Variation Profile of the Orthotospovirus Genome



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
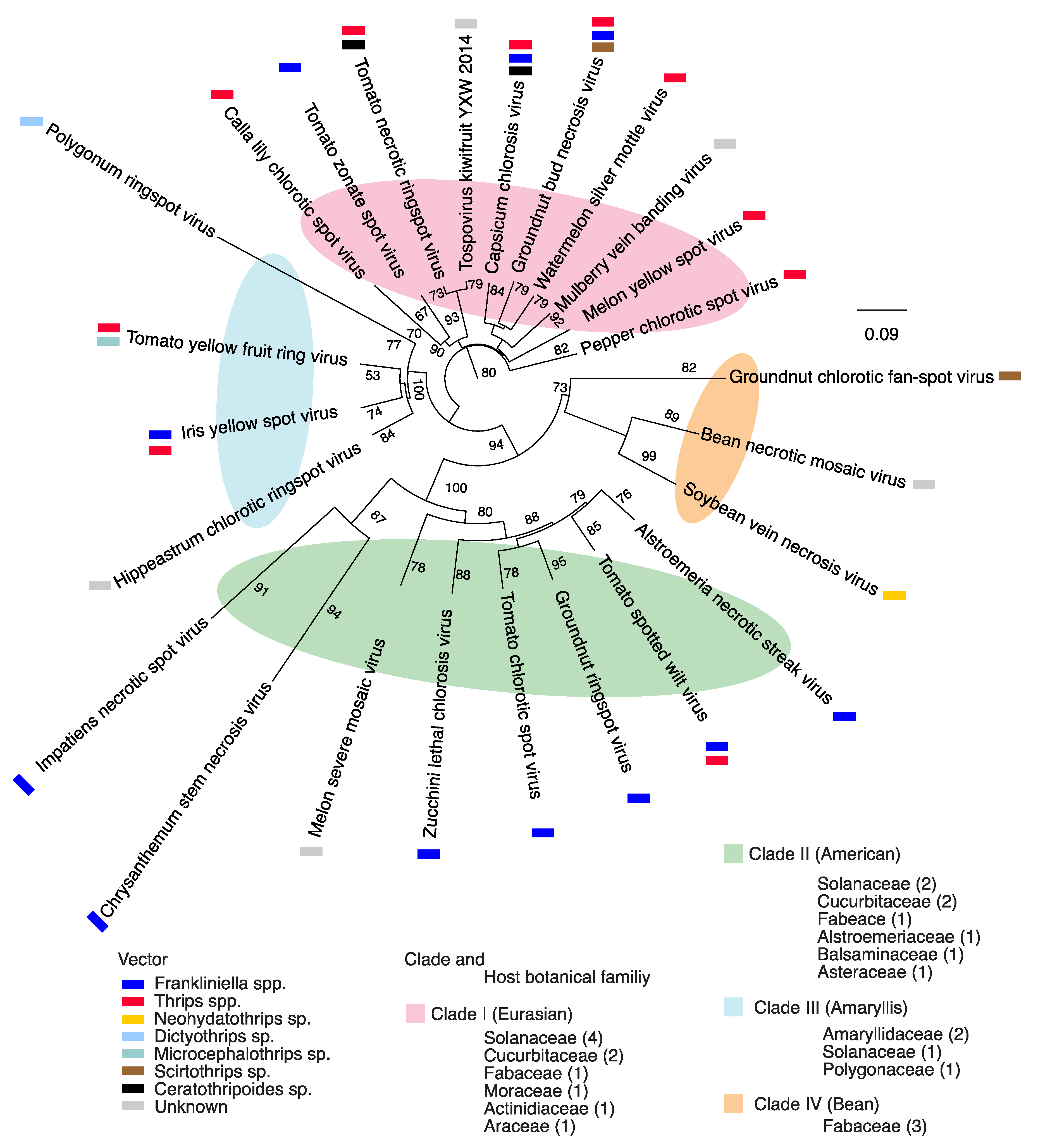
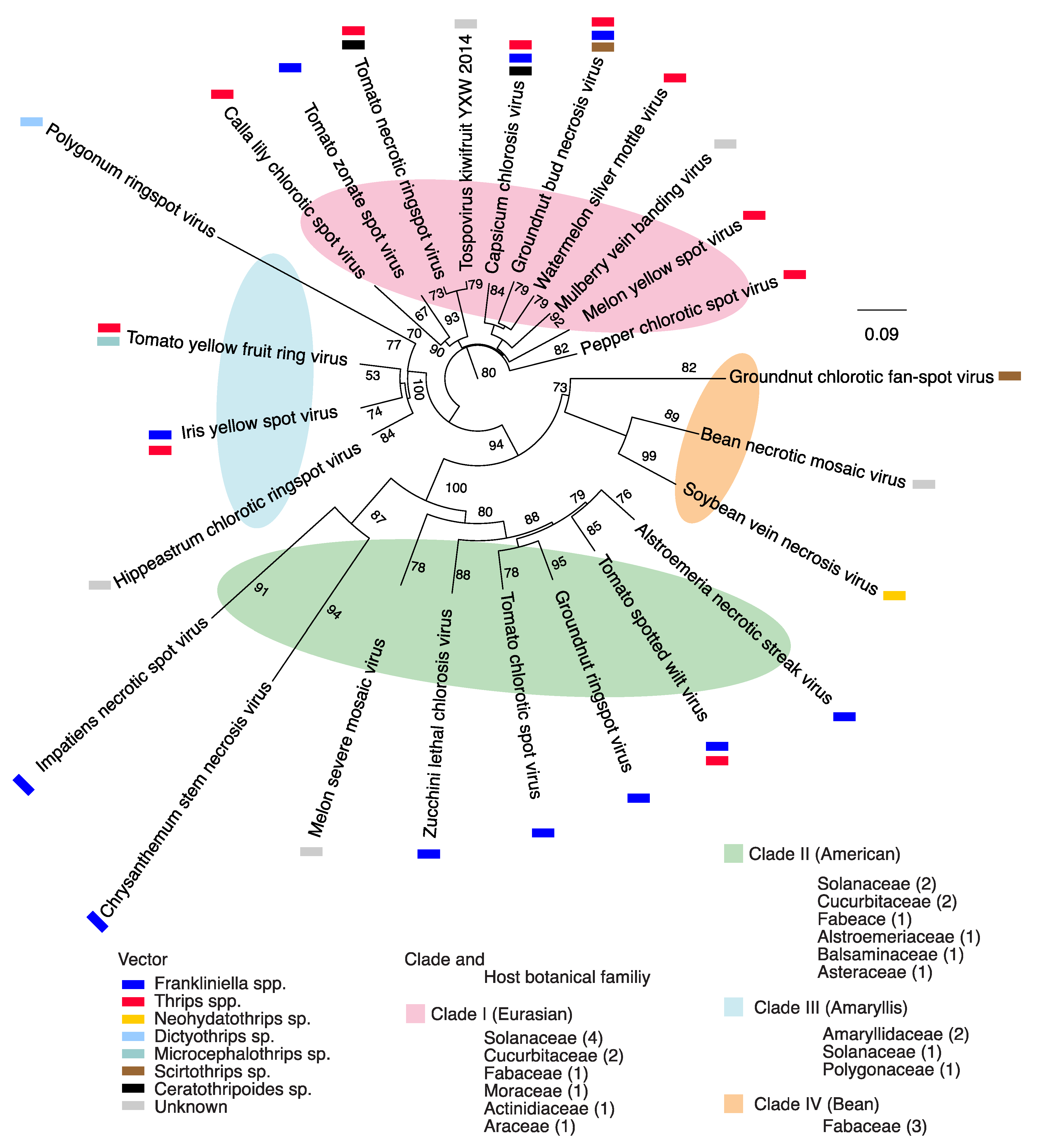
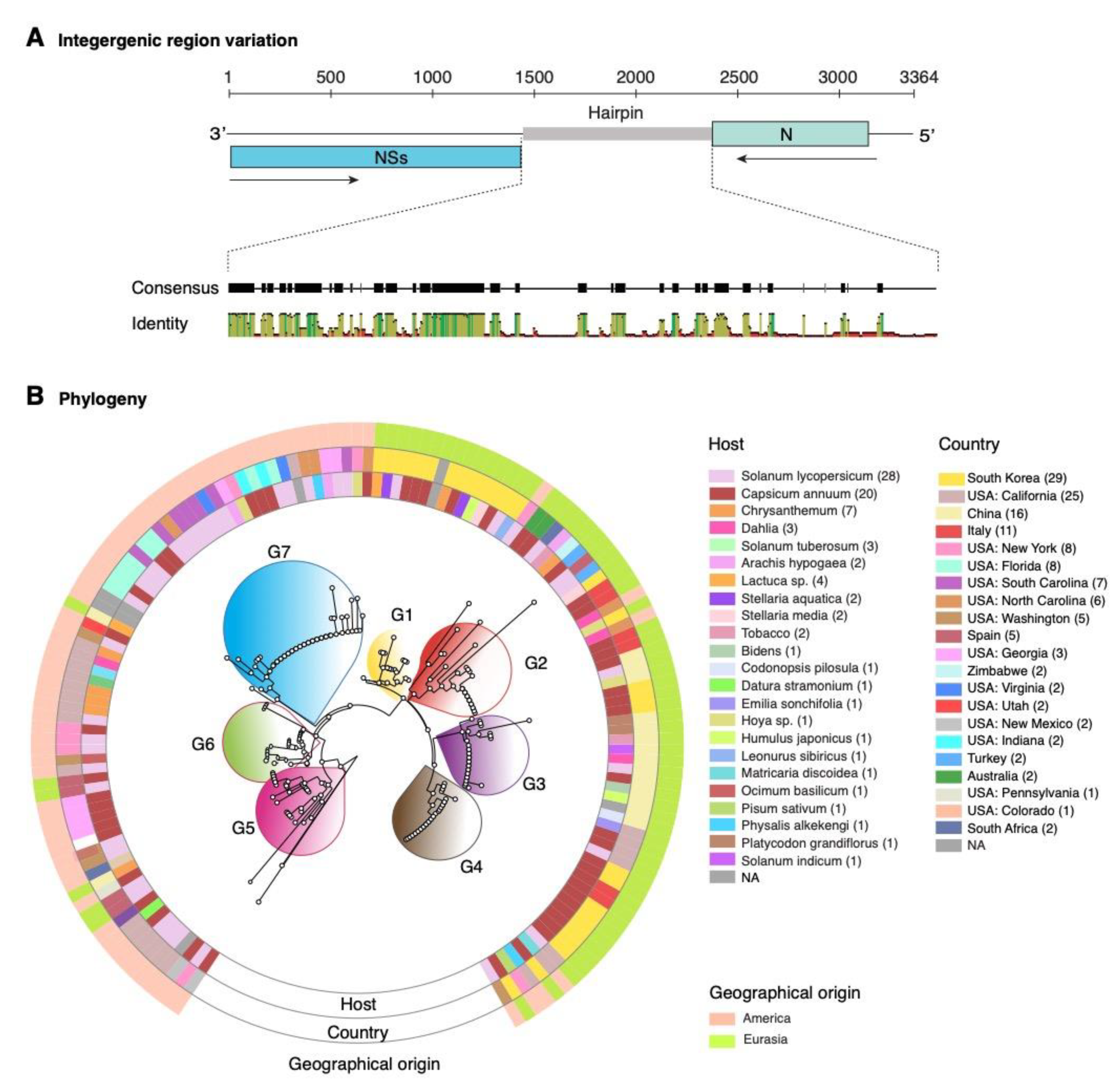
2.1. Orthotospovirus Phylogeny
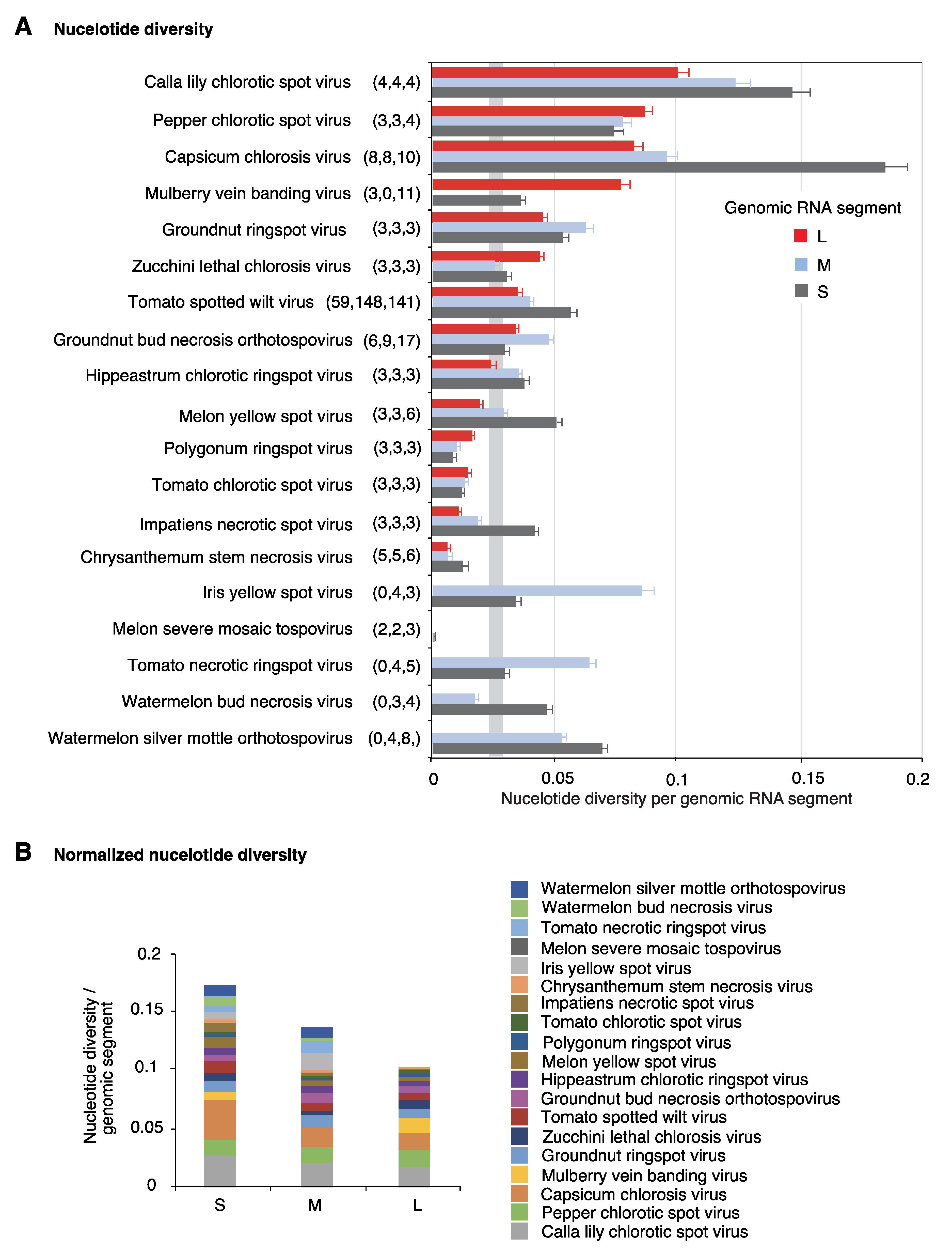
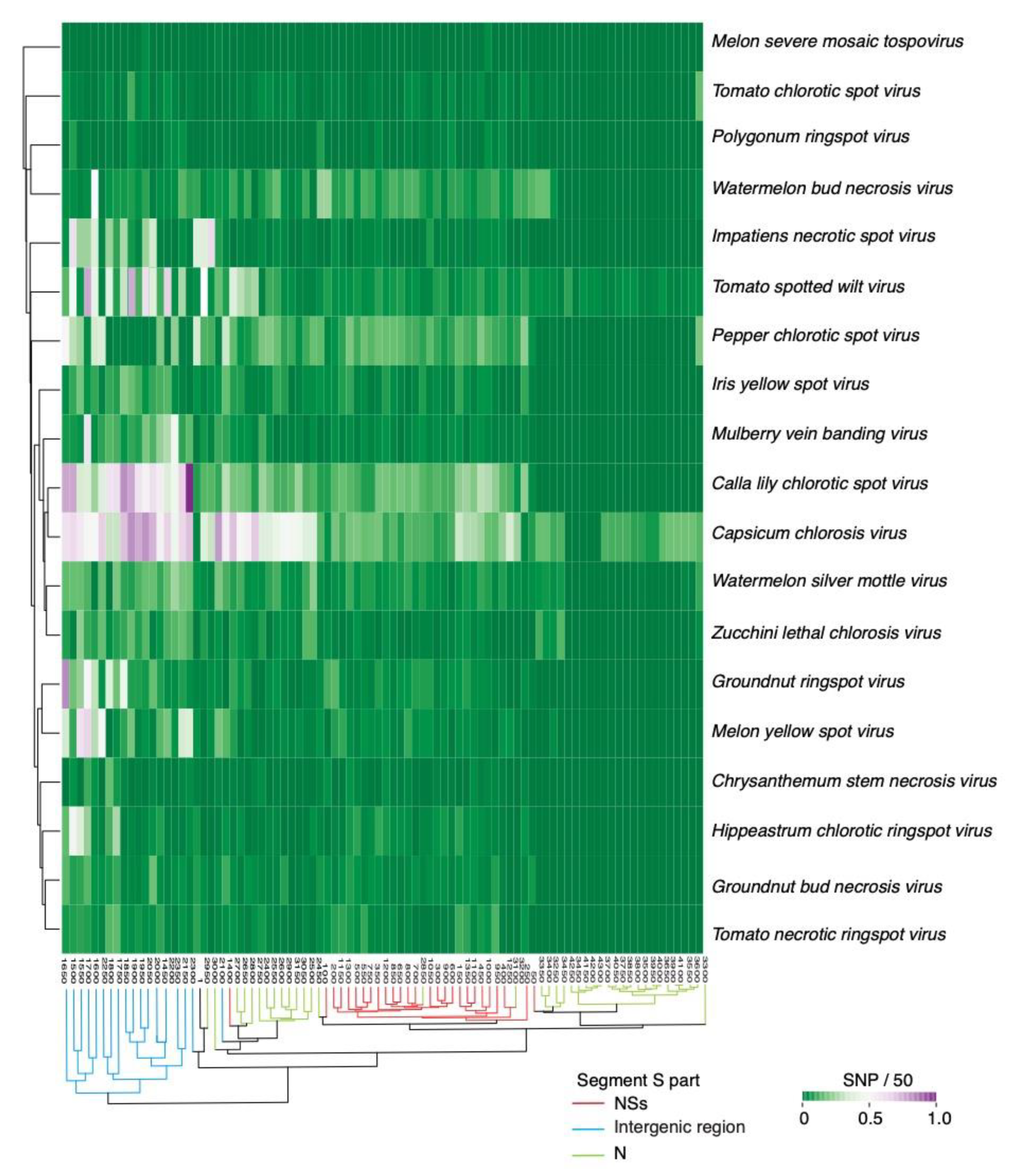
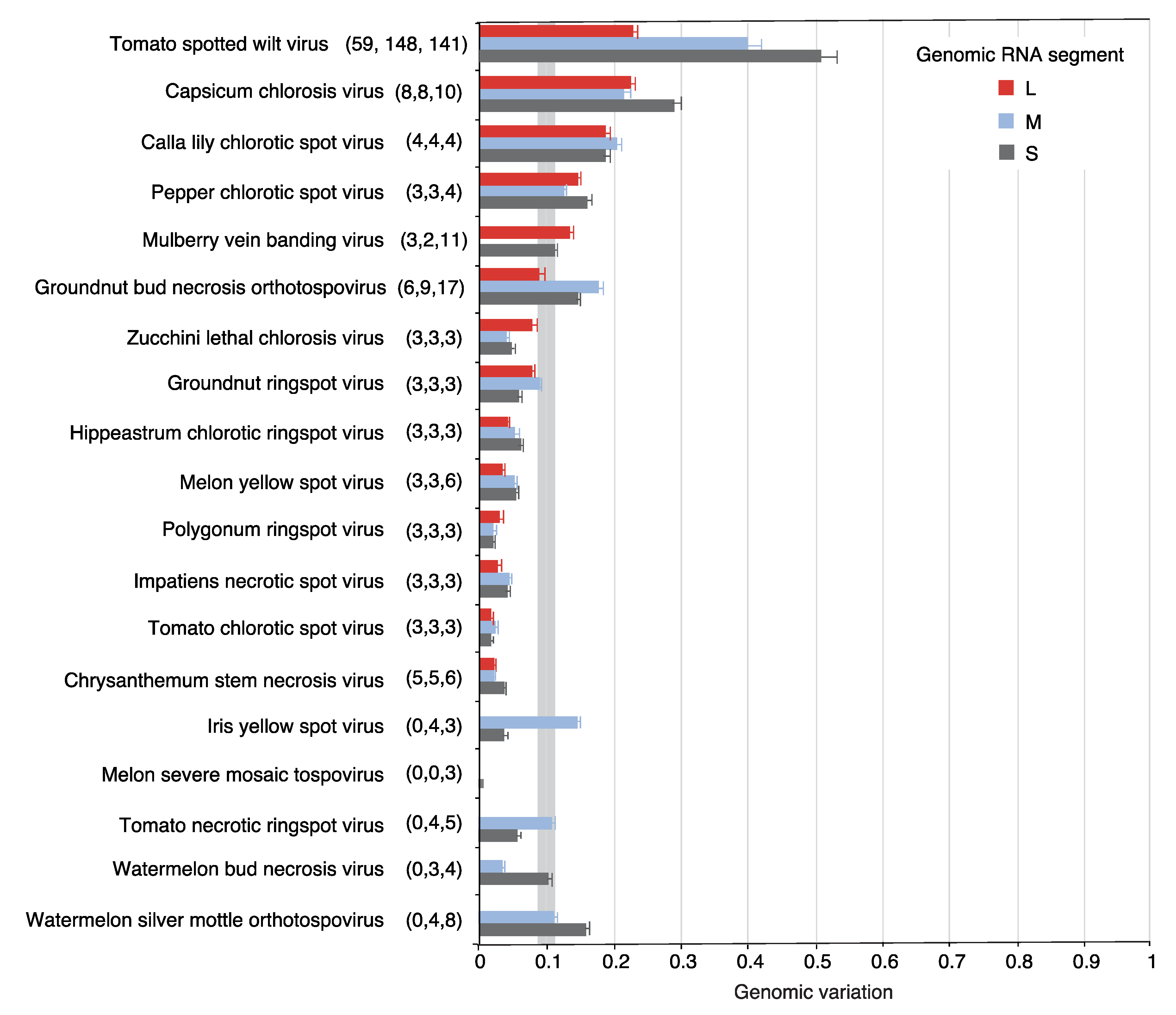
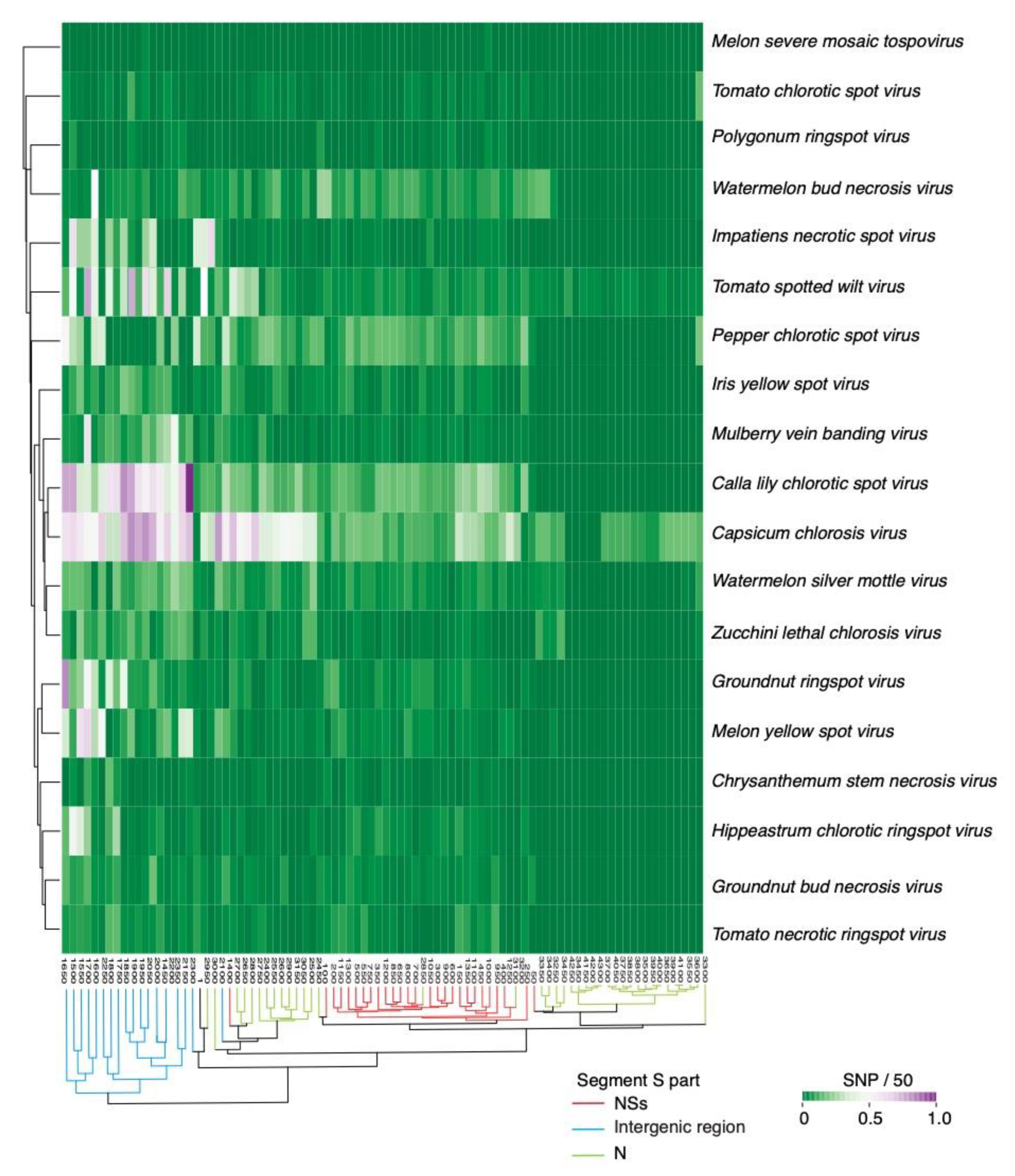
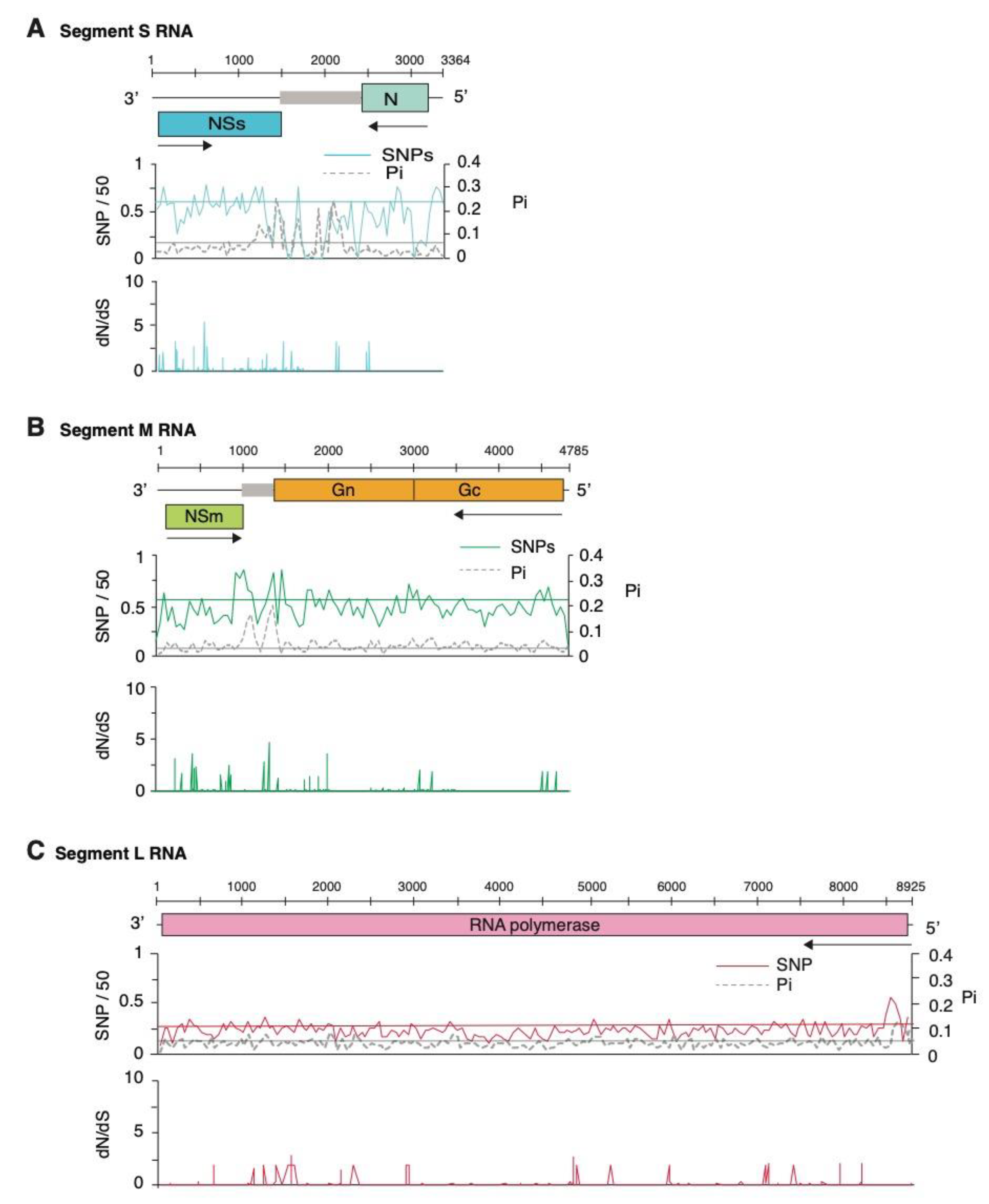
2.2. Segment S Is the Most Variable
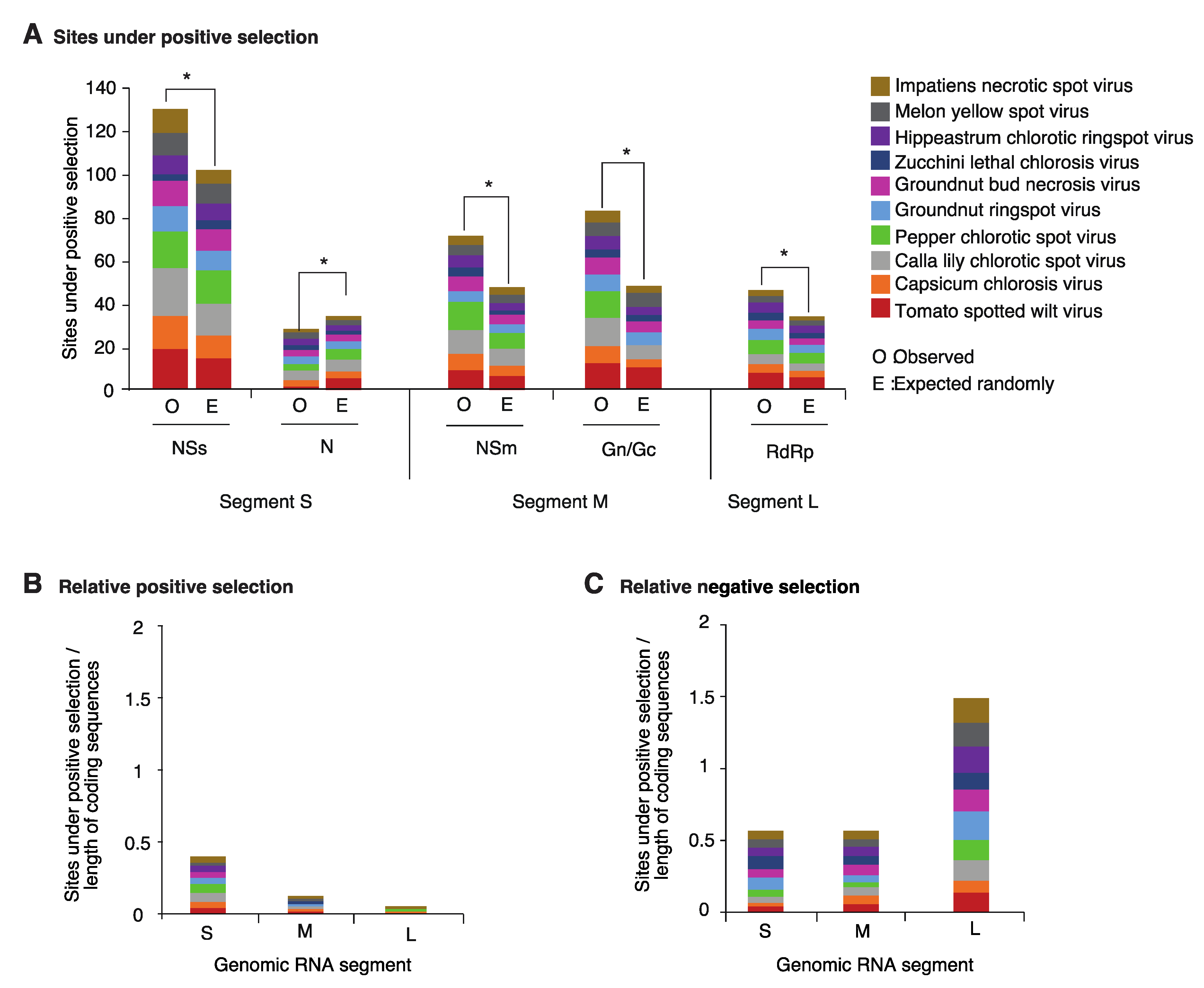
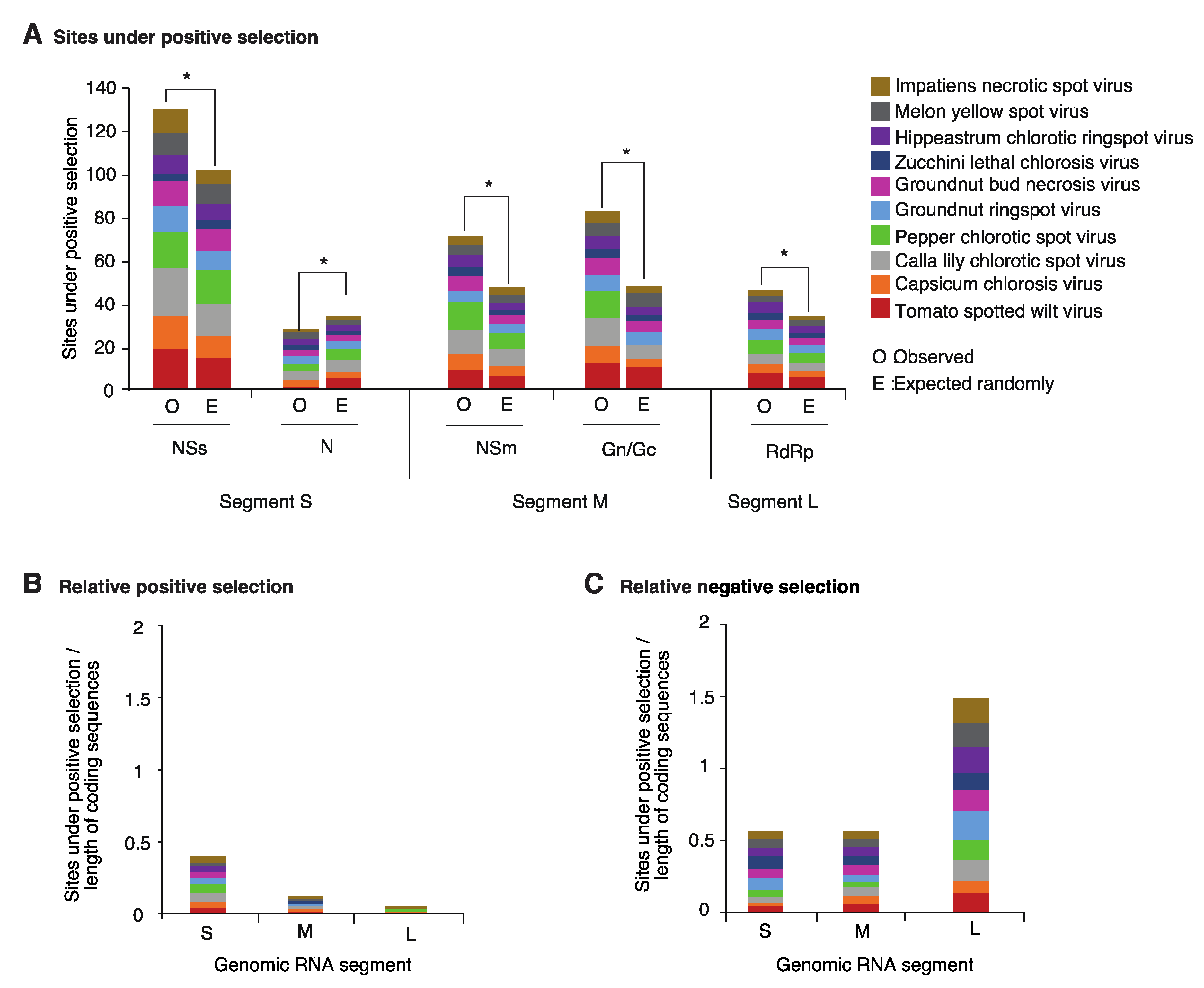
2.3. Positive and Negative Selection on the Orthotospoviral Genome
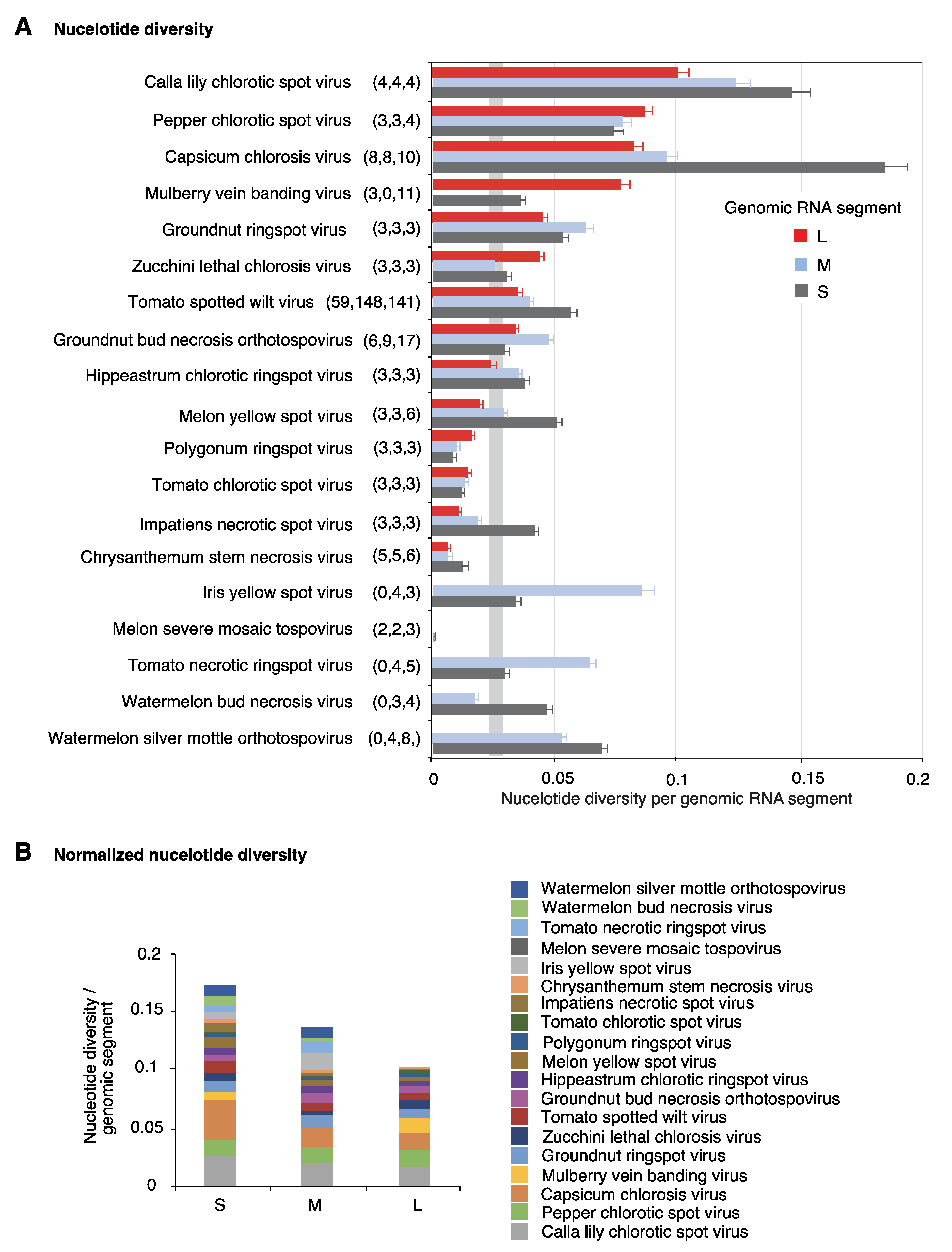
2.4. Nucleotide Variation in Segment S
2.5. Genome-Wide Variation in Tomato Spotted Wilt Virus
2.6. Genome-Wide Variation in Other Orthotospoviruses
2.7. TSWV Genetic Diversity
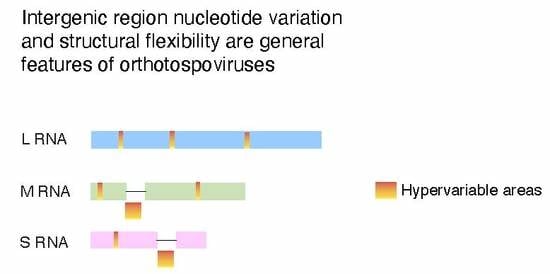
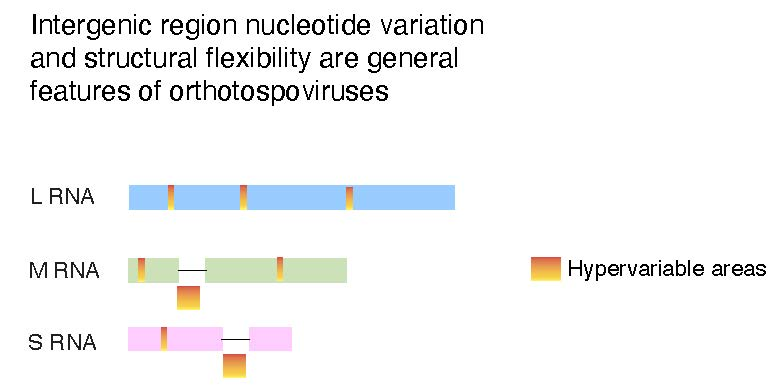
2.8. Nucleotide Variation in Segment S Intergenic Region
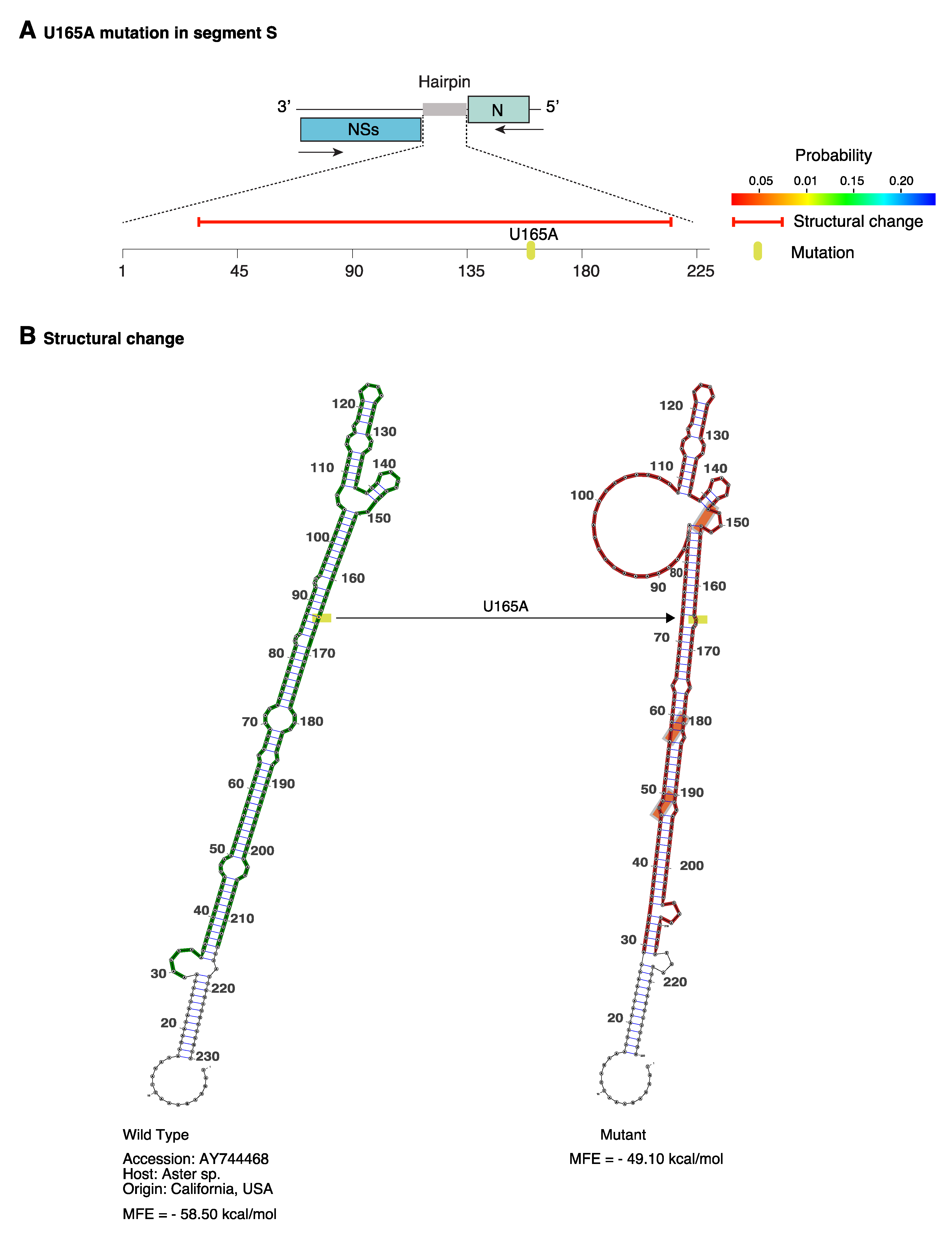
2.9. Structural Flexibility in Segment S Hairpin
2.10. Positive Selection at Intergenic RNA Structures in TSWV
3. Discussion
4. Materials and Methods
4.1. Genomic RNA Sequences
4.2. Removal of Recombinant Sequences
4.3. Complete Genome Sequences and Consensus
4.4. Molecular Phylogeny
4.5. Polymorphism Analysis in L, M and S Segment
4.6. Distribution of Variation in Segment S
4.7. Selection Analysis
4.8. Geographical Origin and Host Range
4.9. Intergenic Region Sequence and Structural Alignment
4.10. Intergenic Region Hairpin RNA Structure Modelling
4.11. Characterization of Polymorphisms in the TSWV Intergenic Region
4.12. Effect of Mutations on Hairpin Secondary Structure
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Abudurexiti, A.; Adkins, S.; Alioto, D.; Alkhovsky, S.V.; Avšič-Županc, T.; Ballinger, M.J.; Bente, D.A.; Beer, M.; Bergeron, É.; Blair, C.D. Taxonomy of the order Bunyavirales: Update 2019. Arch. Virol. 2019, 164, 1949–1965. [Google Scholar] [CrossRef] [Green Version]
- King, A.M.; Adams, M.J.; Carstens, E.B.; Lefkowitz, E.J. Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier: Amsterdam, The Netherlands, 2012; pp. 486–487. [Google Scholar]
- Briese, T.; Alkhovskiy, S.; Beer, M.; Calisher, C.; Charrel, R.; Ebihara, H. Create a New Order, Bunyavirales, to Accommodate Nine Families (Eight New, One Renamed) Comprising Thirteen Genera; International Committee on Taxonomy of Viruses: Budapest, Hungary, 2016. [Google Scholar]
- Oliver, J.E.; Whitfield, A.E. The Genus Tospovirus: Emerging Bunyaviruses that Threaten Food Security. Annu. Rev. Virol. 2016, 3, 101–124. [Google Scholar] [CrossRef]
- Turina, M.; Kormelink, R.; Resende, R.O. Resistance to Tospoviruses in Vegetable Crops: Epidemiological and Molecular Aspects. Annu. Rev. Phytopathol. 2016, 54, 347–371. [Google Scholar] [CrossRef] [PubMed]
- Pappu, H.R.; Jones, R.A.; Jain, R.K. Global status of tospovirus epidemics in diverse cropping systems: Successes achieved and challenges ahead. Virus Res. 2009, 141, 219–236. [Google Scholar] [CrossRef] [PubMed]
- Parrella, G.; Gognalons, P.; Gebre-Selassie, K.; Vovlas, C.; Marchoux, G. An update of the host range of tomato spotted wilt virus. J. Plant Pathol. 2003, 85, 227–264. [Google Scholar]
- Health, E.P.O.P. Scientific Opinion on the pest categorisation of the tospoviruses. Efsa J. 2012, 10, 2772. [Google Scholar]
- Wijkamp, I.; van Lent, J.; Kormelink, R.; Goldbach, R.; Peters, D. Multiplication of tomato spotted wilt virus in its insect vector, Frankliniella occidentalis. J. Gen. Virol. 1993, 74 Pt 3, 341–349. [Google Scholar] [CrossRef]
- Riley, D.G.; Joseph, S.V.; Srinivasan, R.; Diffie, S. Thrips Vectors of Tospoviruses. J. Integr. Pest Manag. 2011, 2, I1–I10. [Google Scholar] [CrossRef]
- Jones, D.R. Plant viruses transmitted by thrips. Eur. J. Plant Pathol. 2005, 113, 119–157. [Google Scholar] [CrossRef]
- Jacobson, A.L.; Kennedy, G.G. Specific insect-virus interactions are responsible for variation in competency of different Thrips tabaci isolines to transmit different Tomato Spotted Wilt Virus isolates. PLoS ONE 2013, 8, e54567. [Google Scholar] [CrossRef] [PubMed]
- Olendraite, I.; Lukhovitskaya, N.I.; Porter, S.D.; Valles, S.M.; Firth, A.E. Polycipiviridae: A proposed new family of polycistronic picorna-like RNA viruses. J. Gen. Virol. 2017, 98, 2368–2378. [Google Scholar] [CrossRef] [PubMed]
- Iglesias, N.G.; Gamarnik, A.V. Dynamic RNA structures in the dengue virus genome. Rna Biol. 2011, 8, 249–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miras, M.; Miller, W.A.; Truniger, V.; Aranda, M.A. Non-canonical Translation in Plant RNA Viruses. Front. Plant Sci. 2017, 8, 494. [Google Scholar] [CrossRef] [Green Version]
- Mumford, R.A.; Barker, I.; Wood, K.R. The biology of the tospoviruses. Ann. Appl. Biol. 1996, 128, 159–183. [Google Scholar] [CrossRef]
- Kaye, A.C.; Moyer, J.W.; Parks, E.J.; Carbone, I.; Cubeta, M.A. Population genetic analysis of Tomato spotted wilt virus on peanut in North Carolina and Virginia. Phytopathology 2011, 101, 147–153. [Google Scholar] [CrossRef] [Green Version]
- Feng, Z.; Xue, F.; Xu, M.; Chen, X.; Zhao, W.; Garcia-Murria, M.J.; Mingarro, I.; Liu, Y.; Huang, Y.; Jiang, L.; et al. The ER-Membrane Transport System Is Critical for Intercellular Trafficking of the NSm Movement Protein and Tomato Spotted Wilt Tospovirus. PLoS Pathog. 2016, 12, e1005443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Ruiz, H.; Gabriel Peralta, S.M.; Harte-Maxwell, P.A. Tomato Spotted Wilt Virus NSs Protein Supports Infection and Systemic Movement of a Potyvirus and Is a Symptom Determinant. Viruses 2018, 10, 129. [Google Scholar] [CrossRef] [Green Version]
- Hedil, M.; Hassani-Mehraban, A.; Lohuis, D.; Kormelink, R. Analysis of the AU rich hairpin from the intergenic region of tospovirus S RNA as target and inducer of RNA silencing. PLoS ONE 2014, 9, e106027. [Google Scholar] [CrossRef]
- Van Knippenberg, I.; Goldbach, R.; Kormelink, R. Tomato spotted wilt virus S-segment mRNAs have overlapping 3’-ends containing a predicted stem-loop structure and conserved sequence motif. Virus Res. 2005, 110, 125–131. [Google Scholar] [CrossRef]
- Gonzalez-Pacheco, B.E.; Delaye, L.; Ochoa, D.; Rojas, R.; Silva-Rosales, L. Changes in the GN/GCof the M segment show positive selection and recombination of one aggressive isolate and two mild isolates of tomato spotted wilt virus. Virus Genes 2020, 56, 217–227. [Google Scholar] [CrossRef]
- Bono, L.M.; Draghi, J.A.; Turner, P.E. Evolvability Costs of Niche Expansion. Trends Genet. 2020, 36, 14–23. [Google Scholar] [CrossRef]
- Moury, B.; Fabre, F.; Hebrard, E.; Froissart, R. Determinants of host species range in plant viruses. J. Gen. Virol. 2017, 98, 862–873. [Google Scholar] [CrossRef] [PubMed]
- Elena, S.F.; Agudelo-Romero, P.; Lalic, J. The evolution of viruses in multi-host fitness landscapes. Open Virol. J. 2009, 3, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Ferris, M.T.; Joyce, P.; Burch, C.L. High frequency of mutations that expand the host range of an RNA virus. Genetics 2007, 176, 1013–1022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, L.; Seth-Pasricha, M.; Stemate, D.; Crespo-Bellido, A.; Gagnon, J.; Draghi, J.; Duffy, S. Existing Host Range Mutations Constrain Further Emergence of RNA Viruses. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elena, S.F.; Agudelo-Romero, P.; Carrasco, P.; Codoner, F.M.; Martin, S.; Torres-Barcelo, C.; Sanjuan, R. Experimental evolution of plant RNA viruses. Heredity (Edinb) 2008, 100, 478–483. [Google Scholar] [CrossRef] [Green Version]
- Ciuffo, M.; Kurowski, C.; Vivoda, E.; Copes, B.; Masenga, V.; Falk, B.W.; Turina, M. A New Tospovirus sp. in Cucurbit Crops in Mexico. Plant Dis. 2009, 93, 467–474. [Google Scholar] [CrossRef] [Green Version]
- Golnaraghi, A.; Shahraeen, N.; Nguyen, H.D. Characterization and Genetic Structure of a Tospovirus Causing Chlorotic Ring Spots and Chlorosis Disease on Peanut; Comparison with Iranian and Polish Populations of Tomato yellow fruit ring virus. Plant Dis. 2018, 102, 1509–1519. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Tzanetakis, I.E. Soybean vein necrosis virus: An emerging virus in North America. Virus Genes 2019, 55, 12–21. [Google Scholar] [CrossRef]
- Nigam, D.; LaTourrette, K.; Souza, P.F.N.; Garcia-Ruiz, H. Genome-Wide Variation in Potyviruses. Front. Plant Sci. 2019, 10, 1439. [Google Scholar] [CrossRef] [Green Version]
- Obenauer, J.C.; Denson, J.; Mehta, P.K.; Su, X.; Mukatira, S.; Finkelstein, D.B.; Xu, X.; Wang, J.; Ma, J.; Fan, Y.; et al. Large-scale sequence analysis of avian influenza isolates. Science 2006, 311, 1576–1580. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, A.S.; Melo, F.L.; Inoue-Nagata, A.K.; Nagata, T.; Kitajima, E.W.; Resende, R.O. Characterization of bean necrotic mosaic virus: A member of a novel evolutionary lineage within the Genus Tospovirus. PLoS ONE 2012, 7, e38634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, J.H.; Cheng, X.F.; Yin, Y.Y.; Fang, Q.; Ding, M.; Li, T.T.; Zhang, L.Z.; Su, X.X.; McBeath, J.H.; Zhang, Z.K. Characterization of tomato zonate spot virus, a new tospovirus in China. Arch. Virol. 2008, 153, 855–864. [Google Scholar] [CrossRef]
- Ooi, K.; Ohshita, S.; Ishii, I.; Yahara, T. Molecular phylogeny of geminivirus infecting wild plants in Japan. J. Plant Res. 1997, 110, 247–257. [Google Scholar] [CrossRef]
- Tentchev, D.; Verdin, E.; Marchal, C.; Jacquet, M.; Aguilar, J.M.; Moury, B. Evolution and structure of Tomato spotted wilt virus populations: Evidence of extensive reassortment and insights into emergence processes. J. Gen. Virol. 2011, 92, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Sanchez-DelBarrio, J.C.; Messeguer, X.; Rozas, R. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 2003, 19, 2496–2497. [Google Scholar] [CrossRef]
- Sin, S.H.; McNulty, B.C.; Kennedy, G.G.; Moyer, J.W. Viral genetic determinants for thrips transmission of Tomato spotted wilt virus. Proc. Natl. Acad. Sci. USA 2005, 102, 5168–5173. [Google Scholar] [CrossRef] [Green Version]
- Badillo-Vargas, I.E.; Chen, Y.T.; Martin, K.M.; Rotenberg, D.; Whitfield, A.E. Discovery of Novel Thrips Vector Proteins That Bind to the Viral Attachment Protein of the Plant Bunyavirus Tomato Spotted Wilt Virus. J. Virol. 2019, 93, e00699-19. [Google Scholar] [CrossRef] [Green Version]
- Poelwijk, F.v.; Haan, P.d.; Kikkert, M.; Prins, M.; Kormelink, R.; Storms, M.; Lent, J.v.; Peters, D.; Goldbach, R. Replication and expression of the tospoviral genome. Tospoviruses Thrips Flor. Veg. Crops 1995, 431, 201–208. [Google Scholar] [CrossRef] [Green Version]
- Will, S.; Joshi, T.; Hofacker, I.L.; Stadler, P.F.; Backofen, R. LocARNA-P: Accurate boundary prediction and improved detection of structural RNAs. RNA 2012, 18, 900–914. [Google Scholar] [CrossRef] [Green Version]
- Churkin, A.; Barash, D. A Biologically Meaningful Extension of the Efficient Method for Deleterious Mutations Prediction in RNAs: Insertions and Deletions in Addition to Substitution Mutations. In Bioinformatics Research and Applications; Springer: Cham, Switzerland, 2018; pp. 174–178. [Google Scholar]
- Sabarinathan, R.; Tafer, H.; Seemann, S.E.; Hofacker, I.L.; Stadler, P.F.; Gorodkin, J. RNAsnp: Efficient detection of local RNA secondary structure changes induced by SNPs. Hum. Mutat. 2013, 34, 546–556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clabbers, M.T.; Olsthoorn, R.C.; Gultyaev, A.P. Tospovirus ambisense genomic RNA segments use almost complete repertoire of stable tetraloops in the intergenic region. Bioinformatics 2014, 30, 1800–1804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowick, K.; Walter Costa, M.B.; Honer Zu Siederdissen, C.; Stadler, P.F. Selection Pressures on RNA Sequences and Structures. Evol. Bioinform. Online 2019, 15, 1176934319871919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Arenal, F.; Fraile, A.; Malpica, J.M. Variability and genetic structure of plant virus populations. Annu. Rev. Phytopathol. 2001, 39, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ruiz, H.; Diaz, A.; Ahlquist, P. Intermolecular RNA Recombination Occurs at Different Frequencies in Alternate Forms of Brome Mosaic Virus RNA Replication Compartments. Viruses 2018, 10, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, C.G.; Reitz, S.R.; Perry, K.L.; Adkins, S. A natural M RNA reassortant arising from two species of plant- and insect-infecting bunyaviruses and comparison of its sequence and biological properties to parental species. Virology 2011, 413, 216–225. [Google Scholar] [CrossRef] [Green Version]
- Elena, S.F.; Bedhomme, S.; Carrasco, P.; Cuevas, J.M.; de la Iglesia, F.; Lafforgue, G.; Lalic, J.; Prosper, A.; Tromas, N.; Zwart, M.P. The evolutionary genetics of emerging plant RNA viruses. Mol. Plant Microbe Interact. 2011, 24, 287–293. [Google Scholar] [CrossRef] [Green Version]
- Bedhomme, S.; Lafforgue, G.; Elena, S.F. Multihost experimental evolution of a plant RNA virus reveals local adaptation and host-specific mutations. Mol. Biol. Evol. 2012, 29, 1481–1492. [Google Scholar] [CrossRef] [Green Version]
- Elena, S.F.; Fraile, A.; Garcia-Arenal, F. Evolution and emergence of plant viruses. Adv. Virus Res. 2014, 88, 161–191. [Google Scholar] [CrossRef] [Green Version]
- Moury, B.; Simon, V. dN/dS-Based Methods Detect Positive Selection Linked to Trade-Offs between Different Fitness Traits in the Coat Protein of Potato virus Y. Mol. Biol. Evol. 2011, 28, 2707–2717. [Google Scholar] [CrossRef] [Green Version]
- Pan, L.L.; Chi, Y.; Liu, C.; Fan, Y.Y.; Liu, S.S. Mutations in the coat protein of a begomovirus result in altered transmission by different species of whitefly vectors. Virus Evol. 2020, 6, veaa014. [Google Scholar] [CrossRef] [PubMed]
- Moury, B.; Morel, C.; Johansen, E.; Jacquemond, M. Evidence for diversifying selection in Potato virus Y and in the coat protein of other potyviruses. J. Gen. Virol. 2002, 83, 2563–2573. [Google Scholar] [CrossRef] [PubMed]
- Peter, K.A.; Liang, D.; Palukaitis, P.; Gray, S.M. Small deletions in the potato leafroll virus readthrough protein affect particle morphology, aphid transmission, virus movement and accumulation. J. Gen. Virol. 2008, 89, 2037–2045. [Google Scholar] [CrossRef] [PubMed]
- Allison, J.R.; Lechner, M.; Hoeppner, M.P.; Poole, A.M. Positive Selection or Free to Vary? Assessing the Functional Significance of Sequence Change Using Molecular Dynamics. PLoS ONE 2016, 11, e0147619. [Google Scholar] [CrossRef] [Green Version]
- Duffy, S.; Turner, P.E.; Burch, C.L. Pleiotropic costs of niche expansion in the RNA bacteriophage Φ6. Genetics 2006, 172, 751–757. [Google Scholar] [CrossRef] [Green Version]
- Bera, S.; Fraile, A.; Garcia-Arenal, F. Analysis of Fitness Trade-Offs in the Host Range Expansion of an RNA Virus, Tobacco Mild Green Mosaic Virus. J. Virol. 2018, 92, e01268-18. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Arenal, F.; Fraile, A. Trade-offs in host range evolution of plant viruses. Plant Pathol. 2013, 62, 2–9. [Google Scholar] [CrossRef]
- Agrawal, A.A. Host-range evolution: Adaptation and trade-offs in fitness of mites on alternative hosts. Ecology 2000, 81, 500–508. [Google Scholar] [CrossRef]
- Hanssen, I.M.; Lapidot, M.; Thomma, B.P. Emerging viral diseases of tomato crops. Mol. Plant Microbe Interact. 2010, 23, 539–548. [Google Scholar] [CrossRef] [Green Version]
- Emery, V.C.; Bishop, D.H. Characterization of Punta Toro S mRNA species and identification of an inverted complementary sequence in the intergenic region of Punta Toro phlebovirus ambisense S RNA that is involved in mRNA transcription termination. Virology 1987, 156, 1–11. [Google Scholar] [CrossRef]
- Qiu, W.; Geske, S.; Hickey, C.; Moyer, J. Tomato Spotted WiltTospovirusGenome Reassortment and Genome Segment-Specific Adaptation. Virology 1998, 244, 186–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geerts-Dimitriadou, C.; Lu, Y.Y.; Geertsema, C.; Goldbach, R.; Kormelink, R. Analysis of the Tomato spotted wilt virus ambisense S RNA-encoded hairpin structure in translation. PLoS ONE 2012, 7, e31013. [Google Scholar] [CrossRef] [PubMed]
- Woolhouse, M.E.; Taylor, L.H.; Haydon, D.T. Population biology of multihost pathogens. Science 2001, 292, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Baumstark, T.; Ahlquist, P. The brome mosaic virus RNA3 intergenic replication enhancer folds to mimic a tRNA TpsiC-stem loop and is modified in vivo. RNA 2001, 7, 1652–1670. [Google Scholar]
- Watters, K.E.; Choudhary, K.; Aviran, S.; Lucks, J.B.; Perry, K.L.; Thompson, J.R. Probing of RNA structures in a positive sense RNA virus reveals selection pressures for structural elements. Nucleic Acids Res. 2018, 46, 2573–2584. [Google Scholar] [CrossRef] [Green Version]
- Ashton, P.; Wu, B.; D’Angelo, J.; Grigull, J.; White, K.A. Biologically-supported structural model for a viral satellite RNA. Nucleic Acids Res. 2015, 43, 9965–9977. [Google Scholar] [CrossRef] [Green Version]
- Tycowski, K.T.; Guo, Y.E.; Lee, N.; Moss, W.N.; Vallery, T.K.; Xie, M.; Steitz, J.A. Viral noncoding RNAs: More surprises. Genes Dev. 2015, 29, 567–584. [Google Scholar] [CrossRef] [Green Version]
- Gultyaev, A.P.; Franch, T.; Gerdes, K. Coupled nucleotide covariations reveal dynamic RNA interaction patterns. RNA 2000, 6, 1483–1491. [Google Scholar] [CrossRef] [Green Version]
- Gultyaev, A.P.; Tsyganov-Bodounov, A.; Spronken, M.I.; van der Kooij, S.; Fouchier, R.A.; Olsthoorn, R.C. RNA structural constraints in the evolution of the influenza A virus genome NP segment. Rna Biol. 2014, 11, 942–952. [Google Scholar] [CrossRef] [Green Version]
- Lindgreen, S.; Gardner, P.P.; Krogh, A. Measuring covariation in RNA alignments: Physical realism improves information measures. Bioinformatics 2006, 22, 2988–2995. [Google Scholar] [CrossRef]
- Ritz, J.; Martin, J.S.; Laederach, A. Evolutionary evidence for alternative structure in RNA sequence co-variation. PLoS Comput. Biol. 2013, 9, e1003152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brault, A.C.; Powers, A.M.; Ortiz, D.; Estrada-Franco, J.G.; Navarro-Lopez, R.; Weaver, S.C. Venezuelan equine encephalitis emergence: Enhanced vector infection from a single amino acid substitution in the envelope glycoprotein. Proc. Natl. Acad. Sci. USA 2004, 101, 11344–11349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Katoh, K.; Kuma, K.; Toh, H.; Miyata, T. MAFFT version 5: Improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 2005, 33, 511–518. [Google Scholar] [CrossRef]
- Page, A.J.; Taylor, B.; Delaney, A.J.; Soares, J.; Seemann, T.; Keane, J.A.; Harris, S.R. SNP-sites: Rapid efficient extraction of SNPs from multi-FASTA alignments. Microb. Genom. 2016, 2, e000056. [Google Scholar] [CrossRef] [Green Version]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Lefort, V.; Longueville, J.E.; Gascuel, O. SMS: Smart Model Selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Asnicar, F.; Weingart, G.; Tickle, T.L.; Huttenhower, C.; Segata, N. Compact graphical representation of phylogenetic data and metadata with GraPhlAn. PeerJ 2015, 3, e1029. [Google Scholar] [CrossRef]
- Lorenz, R.; Bernhart, S.H.; Honer Zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Hofacker, I.L. RNA consensus structure prediction with RNAalifold. Methods Mol. Biol. 2007, 395, 527–544. [Google Scholar] [CrossRef] [PubMed]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nigam, D.; Garcia-Ruiz, H. Variation Profile of the Orthotospovirus Genome. Pathogens 2020, 9, 521. https://doi.org/10.3390/pathogens9070521
Nigam D, Garcia-Ruiz H. Variation Profile of the Orthotospovirus Genome. Pathogens. 2020; 9(7):521. https://doi.org/10.3390/pathogens9070521
Chicago/Turabian StyleNigam, Deepti, and Hernan Garcia-Ruiz. 2020. "Variation Profile of the Orthotospovirus Genome" Pathogens 9, no. 7: 521. https://doi.org/10.3390/pathogens9070521
APA StyleNigam, D., & Garcia-Ruiz, H. (2020). Variation Profile of the Orthotospovirus Genome. Pathogens, 9(7), 521. https://doi.org/10.3390/pathogens9070521