133. [Google Scholar] [CrossRef]
- Nix, R.J.; Gallardo, C.; Hutchings, G.; Blanco, E.; Dixon, L.K. Molecular epidemiology of African swine fever virus studied by analysis of four variable genome regions. Arch. Virol. 2006, 151, 2475–2494. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, C. African swine fever virus p72 genotype IX in domestic pigs, Congo, 2009. Emerg. Infect. Dis. 2011. [Google Scholar] [CrossRef]
- Wade, A.; Achenbach, J.E.; Gallardo, C.; Settypalli, T.B.K.; Souley, A.; Djonwe, G.; Loitsch, A.; Dauphin, G.; Ngang, J.J.E.; Boyomo, O.; et al. Genetic characterization of African swine fever virus in Cameroon, 2010–2018. J. Microbiol. 2019, 57, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, C.; Fernández-Pinero, J.; Pelayo, V.; Gazaev, I.; Markowska-Daniel, I.; Pridotkas, G.; Nieto, R.; Fernández-Pacheco, P.; Bokhan, S.; Nevolko, O.; et al. Genetic variation among African swine fever genotype II viruses, Eastern and Central Europe. Emerg. Infect. Dis. 2014, 20, 1544–1547. [Google Scholar] [CrossRef]
- Nurmoja, I.; Petrov, A.; Breidenstein, C.; Zani, L.; Forth, J.H.; Beer, M.; Kristian, M.; Viltrop, A.; Blome, S. Biological characterization of African swine fever virus genotype II strains from north-eastern Estonia in European wild boar. Transbound. Emerg. Dis. 2017, 64, 2034–2041. [Google Scholar] [CrossRef]
- Nurmoja, I.; Schulz, K.; Staubach, C.; Sauter-Louis, C.; Depner, K.; Conraths, F.J.; Viltrop, A. Development of African swine fever epidemic among wild boar in Estonia-two different areas in the epidemiological focus. Sci. Rep. 2017, 7, 12562. [Google Scholar] [CrossRef]
- Zani, L.; Forth, J.H.; Forth, L.; Nurmoja, I.; Leidenberger, S.; Henke, J.; Carlson, J.; Breidenstein, C.; Viltrop, A.; Höper, D.; et al. Deletion at the 5’-end of Estonian ASFV strains associated with an attenuated phenotype. Sci. Rep. 2018, 8, 6510. [Google Scholar] [CrossRef]
- Nurmoja, I.; Mõtus, K.; Kristian, M.; Niine, T.; Schulz, K.; Depner, K.; Viltrop, A. Epidemiological analysis of the 2015–2017 African swine fever outbreaks in Estonia. Prev. Vet. Med. 2018, S0167587718303611. [Google Scholar] [CrossRef] [PubMed]
- Schulz, K.; Staubach, C.; Blome, S.; Viltrop, A.; Nurmoja, I.; Conraths, F.J.; Sauter-Louis, C. Analysis of Estonian surveillance in wild boar suggests a decline in the incidence of African swine fever. Sci. Rep. 2019, 9, 8490. [Google Scholar] [CrossRef] [PubMed]
- Tignon, M.; Gallardo, C.; Iscaro, C.; Hutet, E.; Van der Stede, Y.; Kolbasov, D.; De Mia, G.M.; Le Potier, M.-F.; Bishop, R.P.; Arias, M.; et al. Development and inter-laboratory validation study of an improved new real-time PCR assay with internal control for detection and laboratory diagnosis of African swine fever virus. J. Virol. Methods 2011, 178, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Duvigneau, J.C.; Hartl, R.T.; Groiss, S.; Gemeiner, M. Quantitative simultaneous multiplex real-time PCR for the detection of porcine cytokines. J. Immunol. Methods 2005, 306, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Chapman, D.A.G.; Darby, A.C.; Da Silva, M.; Upton, C.; Radford, A.D.; Dixon, L.K. Genomic analysis of highly virulent Georgia 2007/1 isolate of African swine fever virus. Emerg. Infect. Dis. 2011, 17, 599–605. [Google Scholar] [CrossRef] [PubMed]
- QGIS.org (2020). QGIS Geographic Information System. Open Source Geospatial Foundation Project. Available online: https://qgis.org/en/site/ (accessed on 9 June 2020).
- Estonian Land Board. Map of Municipal Counties before Public Administration Reform: 2020. Available online: https://geoportaal.maaamet.ee/docs/haldus_asustus/maakond_20171015_shp.zip?t=20171015133716 (accessed on 8 June 2020).
- Pisati, M. Spmap: Stata module to visualize spatial data, Statistical Software Components S456812: Boston College Department of Economics. 2007. revised 18 January 2018. Available online: https://ideas.repec.org/c/boc/bocode/s456812.html (accessed on 9 June 2020).
- Pisati, M. Tools for Spatial Data Analysis. Stata Technical Bulletin. No 60. 2001, pp. 21–37. Available online: https://www.stata.com/products/stb/journals/stb60.pdf (accessed on 14 July 2020).
- Gallardo, C. EURL Activities 2017–2018. In Proceedings of the ASF/CSF Annual Meeting, Hannover, Germany, 29–30 May 2018. [Google Scholar]
- Irusta, P.M.; Borca, M.V.; Kutish, G.F.; Lu, Z.; Caler, E.; Carrillo, C.; Rock, D.L. Amino acid tandem repeats within a late viral gene define the central variable region of African swine fever virus. Virology 1996, 220, 20–27. [Google Scholar] [CrossRef]
- Garigliany, M.; Desmecht, D.; Tignon, M.; Cassart, D.; Lesenfant, C.; Paternostre, J.; Volpe, R.; Cay, A.B.; van den Berg, T.; Linden, A. Phylogeographic analysis of African swine fever virus, Western Europe, 2018. Emerg. Infect. Dis. 2019, 25, 184–186. [Google Scholar] [CrossRef]
- Elsukova, A.; Shevchenko, I.; Varentsova, A.; Zinyakov, N.; Igolkin, A.; Vlasova, N. Tandem repeat sequence in the intergenic region MGF 505 9R/10R is a new marker of the genetic variability among ASF Genotype II viruses. In Proceedings of the Epizone 10th Annual Meeting, Madrid, Spain, 27–29 September 2016. [Google Scholar]
- Norbert Mwiine, F.; Nkamwesiga, J.; Ndekezi, C.; Ochwo, S. Molecular characterization of African swine fever viruses from outbreaks in Peri-Urban Kampala, Uganda. Adv. Virol. 2019, 2019, 1–8. [Google Scholar] [CrossRef]
- Giammarioli, M.; Gallardo, C.; Oggiano, A.; Iscaro, C.; Nieto, R.; Pellegrini, C.; Dei Giudici, S.; Arias, M.; De Mia, G.M. Genetic characterisation of African swine fever viruses from recent and historical outbreaks in Sardinia (1978–2009). Virus Genes 2011, 42, 377–387. [Google Scholar] [CrossRef]
- Sanna, G.; Dei Giudici, S.; Bacciu, D.; Angioi, P.P.; Giammarioli, M.; De Mia, G.M.; Oggiano, A. Improved strategy for molecular characterization of African swine fever viruses from Sardinia, Based on Analysis of p30, CD2V and I73R / I329L Variable Regions. Transbound. Emerg. Dis. 2017, 64, 1280–1286. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Vizcaíno, P.J.M.; Martínez-López, B.; Martínez-Avilés, M.; Martins, C.; Boinas, F.; Michaud, V.; Jori, F.; Etter, E.; Albina, E.; Roger, F. Scientific review on African Swine Fever. EFSA Supporting Publ. 2009, 6. [Google Scholar] [CrossRef]
- Gallardo, M.C.; Reoyo, A.; de la, T.; Fernández-Pinero, J.; Iglesias, I.; Muñoz, M.J.; Arias, M.L. African swine fever: A global view of the current challenge. Porc. Health Manag. 2015, 1, 21. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Cordón, P.J.; Chapman, D.; Jabbar, T.; Reis, A.L.; Goatley, L.; Netherton, C.L.; Taylor, G.; Montoya, M.; Dixon, L. Different routes and doses influence protection in pigs immunised with the naturally attenuated African swine fever virus isolate OURT88/3. Antivir. Res. 2017, 138, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Blome, S.; Gabriel, C.; Dietze, K.; Breithaupt, A.; Beer, M. High virulence of African swine fever virus Caucasus isolate in European wild boars of all ages. Emerg. Infect. Dis. 2012, 18. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, C.; Blome, S.; Malogolovkin, A.; Parilov, S.; Kolbasov, D.; Teifke, J.P.; Beer, M. Characterization of African swine fever virus Caucasus isolate in European wild boars. Emerg. Infect. Dis. 2011, 17, 2342–2345. [Google Scholar] [CrossRef] [PubMed]
- Gallardo, C.; Soler, A.; Rodze, I.; Nieto, R.; Cano-Gómez, C.; Fernandez-Pinero, J.; Arias, M. Attenuated and non-haemadsorbing (non-HAD) genotype II African swine fever virus (ASFV) isolated in Europe, Latvia 2017. Transbound. Emerg. Dis. 2019, 66, 1399–1404. [Google Scholar] [CrossRef]
- Pietschmann, J.; Guinat, C.; Beer, M.; Pronin, V.; Tauscher, K.; Petrov, A.; Keil, G.; Blome, S. Course and transmission characteristics of oral low-dose infection of domestic pigs and European wild boar with a Caucasian African swine fever virus isolate. Arch. Virol. 2015, 160, 1657–1667. [Google Scholar] [CrossRef]
- Fernández-Pinero, J. Molecular analysis of Eastern European African swine fever viruses. In Proceedings of the CSF/ASF Annual Meeting, Hannover, Germany, 29–30 May 2018. [Google Scholar]




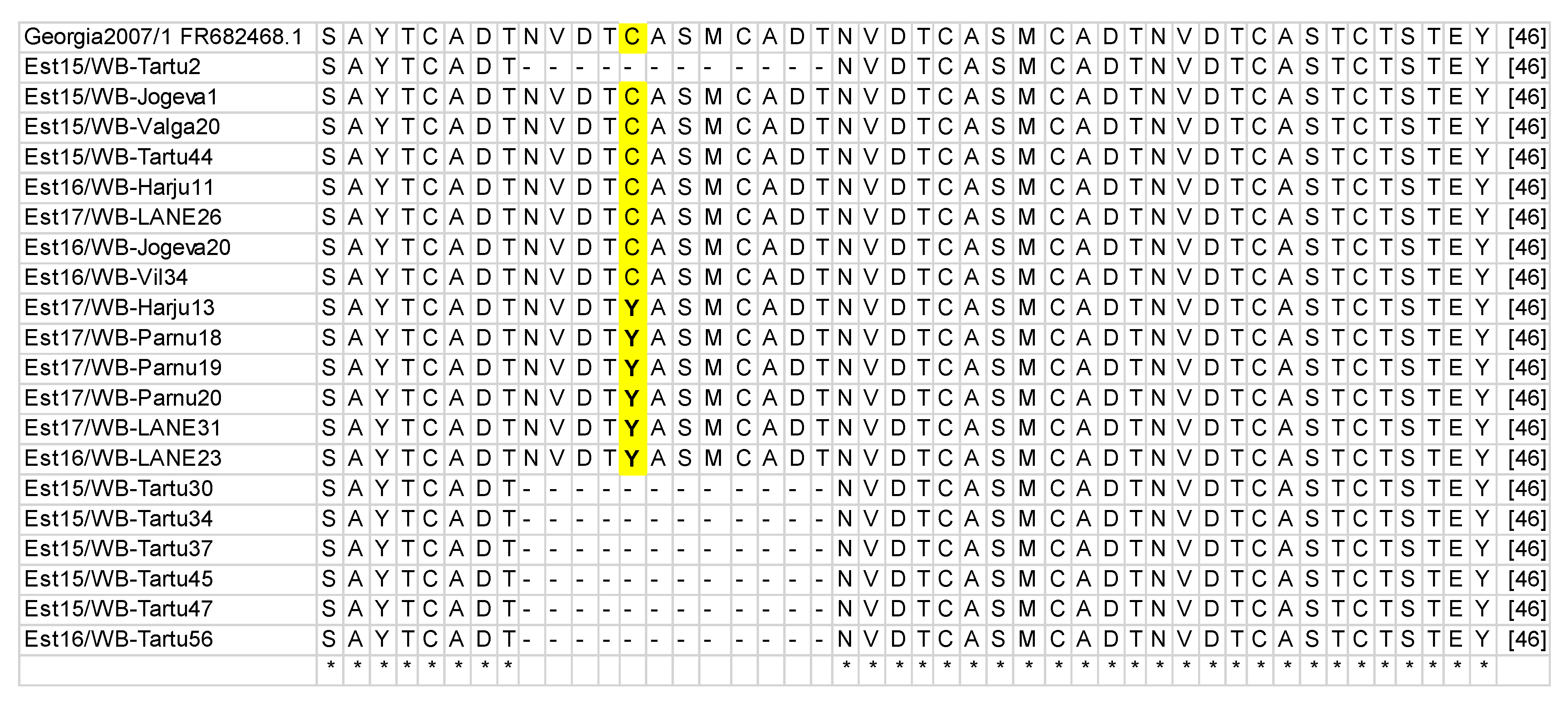{kind=link}
{kind=link}
{kind=link}
| Year | 2014 | 2015 | 2016 | 2017 | 2018 | 2019 | CVR% * | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| County | PCR | CVR | PCR | CVR | PCR | CVR | PCR | CVR | PCR | CVR | PCR | CVR | |
| Harju | 0 | 0 | 0 | 0 | 35 | 6 | 66 | 12 | 17 | 2 | 0 | 0 | 16.9 |
| Ida-Viru | 4 | 3 | 10 | 2 | 22 | 3 | 3 | 1 | 4 | 4 | 4 | 2 | 31.9 |
| Jõgeva | 0 | 0 | 55 | 6 | 146 | 14 | 0 | 0 | 1 | 0 | 0 | 0 | 9.9 |
| Järva | 0 | 0 | 98 | 9 | 95 | 5 | 0 | 0 | 0 | 0 | 0 | 0 | 7.3 |
| Lääne | 0 | 0 | 0 | 0 | 57 | 13 | 97 | 16 | 10 | 2 | 1 | 0 | 18.8 |
| L-Viru | 0 | 0 | 82 | 6 | 171 | 9 | 42 | 4 | 0 | 0 | 0 | 0 | 6.4 |
| Põlva | 0 | 0 | 227 | 6 | 166 | 2 | 9 | 4 | 3 | 1 | 0 | 0 | 3.2 |
| Pärnu | 0 | 0 | 23 | 3 | 76 | 6 | 68 | 13 | 5 | 0 | 0 | 0 | 12.8 |
| Rapla | 0 | 0 | 6 | 2 | 181 | 6 | 51 | 7 | 1 | 0 | 0 | 0 | 6.3 |
| Saare | 0 | 0 | 0 | 0 | 90 | 8 | 236 | 16 | 15 | 1 | 1 | 0 | 7.3 |
| Tartu | 0 | 0 | 117 | 38 | 151 | 26 | 13 | 6 | 1 | 1 | 0 | 0 | 25.2 |
| Valga | 13 | 6 | 113 | 17 | 10 | 3 | 0 | 0 | 0 | 0 | 0 | 0 | 19.1 |
| Viljandi | 47 | 9 | 166 | 22 | 37 | 6 | 3 | 1 | 0 | 0 | 0 | 0 | 15.0 |
| Võru | 9 | 5 | 108 | 11 | 40 | 1 | 3 | 2 | 1 | 0 | 0 | 0 | 11.8 |
| TOTAL | 73 | 25 | 1005 | 122 | 1277 | 108 | 591 | 82 | 58 | 11 | 6 | 2 | 11.6 |
| Size of an Amplicon (bp) | No of WB 1 | No of DP 2 | No of aa * Tetrameric Repeats | Difference From Reference Strain | CVR Variant |
|---|---|---|---|---|---|
| 400 | 300 | 42 | 10 | no | CVR1 |
| 400 | 18 | 6 | 10 | G instead of A resulted in aa change Y instead of C | CVR1/SNP1 |
| ~350 | 17 | 0 | 7 | deletion of 3 aa tetramer repeats CASMCADTNVDT | CVR2 |
| Number of Findings | |||||
|---|---|---|---|---|---|
| Municipality | First Finding | Last Finding | 2015 | 2016 | Total |
| Rannu | 17th of July 2015 | 22nd of Feb 2016 | 5 | 2 | 7 |
| Konguta | 29th of July 2015 | 6th of Nov 2015 | 4 | 0 | 4 |
| Tähtvere | 29th of Sept 2015 | 29th of Mar 2016 | 2 | 1 | 3 |
| Nõo | 14th of Oct 2015 | 15th of Mar 2016 | 2 | 1 | 3 |
| Total | 13 | 4 | 17 | ||
| No of Findings | ||||||
|---|---|---|---|---|---|---|
| County | Municipality | First Finding | Last Finding | 2016 | 2017 | Total |
| Lääne | Hanila | 30th of Nov 2016 | 24th of Jan 2017 | 2 | 1 | 3 |
| Nõva | 10th of Aug 2017 | 10th of Aug 2017 | 0 | 1 | 1 | |
| Noarootsi | 12th of Dec 2017 | 12th of Dec 2017 | 0 | 1 | 1 | |
| Harju | Keila | 5th of Jan 2017 | 4th of July 2017 | 0 | 3 | 3 |
| Pärnu | Varbla | 18th of Jan 2017 | 16th of June 2017 | 0 | 2 | 2 |
| Tõstamaa | 20th of Feb 2017 | 12th of Dec 2017 | 0 | 6 | 6 | |
| Audru | 25th of May 2017 | 16th of June 2017 | 0 | 2 | 2 | |
| Total | 2 | 16 | 18 | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vilem, A.; Nurmoja, I.; Niine, T.; Riit, T.; Nieto, R.; Viltrop, A.; Gallardo, C. Molecular Characterization of African Swine Fever Virus Isolates in Estonia in 2014–2019. Pathogens 2020, 9, 582. https://doi.org/10.3390/pathogens9070582
Vilem A, Nurmoja I, Niine T, Riit T, Nieto R, Viltrop A, Gallardo C. Molecular Characterization of African Swine Fever Virus Isolates in Estonia in 2014–2019. Pathogens. 2020; 9(7):582. https://doi.org/10.3390/pathogens9070582
Chicago/Turabian StyleVilem, Annika, Imbi Nurmoja, Tarmo Niine, Taavi Riit, Raquel Nieto, Arvo Viltrop, and Carmina Gallardo. 2020. "Molecular Characterization of African Swine Fever Virus Isolates in Estonia in 2014–2019" Pathogens 9, no. 7: 582. https://doi.org/10.3390/pathogens9070582
APA StyleVilem, A., Nurmoja, I., Niine, T., Riit, T., Nieto, R., Viltrop, A., & Gallardo, C. (2020). Molecular Characterization of African Swine Fever Virus Isolates in Estonia in 2014–2019. Pathogens, 9(7), 582. https://doi.org/10.3390/pathogens9070582