High-Resolution Melting (HRM) Curve Assay for the Identification of Eight Fusarium Species Causing Ear Rot in Maize


 , ,
, , 
Abstract
1. Introduction
2. Results
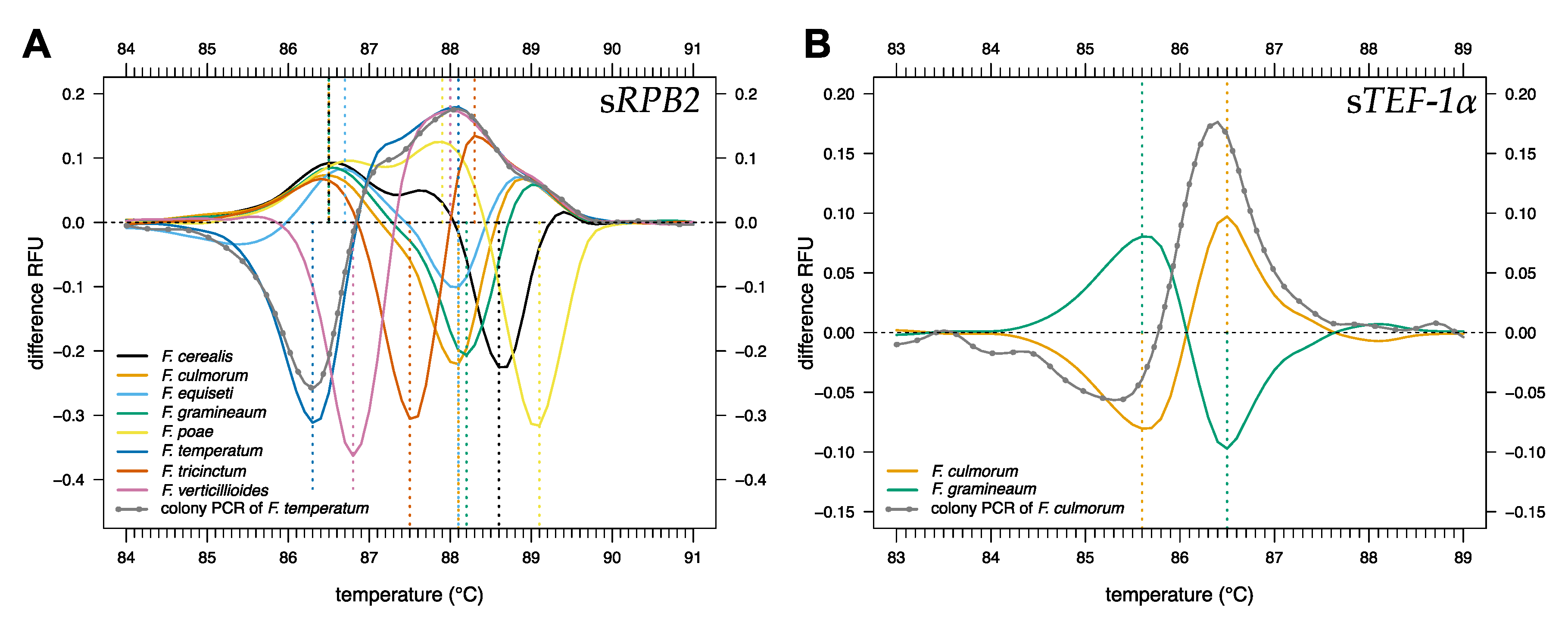
2.1. HRM Analysis of sRPB2 and sTEF-1α for the Identification of Fusarium Species
2.2. Identification of Fusarium Species in Naturally Infected Maize Ears
2.3. Fungal Colony PCR
3. Discussion
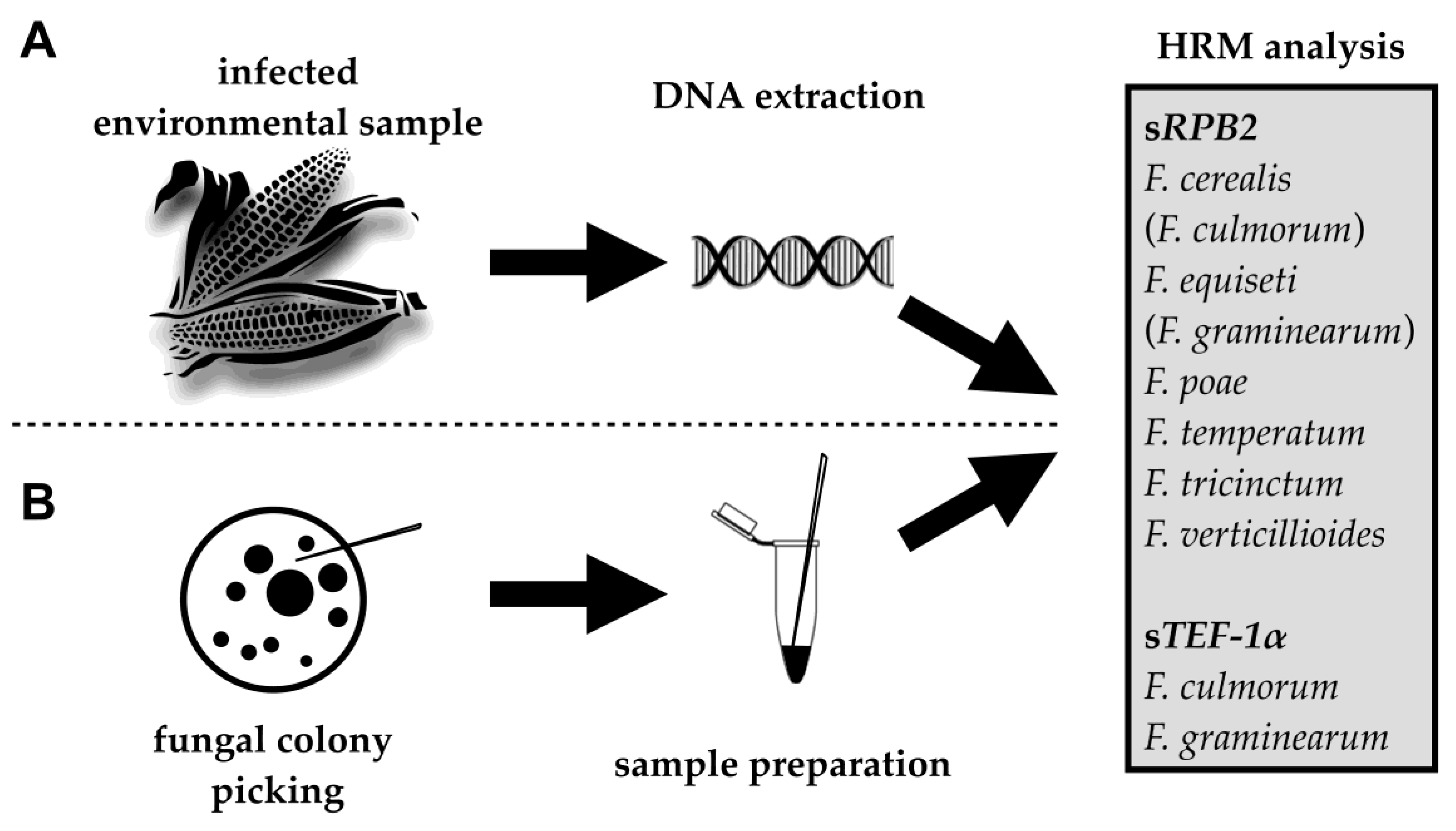
4. Material and Methods
4.1. Reference Strains, Sample Collection, and DNA Extraction
4.2. Fungal Colony PCR
4.3. Primer Design and Maximum Likelihood Tree Analysis
4.4. HRM Analysis
4.5. Fluorescence Data Processing and Taxonomic Assignment
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Xu, X.; Nicholson, P. Community ecology of fungal pathogens causing wheat head blight. Annu. Rev. Phytopathol. 2009, 47, 83–103. [Google Scholar] [CrossRef] [PubMed]
- D’Mello, J.P.F.; Placinta, C.M.; Macdonald, A.M.C. Fusarium mycotoxins: A review of global implications for animal health, welfare and productivity. Anim. Feed Sci. Technol. 1999, 80, 183–205. [Google Scholar] [CrossRef]
- Ferrigo, D.; Raiola, A.; Causin, R. Fusarium toxins in cereals: Occurrence, legislation, factors promoting the appearance and their management. Molecules 2016, 21, 627. [Google Scholar] [CrossRef] [PubMed]
- Duncan, K.E.; Howard, R.J. Biology of maize kernel infection by Fusarium verticillioides. Mol. Plant Microbe Interact. 2010, 23, 6–16. [Google Scholar] [CrossRef]
- Thompson, M.E.H.; Raizada, M.N. Fungal pathogens of maize gaining free passage along the silk road. Pathogens 2018, 7, 81. [Google Scholar] [CrossRef]
- Munkvold, G.P.; McGee, D.C.; Carlton, W.M. Importance of different pathways for maize kernel infection by Fusarium moniliforme. Phytopathology 1997, 87, 209–217. [Google Scholar] [CrossRef]
- Munkvold, G.P.; Carlton, W.M. Influence of inoculation method on systemic Fusarium moniliforme infection of maize plants grown from infected seeds. Plant Dis. 1997, 81, 211–216. [Google Scholar] [CrossRef]
- Murillo-Williams, A.; Munkvold, G.P. Systemic infection by Fusarium verticillioides in maize plants grown under three temperature regimes. Plant Dis. 2008, 92, 1695–1700. [Google Scholar] [CrossRef]
- Gai, X.; Dong, H.; Wang, S.; Liu, B.; Zhang, Z.; Li, X.; Gao, Z. Infection cycle of maize stalk rot and ear rot caused by Fusarium verticillioides. PLoS ONE 2018, 13, 7. [Google Scholar] [CrossRef]
- Alheeti, A.; Caldwell, R.; Smalley, E. Pathogenicity of Fusarium tricinctum, Fusarium poae and Fusarium sporotrichioides in maize ears. Phytopathology 1984, 74, 868. [Google Scholar]
- Logrieco, A.; Mulè, G.; Moretti, A.; Bottalico, A. Toxigenic Fusarium species and mycotoxins associated with maize ear rot in Europe. Eur. J. Plant Pathol. 2002, 108, 597–609. [Google Scholar] [CrossRef]
- Munkvold, G.P. Epidemiology of Fusarium diseases and their mycotoxins in maize ears. Eur. J. Plant Pathol. 2003, 109, 705–713. [Google Scholar] [CrossRef]
- Scauflaire, J.; Gourgue, M.; Munaut, F. Fusarium temperatum sp. nov. from maize, an emergent species closely related to Fusarium subglutinans. Mycologia 2011, 103, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Czembor, E.; Stępień, Ł.; Waśkiewicz, A. Effect of environmental factors on Fusarium species and associated mycotoxins in maize grain grown in Poland. PLoS ONE 2015, 10, 7. [Google Scholar] [CrossRef]
- Fallahi, M.; Saremi, H.; Javan-Nikkhah, M.; Somma, S.; Haidukowski, M.; Logrieco, A.F.; Moretti, A. Isolation, molecular identification and mycotoxin profile of Fusarium species isolated from maize kernels in Iran. Toxins (Basel). 2019, 11, 297. [Google Scholar] [CrossRef]
- Busman, M.; Desjardins, A.E.; Proctor, R.H. Analysis of fumonisin contamination and the presence of Fusarium in wheat with kernel black point disease in the United States. Food Addit. Contam. - Part A Chem. Anal. Control. Expo. Risk Assess. 2012, 29, 1092–1100. [Google Scholar] [CrossRef]
- Amato, B.; Pfohl, K.; Tonti, S.; Nipoti, P.; Dastjerdi, R.; Pisi, A.; Karlovsky, P.; Prodi, A.; Chiara, M.; Fanelli, F.; et al. Fusarium proliferatum and fumonisin B1 co-occur with Fusarium species causing Fusarium head blight in durum wheat in Italy. J. Appl. Bot. Food Qual. 2015, 88, 228–233. [Google Scholar]
- von Bargen, S.; Martinez, O.; Schadock, I.; Eisold, A.M.; Gossmann, M.; Büttner, C. Genetic variability of phytopathogenic Fusarium proliferatum associated with crown rot in asparagus officinalis. J. Phytopathol. 2009, 157, 446–456. [Google Scholar] [CrossRef]
- Dastjerdi, R.; Karlovsky, P. Systemic infection of maize, sorghum, rice, and beet seedlings with fumonisin-producing and nonproducing Fusarium verticillioides strains. Plant Pathol. J. 2015, 31, 334–342. [Google Scholar] [CrossRef]
- Dastjerdi, R. High fumonisin content in maize: search for source of infection and biological function. Ph.D. thesis. 2014. Available online: https//ediss.uni-goettingen.de/handle/11858/00-1735-0000-0022-5EC0-E (accessed on 29 February 2020).
- Sobek, E.A.; Munkvold, G.P. European corn borer (Lepidoptera: Pyralidae) larvae as vectors of Fusarium moniliforme, causing kernel rot and symptomless infection of maize kernels. J. Econ. Entomol. 1999, 92, 503–509. [Google Scholar] [CrossRef]
- Kurtz, B.; Karlovsky, P.; Vidal, S. Interaction between western corn rootworm (Coleoptera: Chrysomelidae) larvae and root-infecting Fusarium verticillioides. Environ. Entomol. 2010, 39, 1532–1538. [Google Scholar] [CrossRef] [PubMed]
- Leslie, J.F.; Summerell, B.A. The Fusarium Laboratory Manual; Department of Plant Pathology, Kansas State University: Manhattan, NY, USA; Blackwell Publishing Professional: Ames, IA, USA, 2006; ISBN 9780813819198. [Google Scholar] [CrossRef]
- Hsieh, W.H. Mating Groups in Fusarium moniliforme. Phytopathology 1977, 77, 1041. [Google Scholar] [CrossRef]
- Kerényi, Z.; Zeller, K.; Hornok, L.; Leslie, J.F. Molecular standardization of mating type terminology in the Gibberella fujikuroi species complex. Appl. Environ. Microbiol. 1999, 65, 4071–4076. [Google Scholar] [CrossRef]
- Watanabe, M.; Yonezawa, T.; Lee, K.; Kumagai, S.; Sugita-Konishi, Y.; Goto, K.; Hara-Kudo, Y. Molecular phylogeny of the higher and lower taxonomy of the Fusarium genus and differences in the evolutionary histories of multiple genes. BMC Evol. Biol. 2011, 11, 322. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, R.; Berdal, K.G.; Holst-Jensen, A. Simultaneous detection and identification of trichothecene- and moniliformin-producing Fusarium species based on multiplex SNP analysis. J. Appl. Microbiol. 2007, 102, 1071–1081. [Google Scholar] [CrossRef]
- O’Donnell, K.; McCormick, S.P.; Busman, M.; Proctor, R.H.; Ward, T.J.; Doehring, G.; Geiser, D.M.; Alberts, J.F.; Rheeder, J.P. Marasas et al. 1984 “Toxigenic Fusarium Species: Identity and mycotoxicology” revisited. Mycologia 2018, 110, 1058–1080. [Google Scholar]
- Thrane, U.; Hansen, U. Chemical and physiological characterization of taxa in the Fusarium sambucinum complex. Mycopathologia 1995, 129, 183–190. [Google Scholar] [CrossRef]
- Zainudin, N.A.I.; Razak, A.A.; Salleh, B. Secondary metabolite profiles and mating populations of Fusarium species in section Liseola associated with bakanae disease of rice. Malays. J. Microbiol. 2008, 4, 6–13. [Google Scholar]
- Nutz, S.; Döll, K.; Karlovsky, P. Determination of the LOQ in real-time PCR by receiver operating characteristic curve analysis: Application to qPCR assays for Fusarium verticillioides and F. proliferatum. Anal. Bioanal. Chem. 2011, 401, 717–726. [Google Scholar] [CrossRef]
- Aoki, T.; O’Donnell, K.; Geiser, D.M. Systematics of key phytopathogenic Fusarium species: Current status and future challenges. J. Gen. Plant Pathol. 2014, 80, 189–201. [Google Scholar] [CrossRef]
- Nicholson, P.; Simpson, D.R.; Weston, G.; Rezanoor, H.N.; Lees, A.K.; Parry, D.W.; Joyce, D. Detection and quantification of Fusarium culmorum and Fusarium graminearum in cereals using PCR assays. Physiol. Mol. Plant Pathol. 1998, 53, 17–37. [Google Scholar] [CrossRef]
- Turner, A.S.; Lees, A.K.; Rezanoor, H.N.; Nicholson, P. Refinement of PCR-detection of Fusarium avenaceum and evidence from DNA marker studies for phenetic relatedness to Fusarium tricinctum. Plant Pathol. 1998, 47, 278–288. [Google Scholar] [CrossRef]
- Wilson, A.; Simpson, D.; Chandler, E.; Jennings, P.; Nicholson, P. Development of PCR assays for the detection and differentiation of Fusarium sporotrichioides and Fusarium langsethiae. FEMS Microbiol. Lett. 2004, 233, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Waalwijk, C.; Van Der Heide, R.; De Vries, I.; Van Der Lee, T.; Schoen, C.; Costrel-de Corainville, G.; Häuser-Hahn, I.; Kastelein, P.; Köhl, J.; Lonnet, P.; et al. Quantitative detection of Fusarium species in wheat using TaqMan. Eur. J. Plant Pathol. 2004, 110, 481–494. [Google Scholar] [CrossRef]
- Brandfass, C.; Karlovsky, P. Upscaled CTAB-based DNA extraction and real-time PCR assays for Fusarium culmorum and F. graminearum DNA in plant material with reduced sampling error. Int. J. Mol. Sci. 2008, 9, 2306–2321. [Google Scholar] [CrossRef] [PubMed]
- Scauflaire, J.; Godet, M.; Gourgue, M.; Liénard, C.; Munaut, F. A multiplex real-time PCR method using hybridization probes for the detection and the quantification of Fusarium proliferatum, F. subglutinans, F. temperatum, and F. verticillioides. Fungal Biol. 2012, 116, 1073–1080. [Google Scholar] [CrossRef]
- Stakheev, A.A.; Khairulina, D.R.; Riazantsev, D.I.; Zavriev, S.K. Phosphate permease gene as a marker for the specific identification of toxigenic fungus Fusarium cerealis. Russ. J. Bioorganic Chem. 2013, 39, 175–183. [Google Scholar] [CrossRef]
- Gräfenhan, T.; Schroers, H.J.; Nirenberg, H.I.; Seifert, K.A. An overview of the taxonomy, phylogeny, and typification of nectriaceous fungi in Cosmospora, Acremonium, Fusarium, Stilbella, and Volutella. Stud. Mycol. 2011, 68, 79–113. [Google Scholar] [CrossRef]
- Moussa, T.A.A.; Al-Zahrani, H.S.; Kadasa, N.M.S.; Ahmed, S.A.; de Hoog, G.S.; Al-Hatmi, A.M.S. Two new species of the Fusarium fujikuroi species complex isolated from the natural environment. Antonie van Leeuwenhoek, Int. J. Gen. Mol. Microbiol. 2017, 110, 819–832. [Google Scholar] [CrossRef]
- Słomka, M.; Sobalska-Kwapis, M.; Wachulec, M.; Bartosz, G.; Strapagiel, D. High resolution melting (HRM) for high-throughput genotyping—limitations and caveats in practical case studies. Int. J. Mol. Sci. 2017, 18, 2316. [Google Scholar] [CrossRef]
- Mao, F.; Leung, W.Y.; Xin, X. Characterization of EvaGreen and the implication of its physicochemical properties for qPCR applications. BMC Biotechnol. 2007, 7, 76. [Google Scholar] [CrossRef]
- Garritano, S.; Gemignani, F.; Voegele, C.; Nguyen-Dumont, T.; Le Calvez-Kelm, F.; De Silva, D.; Lesueur, F.; Landi, S.; Tavtigian, S.V. Determining the effectiveness of high resolution melting analysis for SNP genotyping and mutation scanning at the TP53 locus. BMC Genet. 2009, 10, 5. [Google Scholar] [CrossRef] [PubMed]
- Gelaye, E.; Mach, L.; Kolodziejek, J.; Grabherr, R.; Loitsch, A.; Achenbach, J.E.; Nowotny, N.; Diallo, A.; Lamien, C.E. A novel HRM assay for the simultaneous detection and differentiation of eight poxviruses of medical and veterinary importance. Sci. Rep. 2017, 7, 42892. [Google Scholar] [CrossRef] [PubMed]
- Solano, J.; Anabalón, L.; Encina, F.; Esse, C.; Penneckamp, D. Hybrid identification in Nothofagus subgenus using high resolution melting with ITS and trnL approach. PeerJ 2019, 7, e6779. [Google Scholar] [CrossRef] [PubMed]
- Chabi, J.; Van’t Hof, A.; N’dri, L.K.; Datsomor, A.; Okyere, D.; Njoroge, H.; Pipini, D.; Hadi, M.P.; De Souza, D.K.; Suzuki, T.; et al. Rapid high throughput SYBR green assay for identifying the malaria vectors Anopheles arabiensis, Anopheles coluzzii and Anopheles gambiae s.s. Giles. PLoS ONE 2019, 14, 4. [Google Scholar] [CrossRef]
- Brandfass, C.; Karlovsky, P. Simultaneous detection of Fusarium culmorum and F. graminearum in plant material by duplex PCR with melting curve analysis. BMC Microbiol. 2006, 6, 4. [Google Scholar] [CrossRef]
- Ganopoulos, I.; Madesis, P.; Zambounis, A.; Tsaftaris, A. High-resolution melting analysis allowed fast and accurate closed-tube genotyping of Fusarium oxysporum formae speciales complex. FEMS Microbiol. Lett. 2012, 334, 16–21. [Google Scholar] [CrossRef]
- Pfordt, A.; Ramos Romero, L.; Schiwek, S.; Karlovsky, P.; von Tiedemann, A. Impact of environmental conditions and agronomic practices on the prevalence of Fusarium species associated with ear- and stalk rot in maize. Pathogens 2020, 9, 236. [Google Scholar] [CrossRef]
- Lu, S.; Mirchevska, G.; Phatak, S.S.; Li, D.; Luka, J.; Calderone, R.A.; Fonzi, W.A. Dynamic time warping assessment of high-resolution melt curves provides a robust metric for fungal identification. PLoS ONE 2017, 12, e0173320. [Google Scholar] [CrossRef]
- Papavasileiou, A.; Madesis, P.; Karaoglanidis, G. Identification and differentiation of Monilinia species causing brown rot of pome and stone fruit using high-resolution melting (HRM) analysis. Phytopathology 2016, 106, 1055–1064. [Google Scholar] [CrossRef]
- Perrone, G.; Bottalico, A. Toxigenic Fusarium species and mycotoxins associated with head blight in small-grain cereals in Europe. Eur. J. Plant Pathol. 2002, 108, 611–624. [Google Scholar]
- Zhang, N.; Geiser, D.M.; Smart, C.D. Macroarray detection of solanaceous plant pathogens in the Fusarium solani species complex. Plant Dis. 2007, 91, 1612–1620. [Google Scholar] [CrossRef] [PubMed]
- Tambong, J.T.; De Cock, A.W.A.M.; Tinker, N.A.; Lévesque, C.A. Oligonucleotide array for identification and detection of Pythium species. Appl. Environ. Microbiol. 2006, 72, 2691–2706. [Google Scholar] [CrossRef] [PubMed]
- van Doorn, R.; Szemes, M.; Bonants, P.; Kowalchuk, G.A.; Salles, J.F.; Ortenberg, E.; Schoen, C.D. Quantitative multiplex detection of plant pathogens using a novel ligation probe-based system coupled with universal, high-throughput real-time PCR on OpenArraysTM. BMC Genomics 2007, 8, 276. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.J.; Whelen, S.; Hall, B.D. Phylogenetic relationships among ascomycetes: Evidence from an RNA polymerse II subunit. Mol. Biol. Evol. 1999, 16, 1799–1808. [Google Scholar] [CrossRef]
- Stielow, J.B.; Lévesque, C.A.; Seifert, K.A.; Meyer, W.; Irinyi, L.; Smits, D.; Renfurm, R.; Verkley, G.J.M.; Groenewald, M.; Chaduli, D.; et al. One fungus, which genes? Development and assessment of universal primers for potential secondary fungal DNA barcodes. Persoonia Mol. Phylogeny Evol. Fungi 2015, 35, 242–263. [Google Scholar] [CrossRef] [PubMed]
- Lofgren, L.A.; LeBlanc, N.R.; Certano, A.K.; Nachtigall, J.; LaBine, K.M.; Riddle, J.; Broz, K.; Dong, Y.; Bethan, B.; Kafer, C.W.; et al. Fusarium graminearum: pathogen or endophyte of North American grasses? New Phytol. 2018, 217, 1203–1212. [Google Scholar] [CrossRef]
- O’Donnell, K.; Kistler, H.C.; Cigelnik, E.; Ploetz, R.C. Multiple evolutionary origins of the fungus causing Panama disease of banana: concordant evidence from nuclear and mitochondrial gene genealogies. Proc. Natl. Acad. Sci. USA 1998, 95, 2044–2049. [Google Scholar] [CrossRef]
- Thompson, J.D.; Higgins, D.G.; Gibson, T.J. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994, 22, 4673–4680. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Notredame, C.; Higgins, D.G.; Heringa, J. T-coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 2000, 302, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, 320–324. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: https://www.r-project.org/ (accessed on 10 March 2020).




{kind=link}
{kind=link}
{kind=link}
| Name | Sequence (5‘–3‘) | Gene | Amplicon Length (bp) | Reference |
|---|---|---|---|---|
| RPB2-5F2 | GGGGWGAYCAGAAGAAGGC | RPB2 | 1200 | [59] |
| RPB2-7CR | CCCATRGCTTGYTTRCCCAT | |||
| EF1αF | ATGGGTAAGGARGACAAGAC | TEF-1α | 694 | [60] |
| EF1αR | GGARGTACCAGTRATCATGTT | |||
| RPB2-5R1s | TCAACVACTTCCATACCTC | sRPB2 * | 304 (with RPB2-5F2) | This study |
| TEF-1aFs2 | CAATAGGAAGCCGCYGAG | sTEF-1α * | 247 (with EF1αR) | This study |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schiwek, S.; Beule, L.; Vinas, M.; Pfordt, A.; von Tiedemann, A.; Karlovsky, P. High-Resolution Melting (HRM) Curve Assay for the Identification of Eight Fusarium Species Causing Ear Rot in Maize. Pathogens 2020, 9, 270. https://doi.org/10.3390/pathogens9040270
Schiwek S, Beule L, Vinas M, Pfordt A, von Tiedemann A, Karlovsky P. High-Resolution Melting (HRM) Curve Assay for the Identification of Eight Fusarium Species Causing Ear Rot in Maize. Pathogens. 2020; 9(4):270. https://doi.org/10.3390/pathogens9040270
Chicago/Turabian StyleSchiwek, Simon, Lukas Beule, Maria Vinas, Annette Pfordt, Andreas von Tiedemann, and Petr Karlovsky. 2020. "High-Resolution Melting (HRM) Curve Assay for the Identification of Eight Fusarium Species Causing Ear Rot in Maize" Pathogens 9, no. 4: 270. https://doi.org/10.3390/pathogens9040270
APA StyleSchiwek, S., Beule, L., Vinas, M., Pfordt, A., von Tiedemann, A., & Karlovsky, P. (2020). High-Resolution Melting (HRM) Curve Assay for the Identification of Eight Fusarium Species Causing Ear Rot in Maize. Pathogens, 9(4), 270. https://doi.org/10.3390/pathogens9040270