A Review on SARS-CoV-2 Virology, Pathophysiology, Animal Models, and Anti-Viral Interventions


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Virology
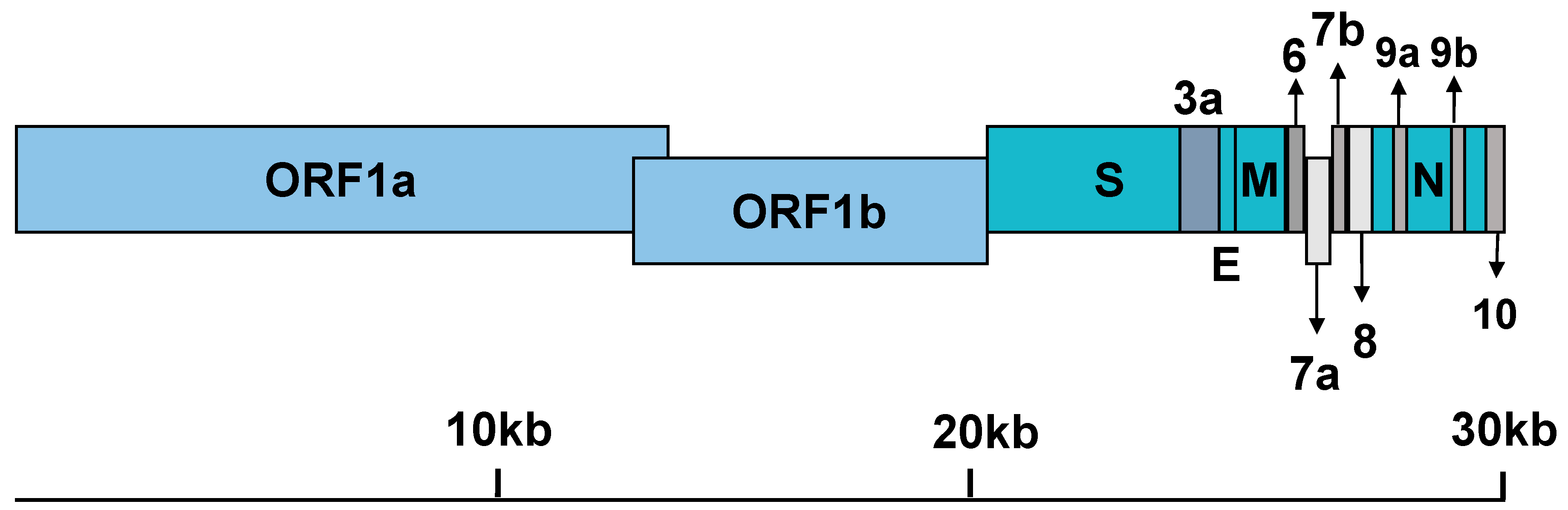
2.1. Classification and Origin
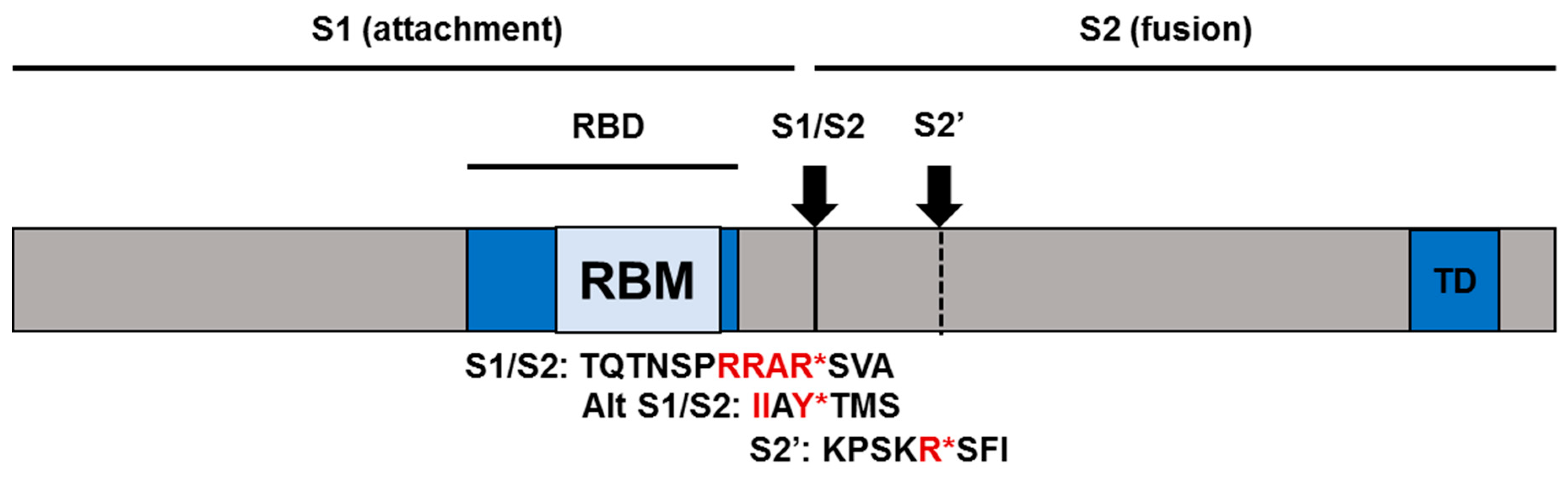
2.2. Virus Structure and Stability
2.3. SARS-CoV-2 Receptor and Entry
2.4. General Features of Genome Replication and Particle Production
3. Clinical Features and Pathophysiology
4. Animals Models of COVID-19
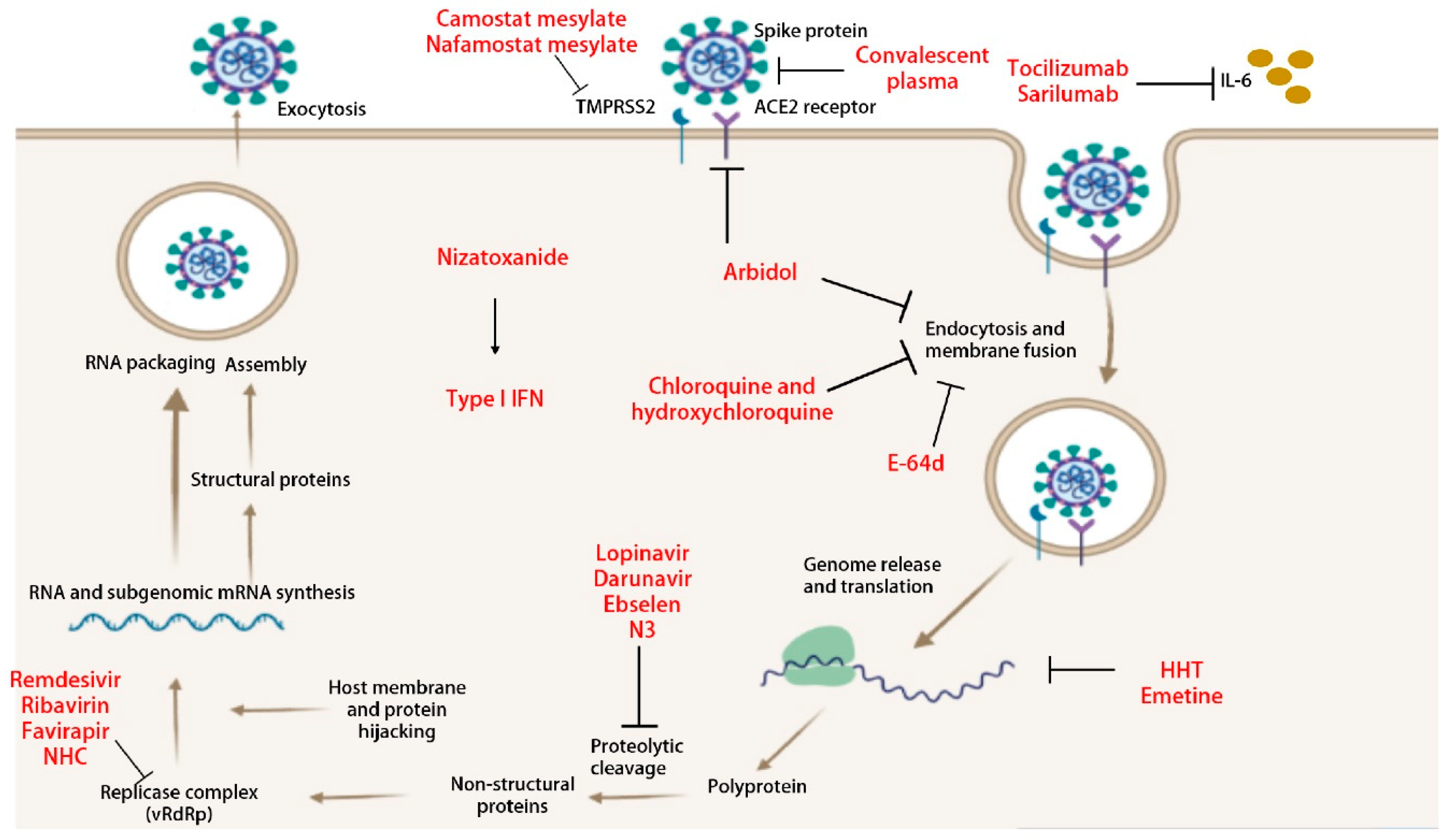
5. Anti-Viral Interventions
5.1. Anti-Viral Therapeutics
5.1.1. β-D-N4-hydroxycytidine (NHC)
5.1.2. Remdesivir
5.1.3. Ivermectin
5.1.4. N3
5.1.5. Lopinavir/Ritonavir
5.1.6. Chloroquine and Hydroxychloroquine
5.1.7. Type I IFN
5.1.8. Arbidol
5.1.9. Ribavirin
5.1.10. Nizatoxanide
5.1.11. Homoharringtonine (HHT)
5.1.12. Emetine
5.2. Adjunctive Therapies
5.2.1. Corticosteroids
5.2.2. Tocilizumab and Sarilumab
5.2.3. Convalescent Plasma
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef]
- Jin, Y.; Yang, H.; Ji, W.; Wu, W.; Chen, S.; Zhang, W.; Duan, G. Virology, epidemiology, pathogenesis, and control of COVID-19. Viruses 2020, 12, 372. [Google Scholar] [CrossRef]
- Zhang, T.; Wu, Q.; Zhang, Z. Probable pangolin origin of SARS-CoV-2 associated with the COVID-19 outbreak. Curr. Biol. 2020, 30, 1346–1351.e1342. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Chin, A.; Chu, J.; Perera, M.; Hui, K.; Yen, H.-L.; Chan, M.; Peiris, M.; Poon, L. Stability of SARS-CoV-2 in different environmental conditions. medRxiv 2020. [Google Scholar] [CrossRef]
- Van Doremalen, N.; Bushmaker, T.; Morris, D.H.; Holbrook, M.G.; Gamble, A.; Williamson, B.N.; Tamin, A.; Harcourt, J.L.; Thornburg, N.J.; Gerber, S.I.; et al. Aerosol and surface stability of SARS-CoV-2 as compared with SARS-CoV-1. N. Engl. J. Med. 2020, 382, 1564–1567. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [PubMed]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef]
- Götz, V.; Magar, L.; Dornfeld, D.; Giese, S.; Pohlmann, A.; Höper, D.; Kong, B.-W.; Jans, D.A.; Beer, M.; Haller, O.; et al. Influenza A viruses escape from MxA restriction at the expense of efficient nuclear vRNP import. Sci. Rep. 2016, 6, 23138. [Google Scholar] [CrossRef]
- Sanders, J.M.; Monogue, M.L.; Jodlowski, T.Z.; Cutrell, J.B. Pharmacologic Treatments for Coronavirus Disease 2019 (COVID-19): A review. JAMA 2020. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292.e286. [Google Scholar] [CrossRef]
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 2020, 176, 104742. [Google Scholar] [CrossRef]
- Hoffmann, M.; Schroeder, S.; Kleine-Weber, H.; Müller, M.A.; Drosten, C.; Pöhlmann, S. Nafamostat mesylate blocks activation of SARS-CoV-2: New treatment option for COVID-19. In Antimicrob. Agents Chemother.; 2020; 64, p. e00754-20. Available online: https://pubmed.ncbi.nlm.nih.gov/32312781/ (accessed on 28 May 2020).
- Matsuyama, S.; Nao, N.; Shirato, K.; Kawase, M.; Saito, S.; Takayama, I.; Nagata, N.; Sekizuka, T.; Katoh, H.; Kato, F.; et al. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc. Natl. Acad. Sci. USA 2020, 117, 7001–7003. [Google Scholar] [CrossRef]
- Iwata-Yoshikawa, N.; Okamura, T.; Shimizu, Y.; Hasegawa, H.; Takeda, M.; Nagata, N. TMPRSS2 contributes to virus spread and immunopathology in the airways of murine models after coronavirus infection. J. Virol. 2019, 93, e01815–e01818. [Google Scholar] [CrossRef]
- Sungnak, W.; Huang, N.; Bécavin, C.; Berg, M.; Queen, R.; Litvinukova, M.; Talavera-López, C.; Maatz, H.; Reichart, D.; Sampaziotis, F.; et al. SARS-CoV-2 entry factors are highly expressed in nasal epithelial cells together with innate immune genes. Nat. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. In Coronaviruses: Methods and Protocols; Maier, H.J., Bickerton, E., Britton, P., Eds.; Springer: New York, NY, USA, 2015; pp. 1–23. [Google Scholar]
- Cascella, M.; Rajnik, M.; Cuomo, A.; Dulebohn, S.C.; Di Napoli, R. Features, evaluation and treatment coronavirus (COVID-19). In Statpearls [Internet]; StatPearls Publishing: Treasure island, NV, USA, 2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK554776/ (accessed on 6 April 2020).
- Zheng, M.; Gao, Y.; Wang, G.; Song, G.; Liu, S.; Sun, D.; Xu, Y.; Tian, Z. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell. Mol. Immunol. 2020. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Zhou, Y.; Fu, B.; Zheng, X.; Wang, D.; Zhao, C.; Qi, Y.; Sun, R.; Tian, Z.; Xu, X.; Wei, H. Pathogenic T cells and inflammatory monocytes incite inflammatory storm in severe COVID-19 patients. Natl. Sci. Rev. 2020. [Google Scholar] [CrossRef]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of immune response in patients with COVID-19 in Wuhan, China. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Boudewijns, R.; Thibaut, H.J.; Kaptein, S.J.F.; Li, R.; Vergote, V.; Seldeslachts, L.; De Keyzer, C.; Sharma, S.; Jansen, S.; Van Weyenbergh, J.; et al. STAT2 signaling as double-edged sword restricting viral dissemination but driving severe pneumonia in SARS-CoV-2 infected hamsters. bioRxiv 2020. [Google Scholar] [CrossRef]
- Bao, L.; Deng, W.; Huang, B.; Gao, H.; Ren, L.; Wei, Q.; Yu, P.; Xu, Y.; Liu, J.; Qi, F.; et al. The pathogenicity of 2019 novel coronavirus in hACE2 transgenic mice. bioRxiv 2020. [Google Scholar] [CrossRef]
- Dinnon, K.H.; Leist, S.R.; Schäfer, A.; Edwards, C.E.; Martinez, D.R.; Montgomery, S.A.; West, A.; Yount, B.L.; Hou, Y.J.; Adams, L.E.; et al. A mouse-adapted SARS-CoV-2 model for the evaluation of COVID-19 medical countermeasures. bioRxiv 2020. [Google Scholar] [CrossRef]
- Enkirch, T.; von Messling, V. Ferret models of viral pathogenesis. Virology 2015, 479–480, 259–270. [Google Scholar] [CrossRef]
- Shi, J.; Wen, Z.; Zhong, G.; Yang, H.; Wang, C.; Huang, B.; Liu, R.; He, X.; Shuai, L.; Sun, Z.; et al. Susceptibility of ferrets, cats, dogs, and other domesticated animals to SARS–coronavirus 2. Science 2020. [Google Scholar] [CrossRef]
- McNamara, T.; Richt, J.A.; Glickman, L. A critical needs assessment for research in companion animals and livestock following the pandemic of COVID-19 in humans. Vector-Borne Zoonotic Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Sit, T.H.C.; Brackman, C.J.; Ip, S.M.; Tam, K.W.S.; Law, P.Y.T.; To, E.M.W.; Yu, V.Y.T.; Sims, L.D.; Tsang, D.N.C.; Chu, D.K.W.; et al. Infection of dogs with SARS-CoV-2. Nature 2020. [Google Scholar] [CrossRef] [PubMed]
- Bao, L.; Deng, W.; Gao, H.; Xiao, C.; Liu, J.; Xue, J.; Lv, Q.; Liu, J.; Yu, P.; Xu, Y.; et al. Reinfection could not occur in SARS-CoV-2 infected rhesus macaques. bioRxiv 2020. [Google Scholar] [CrossRef]
- Munster, V.J.; Feldmann, F.; Williamson, B.N.; van Doremalen, N.; Pérez-Pérez, L.; Schulz, J.; Meade-White, K.; Okumura, A.; Callison, J.; Brumbaugh, B.; et al. Respiratory disease in rhesus macaques inoculated with SARS-CoV-2. Nature 2020. [Google Scholar] [CrossRef] [PubMed]
- Rockx, B.; Kuiken, T.; Herfst, S.; Bestebroer, T.; Lamers, M.M.; Oude Munnink, B.B.; de Meulder, D.; van Amerongen, G.; van den Brand, J.; Okba, N.M.A.; et al. Comparative pathogenesis of COVID-19, MERS, and SARS in a nonhuman primate model. Science 2020. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Tostanoski, L.H.; Peter, L.; Mercado, N.B.; McMahan, K.; Mahrokhian, S.H.; Nkolola, J.P.; Liu, J.; Li, Z.; Chandrashekar, A.; et al. DNA vaccine protection against SARS-CoV-2 in rhesus macaques. Science 2020. [Google Scholar] [CrossRef]
- Roper, R.L.; Rehm, K.E. SARS vaccines: Where are we? Expert Rev. Vaccines 2009, 8, 887–898. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.-T.; Sbrana, E.; Iwata-Yoshikawa, N.; Newman, P.C.; Garron, T.; Atmar, R.L.; Peters, C.J.; Couch, R.B. Immunization with SARS coronavirus vaccines leads to pulmonary immunopathology on challenge with the SARS virus. PLoS ONE 2012, 7, e35421. [Google Scholar] [CrossRef]
- Bolles, M.; Deming, D.; Long, K.; Agnihothram, S.; Whitmore, A.; Ferris, M.; Funkhouser, W.; Gralinski, L.; Totura, A.; Heise, M.; et al. A double-inactivated severe acute respiratory syndrome coronavirus vaccine provides incomplete protection in mice and induces increased eosinophilic proinflammatory pulmonary response upon challenge. J. Virol. 2011, 85, 12201–12215. [Google Scholar] [CrossRef]
- Amanat, F.; Krammer, F. SARS-CoV-2 vaccines: Status report. Immunity 2020, 52, 583–589. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Choy, K.T.; Wong, A.Y.; Kaewpreedee, P.; Sia, S.F.; Chen, D.; Hui, K.P.Y.; Chu, D.K.W.; Chan, M.C.W.; Cheung, P.P.; Huang, X.; et al. Remdesivir, lopinavir, emetine, and homoharringtonine inhibit SARS-CoV-2 replication in vitro. Antivir. Res. 2020, 178, 104786. [Google Scholar] [CrossRef] [PubMed]
- Sheahan, T.P.; Sims, A.C.; Graham, R.L.; Menachery, V.D.; Gralinski, L.E.; Case, J.B.; Leist, S.R.; Pyrc, K.; Feng, J.Y.; Trantcheva, I.; et al. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef]
- Yin, W.; Mao, C.; Luan, X.; Shen, D.-D.; Shen, Q.; Su, H.; Wang, X.; Zhou, F.; Zhao, W.; Gao, M.; et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science 2020. [Google Scholar] [CrossRef]
- Agostini, M.L.; Andres, E.L.; Sims, A.C.; Graham, R.L.; Sheahan, T.P.; Lu, X.; Smith, E.C.; Case, J.B.; Feng, J.Y.; Jordan, R.; et al. Coronavirus susceptibility to the antiviral remdesivir (GS-5734) is mediated by the viral polymerase and the proofreading exoribonuclease. mBio 2018, 9. [Google Scholar] [CrossRef]
- Sheahan, T.P.; Sims, A.C.; Leist, S.R.; Schäfer, A.; Won, J.; Brown, A.J.; Montgomery, S.A.; Hogg, A.; Babusis, D.; Clarke, M.O.; et al. Comparative therapeutic efficacy of remdesivir and combination lopinavir, ritonavir, and interferon beta against MERS-CoV. Nat. Commun. 2020, 11, 222. [Google Scholar] [CrossRef]
- De Wit, E.; Feldmann, F.; Cronin, J.; Jordan, R.; Okumura, A.; Thomas, T.; Scott, D.; Cihlar, T.; Feldmann, H. Prophylactic and therapeutic remdesivir (GS-5734) treatment in the rhesus macaque model of MERS-CoV infection. Proc. Natl. Acad. Sci. USA 2020, 117, 6771–6776. [Google Scholar] [CrossRef]
- Holshue, M.L.; DeBolt, C.; Lindquist, S.; Lofy, K.H.; Wiesman, J.; Bruce, H.; Spitters, C.; Ericson, K.; Wilkerson, S.; Tural, A.; et al. First case of 2019 novel coronavirus in the United States. N. Engl. J. Med. 2020, 382, 929–936. [Google Scholar] [CrossRef]
- Kujawski, S.A.; Wong, K.K.; Collins, J.P.; Epstein, L.; Killerby, M.E.; Midgley, C.M.; Abedi, G.R.; Ahmed, N.S.; Almendares, O.; Alvarez, F.N.; et al. First 12 patients with coronavirus disease 2019 (COVID-19) in the United States. medRxiv 2020. [Google Scholar] [CrossRef]
- Hendaus, M.A. Remdesivir in the treatment of coronavirus disease 2019 (COVID-19): A simplified summary. J. Biomol. Struct. Dyn. 2020, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, L.; Pinkham, C.; Baer, A.; Amaya, M.; Narayanan, A.; Wagstaff, K.M.; Jans, D.A.; Kehn-Hall, K. Nuclear import and export inhibitors alter capsid protein distribution in mammalian cells and reduce Venezuelan Equine Encephalitis Virus replication. Antivir. Res. 2013, 100, 662–672. [Google Scholar] [CrossRef] [PubMed]
- Tay, M.Y.; Fraser, J.E.; Chan, W.K.; Moreland, N.J.; Rathore, A.P.; Wang, C.; Vasudevan, S.G.; Jans, D.A. Nuclear localization of dengue virus (DENV) 1-4 non-structural protein 5; protection against all 4 DENV serotypes by the inhibitor Ivermectin. Antivir. Res. 2013, 99, 301–306. [Google Scholar] [CrossRef]
- Mastrangelo, E.; Pezzullo, M.; De Burghgraeve, T.; Kaptein, S.; Pastorino, B.; Dallmeier, K.; de Lamballerie, X.; Neyts, J.; Hanson, A.M.; Frick, D.N.; et al. Ivermectin is a potent inhibitor of flavivirus replication specifically targeting NS3 helicase activity: New prospects for an old drug. J. Antimicrob. Chemother. 2012, 67, 1884–1894. [Google Scholar] [CrossRef]
- Yang, S.N.Y.; Atkinson, S.C.; Wang, C.; Lee, A.; Bogoyevitch, M.A.; Borg, N.A.; Jans, D.A. The broad spectrum antiviral ivermectin targets the host nuclear transport importin alpha/beta1 heterodimer. Antivir. Res. 2020, 177, 104760. [Google Scholar] [CrossRef]
- Caly, L.; Wagstaff, K.M.; Jans, D.A. Nuclear trafficking of proteins from RNA viruses: Potential target for antivirals? Antivir. Res. 2012, 95, 202–206. [Google Scholar] [CrossRef] [PubMed]
- Caly, L.; Druce, J.D.; Catton, M.G.; Jans, D.A.; Wagstaff, K.M. The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2 in vitro. Antivir. Res. 2020, 178, 104787. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature 2020. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.M.; Cheng, V.C.C.; Hung, I.F.N.; Wong, M.M.L.; Chan, K.H.; Chan, K.S.; Kao, R.Y.T.; Poon, L.L.M.; Wong, C.L.P.; Guan, Y.; et al. Role of lopinavir/ritonavir in the treatment of SARS: Initial virological and clinical findings. Thorax 2004, 59, 252–256. [Google Scholar] [CrossRef]
- De Wilde, A.H.; Jochmans, D.; Posthuma, C.C.; Zevenhoven-Dobbe, J.C.; van Nieuwkoop, S.; Bestebroer, T.M.; van den Hoogen, B.G.; Neyts, J.; Snijder, E.J. Screening of an FDA-approved compound library identifies four small-molecule inhibitors of middle east respiratory syndrome coronavirus replication in cell culture. Antimicrob. Agents Chemother. 2014, 58, 4875–4884. [Google Scholar] [CrossRef]
- McCreary, E.K.; Pogue, J.M. Coronavirus disease 2019 treatment: A review of early and emerging options. Open Forum Infect. Dis. 2020, 7. [Google Scholar] [CrossRef]
- Ratia, K.; Pegan, S.; Takayama, J.; Sleeman, K.; Coughlin, M.; Baliji, S.; Chaudhuri, R.; Fu, W.; Prabhakar, B.S.; Johnson, M.E.; et al. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. USA 2008, 105, 16119–16124. [Google Scholar] [CrossRef] [PubMed]
- Arabi, Y.M.; Asiri, A.Y.; Assiri, A.M.; Aziz Jokhdar, H.A.; Alothman, A.; Balkhy, H.H.; AlJohani, S.; Al Harbi, S.; Kojan, S.; Al Jeraisy, M.; et al. Treatment of Middle East respiratory syndrome with a combination of lopinavir/ritonavir and interferon-β1b (MIRACLE trial): Statistical analysis plan for a recursive two-stage group sequential randomized controlled trial. Trials 2020, 21, 8. [Google Scholar] [CrossRef]
- Cao, B.; Wang, Y.; Wen, D.; Liu, W.; Wang, J.; Fan, G.; Ruan, L.; Song, B.; Cai, Y.; Wei, M.; et al. A trial of lopinavir–ritonavir in adults hospitalized with severe COVID-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Young, B.E.; Ong, S.W.X.; Kalimuddin, S.; Low, J.G.; Tan, S.Y.; Loh, J.; Ng, O.-T.; Marimuthu, K.; Ang, L.W.; Mak, T.M.; et al. Epidemiologic features and clinical course of patients infected with SARS-CoV-2 in Singapore. JAMA 2020, 323, 1488–1494. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.; Jeon, S.; Shin, H.Y.; Kim, M.J.; Seong, Y.M.; Lee, W.J.; Choe, K.W.; Kang, Y.M.; Lee, B.; Park, S.J. Case of the index patient who caused tertiary transmission of COVID-19 infection in Korea: The application of lopinavir/ritonavir for the treatment of COVID-19 infected pneumonia monitored by quantitative RT-PCR. J. Korean Med. Sci. 2020, 35, e79. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Quan, B.; Guo, Y.; Zhang, J.; Lu, Y.; Feng, G.; Wu, Q.; Fang, F.; Cheng, L.; Jiao, N.; et al. The course of clinical diagnosis and treatment of a case infected with coronavirus disease 2019. J. Med. Virol. 2020, 92, 461–463. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, X.; Lu, Y.; Chen, F.; Zhang, W. Clinical characteristics and therapeutic procedure for four cases with 2019 novel coronavirus pneumonia receiving combined Chinese and Western medicine treatment. Biosci. Trends 2020, 14, 64–68. [Google Scholar] [CrossRef]
- Costanzo, M.; De Giglio, M.A.R.; Roviello, G.N. SARS-CoV-2: Recent reports on antiviral therapies based on lopinavir/ritonavir, darunavir/umifenovir, hydroxychloroquine, remdesivir, favipiravir and other drugs for the treatment of the new coronavirus. Curr. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Colson, P.; Rolain, J.M.; Lagier, J.C.; Brouqui, P.; Raoult, D. Chloroquine and hydroxychloroquine as available weapons to fight COVID-19. Int. J. Antimicrob. Agents 2020, 55, 105932. [Google Scholar] [CrossRef]
- Gao, J.; Tian, Z.; Yang, X. Breakthrough: Chloroquine phosphate has shown apparent efficacy in treatment of COVID-19 associated pneumonia in clinical studies. Biosci. Trends 2020, 14, 72–73. [Google Scholar] [CrossRef]
- Yao, X.; Ye, F.; Zhang, M.; Cui, C.; Huang, B.; Niu, P.; Liu, X.; Zhao, L.; Dong, E.; Song, C.; et al. In vitro antiviral activity and projection of optimized dosing design of hydroxychloroquine for the treatment of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Gautret, P.; Lagier, J.-C.; Parola, P.; Hoang, V.T.; Meddeb, L.; Mailhe, M.; Doudier, B.; Courjon, J.; Giordanengo, V.; Vieira, V.E.; et al. Hydroxychloroquine and azithromycin as a treatment of COVID-19: Results of an open-label non-randomized clinical trial. Int. J. Antimicrob. Agents 2020. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Liu, D.; Liu, l. A pilot study of hydroxychloroquine in treatment of patients with common coronavirus disease-19 (COVID-19). J. Zhejiang Univ. (Med. Sci.) 2020. [Google Scholar] [CrossRef]
- Geleris, J.; Sun, Y.; Platt, J.; Zucker, J.; Baldwin, M.; Hripcsak, G.; Labella, A.; Manson, D.K.; Kubin, C.; Barr, R.G.; et al. Observational study of hydroxychloroquine in hospitalized patients with COVID-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.P.; Lau, C.C.Y.; Chan, K.H.; Li, C.P.Y.; Chen, H.; Jin, D.Y.; Chan, J.F.W.; Woo, P.C.Y.; Yuen, K.Y. Delayed induction of proinflammatory cytokines and suppression of innate antiviral response by the novel Middle East respiratory syndrome coronavirus: Implications for pathogenesis and treatment. J. Gen. Virol. 2013, 94, 2679–2690. [Google Scholar] [CrossRef] [PubMed]
- Lokugamage, K.G.; Hage, A.; Schindewolf, C.; Rajsbaum, R.; Menachery, V.D. SARS-CoV-2 is sensitive to type I interferon pretreatment. bioRxiv 2020. [Google Scholar] [CrossRef]
- Chan, J.F.-W.; Yao, Y.; Yeung, M.-L.; Deng, W.; Bao, L.; Jia, L.; Li, F.; Xiao, C.; Gao, H.; Yu, P.; et al. Treatment with lopinavir/ritonavir or interferon-β1b improves outcome of MERS-CoV infection in a nonhuman primate model of common marmoset. J. Infect. Dis. 2015, 212, 1904–1913. [Google Scholar] [CrossRef]
- Frieman, M.B.; Chen, J.; Morrison, T.E.; Whitmore, A.; Funkhouser, W.; Ward, J.M.; Lamirande, E.W.; Roberts, A.; Heise, M.; Subbarao, K.; et al. SARS-CoV pathogenesis is regulated by a STAT1 dependent but a type I, II and III interferon receptor independent mechanism. PloS Pathog. 2010, 6, e1000849. [Google Scholar] [CrossRef]
- Dong, L.; Hu, S.; Gao, J. Discovering drugs to treat coronavirus disease 2019 (COVID-19). Drug Discov. Ther. 2020, 14, 58–60. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Yang, B.; Li, Q.; Wen, L.; Zhang, R. Clinical features of 69 cases with coronavirus disease 2019 in Wuhan, China. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.F.W.; Chan, K.-H.; Kao, R.Y.T.; To, K.K.W.; Zheng, B.-J.; Li, C.P.Y.; Li, P.T.W.; Dai, J.; Mok, F.K.Y.; Chen, H.; et al. Broad-spectrum antivirals for the emerging Middle East respiratory syndrome coronavirus. J. Infect. 2013, 67, 606–616. [Google Scholar] [CrossRef] [PubMed]
- Pruijssers, A.J.; George, A.S.; Schäfer, A.; Leist, S.R.; Gralinksi, L.E.; Dinnon, K.H.; Yount, B.L.; Agostini, M.L.; Stevens, L.J.; Chappell, J.D.; et al. Remdesivir potently inhibits SARS-CoV-2 in human lung cells and chimeric SARS-CoV expressing the SARS-CoV-2 RNA polymerase in mice. bioRxiv 2020. [Google Scholar] [CrossRef]
- Falzarano, D.; de Wit, E.; Martellaro, C.; Callison, J.; Munster, V.J.; Feldmann, H. Inhibition of novel β coronavirus replication by a combination of interferon-α2b and ribavirin. Sci. Rep. 2013, 3, 1686. [Google Scholar] [CrossRef]
- Falzarano, D.; de Wit, E.; Rasmussen, A.L.; Feldmann, F.; Okumura, A.; Scott, D.P.; Brining, D.; Bushmaker, T.; Martellaro, C.; Baseler, L.; et al. Treatment with interferon-α2b and ribavirin improves outcome in MERS-CoV–infected rhesus macaques. Nat. Med. 2013, 19, 1313–1317. [Google Scholar] [CrossRef]
- Al-Tawfiq, J.A.; Momattin, H.; Dib, J.; Memish, Z.A. Ribavirin and interferon therapy in patients infected with the Middle East respiratory syndrome coronavirus: An observational study. Int. J. Infect. Dis. 2014, 20, 42–46. [Google Scholar] [CrossRef]
- Rossignol, J.-F. Nitazoxanide: A first-in-class broad-spectrum antiviral agent. Antivir. Res. 2014, 110, 94–103. [Google Scholar] [CrossRef]
- Rossignol, J.F.; La Frazia, S.; Chiappa, L.; Ciucci, A.; Santoro, M.G. Thiazolides, a new class of anti-influenza molecules targeting viral hemagglutinin at the post-translational level. J. Biol. Chem. 2009, 284, 29798–29808. [Google Scholar] [CrossRef]
- Teran, C.G.; Teran-Escalera, C.N.; Villarroel, P. Nitazoxanide vs. probiotics for the treatment of acute rotavirus diarrhea in children: A randomized, single-blind, controlled trial in Bolivian children. Int. J. Infect. Dis. 2009, 13, 518–523. [Google Scholar] [CrossRef]
- Hargest, V.; Sharp, B.; Livingston, B.; Cortez, V.; Schultz-Cherry, S. astrovirus replication is inhibited by nitazoxanide in vitro and in vivo. J. Virol. 2020, 94, e01706–e01719. [Google Scholar] [CrossRef]
- Rossignol, J.-F.; Abu-Zekry, M.; Hussein, A.; Santoro, M.G. Effect of nitazoxanide for treatment of severe rotavirus diarrhoea: Randomised double-blind placebo-controlled trial. Lancet 2006, 368, 124–129. [Google Scholar] [CrossRef]
- Shi, Z.; Wei, J.; Deng, X.; Li, S.; Qiu, Y.; Shao, D.; Li, B.; Zhang, K.; Xue, F.; Wang, X.; et al. Nitazoxanide inhibits the replication of Japanese encephalitis virus in cultured cells and in a mouse model. Virol. J. 2014, 11, 10. [Google Scholar] [CrossRef]
- Perelygina, L.; Hautala, T.; Seppänen, M.; Adebayo, A.; Sullivan, K.E.; Icenogle, J. Inhibition of rubella virus replication by the broad-spectrum drug nitazoxanide in cell culture and in a patient with a primary immune deficiency. Antivir. Res. 2017, 147, 58–66. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, Z.; Brecher, M.; Deng, Y.-Q.; Zhang, J.; Sakamuru, S.; Liu, B.; Huang, R.; Koetzner, C.A.; Allen, C.A.; Jones, S.A.; et al. Existing drugs as broad-spectrum and potent inhibitors for Zika virus by targeting NS2B-NS3 interaction. Cell Res. 2017, 27, 1046–1064. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, J.F.; Elfert, A.; El–Gohary, Y.; Keeffe, E.B. Improved virologic response in chronic hepatitis C genotype 4 treated with nitazoxanide, peginterferon, and ribavirin. Gastroenterology 2009, 136, 856–862. [Google Scholar] [CrossRef]
- Korba, B.E.; Montero, A.B.; Farrar, K.; Gaye, K.; Mukerjee, S.; Ayers, M.S.; Rossignol, J.-F. Nitazoxanide, tizoxanide and other thiazolides are potent inhibitors of hepatitis B virus and hepatitis C virus replication. Antivir. Res. 2008, 77, 56–63. [Google Scholar] [CrossRef]
- Rossignol, J.F. Nitazoxanide, a new drug candidate for the treatment of Middle East respiratory syndrome coronavirus. J. Infect. Public Health 2016, 9, 227–230. [Google Scholar] [CrossRef]
- Shen, L.; Niu, J.; Wang, C.; Huang, B.; Wang, W.; Zhu, N.; Deng, Y.; Wang, H.; Ye, F.; Cen, S.; et al. High-throughput screening and identification of potent broad-spectrum inhibitors of coronaviruses. J. Virol. 2019, 93, e00023-19. [Google Scholar] [CrossRef]
- Haffizulla, J.; Hartman, A.; Hoppers, M.; Resnick, H.; Samudrala, S.; Ginocchio, C.; Bardin, M.; Rossignol, J.F. Effect of nitazoxanide in adults and adolescents with acute uncomplicated influenza: A double-blind, randomised, placebo-controlled, phase 2b/3 trial. Lancet Infect. Dis. 2014, 14, 609–618. [Google Scholar] [CrossRef]
- Gamino-Arroyo, A.E.; Guerrero, M.L.; McCarthy, S.; Ramirez-Venegas, A.; Llamosas-Gallardo, B.; Galindo-Fraga, A.; Moreno-Espinosa, S.; Roldan-Aragon, Y.; Araujo-Melendez, J.; Hunsberger, S.; et al. Efficacy and safety of nitazoxanide in addition to standard of care for the treatment of severe acute respiratory illness. Clin. Infect. Dis. 2019, 69, 1903–1911. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.J.Y.; Wu, A.; Joynt, G.M.; Yuen, K.Y.; Lee, N.; Chan, P.K.S.; Cockram, C.S.; Ahuja, A.T.; Yu, L.M.; Wong, V.W.; et al. Severe acute respiratory syndrome: Report of treatment and outcome after a major outbreak. Thorax 2004, 59, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Chan, K.C.A.; Hui, D.S.; Ng, E.K.O.; Wu, A.; Chiu, R.W.K.; Wong, V.W.S.; Chan, P.K.S.; Wong, K.T.; Wong, E.; et al. Effects of early corticosteroid treatment on plasma SARS-associated Coronavirus RNA concentrations in adult patients. J. Clin. Virol. 2004, 31, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.T.S.; Wing, Y.K.; Leung, H.C.M.; Sung, J.J.Y.; Ng, Y.K.; Yiu, G.C.; Chen, R.Y.L.; Chiu, H.F.K. Factors associated with psychosis among patients with severe acute respiratory syndrome: A case-control study. Clin. Infect. Dis. 2004, 39, 1247–1249. [Google Scholar] [CrossRef] [PubMed]
- Auyeung, T.W.; Lee, J.S.; Lai, W.K.; Choi, C.H.; Lee, H.K.; Lee, J.S.; Li, P.C.; Lok, K.H.; Ng, Y.Y.; Wong, W.M.; et al. The use of corticosteroid as treatment in SARS was associated with adverse outcomes: A retrospective cohort study. J. Infect. 2005, 51, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.C.; Tang, X.P.; Tan, S.Y.; Liang, B.L.; Wan, Z.Y.; Fang, J.Q.; Zhong, N. Treatment of severe acute respiratory syndrome with glucosteroids: The Guangzhou experience. Chest 2006, 129, 1441–1452. [Google Scholar] [CrossRef]
- Arabi, Y.M.; Mandourah, Y.; Al-Hameed, F.; Sindi, A.A.; Almekhlafi, G.A.; Hussein, M.A.; Jose, J.; Pinto, R.; Al-Omari, A.; Kharaba, A.; et al. Corticosteroid therapy for critically ill patients with middle east respiratory syndrome. Am. J. Respir. Crit. Care Med. 2018, 197, 757–767. [Google Scholar] [CrossRef]
- Wu, C.; Chen, X.; Cai, Y.; Xia, J.A.; Zhou, X.; Xu, S.; Huang, H.; Zhang, L.; Zhou, X.; Du, C.; et al. Risk factors associated with acute respiratory distress syndrome and death in patients with coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern. Med. 2020. [Google Scholar] [CrossRef]
- Xu, X.; Han, M.; Li, T. Effective treatment of severe COVID-19 patients with tocilizumab. Chinaxiv. Prepr. 2020. [Google Scholar] [CrossRef]
- Guo, C.; Li, B.; Ma, H.; Wang, X.; Cai, P.; Yu, Q.; Zhu, L.; Jin, L.; Jiang, C.; Fang, J.; et al. Tocilizumab treatment in severe COVID-19 patients attenuates the inflammatory storm incited by monocyte centric immune interactions revealed by single-cell analysis. bioRxiv 2020. [Google Scholar] [CrossRef]
- Chen, L.; Xiong, J.; Bao, L.; Shi, Y. Convalescent plasma as a potential therapy for COVID-19. Lancet Infect. Dis. 2020, 20, 398–400. [Google Scholar] [CrossRef]
- Duan, K.; Liu, B.; Li, C.; Zhang, H.; Yu, T.; Qu, J.; Zhou, M.; Chen, L.; Meng, S.; Hu, Y.; et al. Effectiveness of convalescent plasma therapy in severe COVID-19 patients. Proc. Natl. Acad. Sci. USA 2020. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neerukonda, S.N.; Katneni, U. A Review on SARS-CoV-2 Virology, Pathophysiology, Animal Models, and Anti-Viral Interventions. Pathogens 2020, 9, 426. https://doi.org/10.3390/pathogens9060426
Neerukonda SN, Katneni U. A Review on SARS-CoV-2 Virology, Pathophysiology, Animal Models, and Anti-Viral Interventions. Pathogens. 2020; 9(6):426. https://doi.org/10.3390/pathogens9060426
Chicago/Turabian StyleNeerukonda, Sabari Nath, and Upendra Katneni. 2020. "A Review on SARS-CoV-2 Virology, Pathophysiology, Animal Models, and Anti-Viral Interventions" Pathogens 9, no. 6: 426. https://doi.org/10.3390/pathogens9060426
APA StyleNeerukonda, S. N., & Katneni, U. (2020). A Review on SARS-CoV-2 Virology, Pathophysiology, Animal Models, and Anti-Viral Interventions. Pathogens, 9(6), 426. https://doi.org/10.3390/pathogens9060426