Zika Virus-Induction of the Suppressor of Cytokine Signaling 1/3 Contributes to the Modulation of Viral Replication


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Quantitative Reverse Transcription PCR (qRT-PCR) Analysis
2.3. Knockdown of SOCS1 or SOCS3 Using Small Interfering RNA (siRNA)
2.4. Confocal Microscopy
2.5. Luciferase Reporter Assay
2.6. Enzyme-Linked Immunosorbent Assay (ELISA)
2.7. Western Blot Analysis
2.8. Statistical Analysis
3. Results
3.1. ZIKV Infection Induces SOCS1 and SOCS3 Upregulation in Various Cells
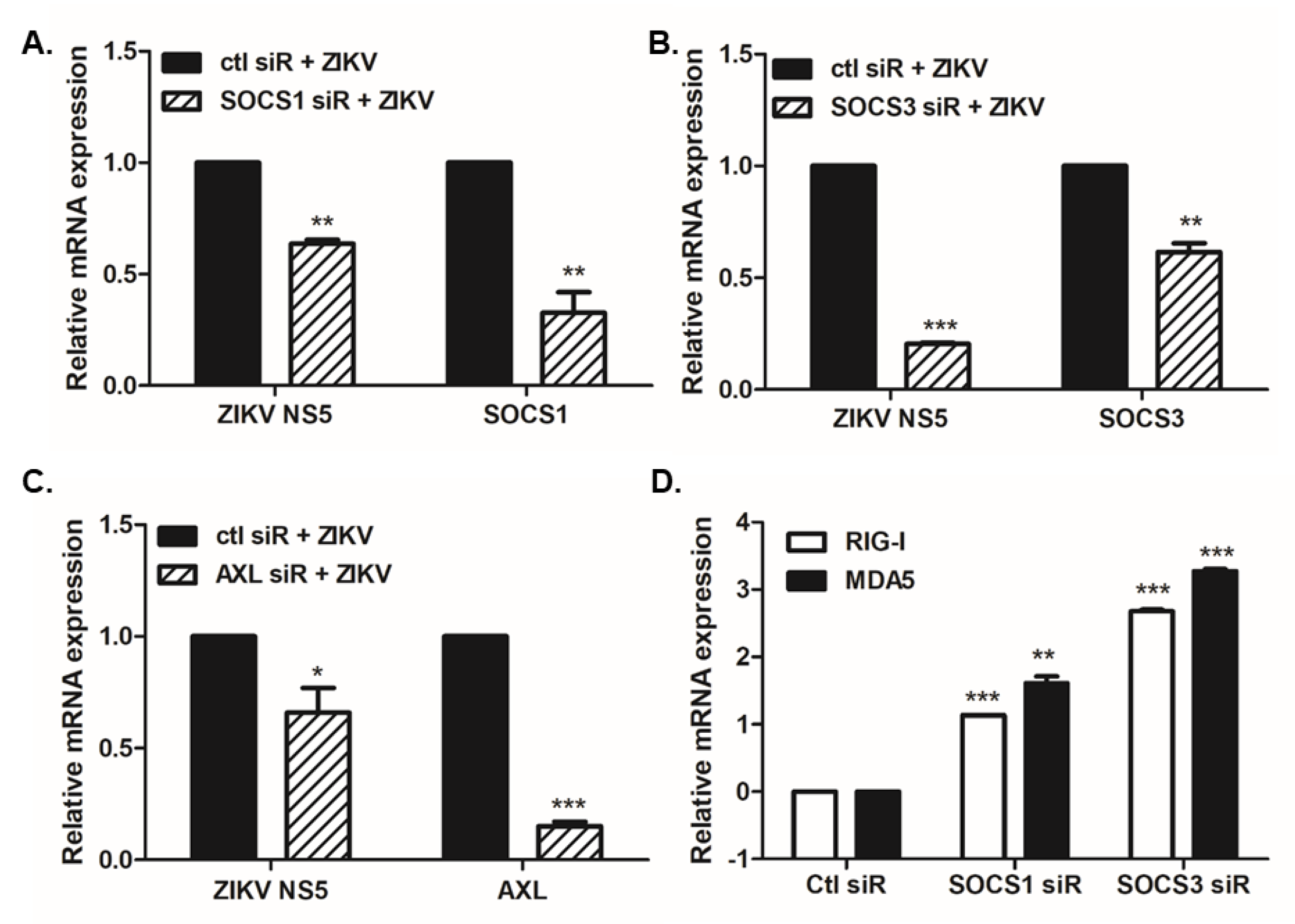
3.2. Overexpression or Knockdown of SOCS1 and SOCS3 Modulates ZIKV Replication
3.3. SOCS1 and SOCS3 Negatively Regulate Type I and III IFN Responses, Which Are Essential for ZIKV Control
4. Discussion
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Dick, G.W.; Kitchen, S.F.; Haddow, A.J. Zika virus. I. Isolations and serological specificity. Trans. R. Soc. Trop. Med. Hyg. 1952, 46, 509–520. [Google Scholar] [CrossRef]
- Leal, M.C.; Muniz, L.F.; Ferreira, T.S.; Santos, C.M.; Almeida, L.C.; Van Der Linden, V.; Ramos, R.C.; Rodrigues, L.C.; Neto, S.S. Hearing loss in infants with microcephaly and evidence of congenital zika virus infection—Brazil, November 2015–May 2016. Morb. Mortal. Wkly. Rep. 2016, 65, 917–919. [Google Scholar] [CrossRef] [PubMed]
- Cao-Lormeau, V.M.; Blake, A.; Mons, S.; Lastere, S.; Roche, C.; Vanhomwegen, J.; Dub, T.; Baudouin, L.; Teissier, A.; Larre, P.; et al. Guillain-barre syndrome outbreak associated with Zika virus infection in French polynesia: A case-control study. Lancet 2016, 387, 1531–1539. [Google Scholar] [CrossRef]
- Araujo, L.M.; Ferreira, M.L.; Nascimento, O.J. Guillain-barre syndrome associated with the Zika virus outbreak in Brazil. Arq. Neuro-Psiquiatr. 2016, 74, 253–255. [Google Scholar] [CrossRef] [PubMed]
- Lazear, H.M.; Diamond, M.S. Zika virus: New clinical syndromes and its emergence in the western hemisphere. J. Virol. 2016, 90, 4864–4875. [Google Scholar] [CrossRef]
- Lee, J.K.; Shin, O.S. Advances in Zika virus-host cell interaction: Current knowledge and future perspectives. Int. J. Mol. Sci. 2019, 20, 1101. [Google Scholar] [CrossRef]
- Elshahawi, H.; Syed Hassan, S.; Balasubramaniam, V. Importance of Zika virus NS5 protein for viral replication. Pathogens 2019, 8, 169. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, Q.; Zhou, J.; Xie, W.; Chen, C.; Wang, Z.; Yang, H.; Cui, J. Zika virus evades interferon-mediated antiviral response through the co-operation of multiple nonstructural proteins in vitro. Cell Discov. 2017, 3, 17006. [Google Scholar] [CrossRef]
- Kim, J.A.; Seong, R.K.; Son, S.W.; Shin, O.S. Insights into ZIKV-mediated innate immune responses in human dermal fibroblasts and epidermal keratinocytes. J. Investig. Dermatol. 2019, 139, 391–399. [Google Scholar] [CrossRef]
- Kumar, A.; Hou, S.; Airo, A.M.; Limonta, D.; Mancinelli, V.; Branton, W.; Power, C.; Hobman, T.C. Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep. 2016, 17, 1766–1775. [Google Scholar] [CrossRef]
- Grant, A.; Ponia, S.S.; Tripathi, S.; Balasubramaniam, V.; Miorin, L.; Sourisseau, M.; Schwarz, M.C.; Sanchez-Seco, M.P.; Evans, M.J.; Best, S.M.; et al. Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host Microbe 2016, 19, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, A.; Naka, T.; Kubo, M. SOCS proteins, cytokine signalling and immune regulation. Nat. Rev. Immunol. 2007, 7, 454–465. [Google Scholar] [CrossRef] [PubMed]
- Hilton, D.J.; Richardson, R.T.; Alexander, W.S.; Viney, E.M.; Willson, T.A.; Sprigg, N.S.; Starr, R.; Nicholson, S.E.; Metcalf, D.; Nicola, N.A. Twenty proteins containing a C-terminal SOCS box form five structural classes. Proc. Natl. Acad. Sci. USA 1998, 95, 114–119. [Google Scholar] [CrossRef]
- Akhtar, L.N.; Benveniste, E.N. Viral exploitation of host SOCS protein functions. J. Virol. 2011, 85, 1912–1921. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, L.N.; Qin, H.; Muldowney, M.T.; Yanagisawa, L.L.; Kutsch, O.; Clements, J.E.; Benveniste, E.N. Suppressor of cytokine signaling 3 inhibits antiviral IFN-beta signaling to enhance HIV-1 replication in macrophages. J. Immunol. 2010, 185, 2393–2404. [Google Scholar] [CrossRef]
- Shao, R.X.; Zhang, L.; Hong, Z.; Goto, K.; Cheng, D.; Chen, W.C.; Jilg, N.; Kumthip, K.; Fusco, D.N.; Peng, L.F.; et al. SOCS1 abrogates IFN’s antiviral effect on hepatitis C virus replication. Antivir. Res. 2013, 97, 101–107. [Google Scholar] [CrossRef][Green Version]
- Li, X.; Zhu, Q.; Cao, Q.; Chen, H.; Qian, P. Japanese encephalitis virus upregulates the expression of SOCS3 in mouse brain and Raw264.7 cells. Viruses 2014, 6, 4280–4293. [Google Scholar] [CrossRef]
- Choi, E.J.; Lee, C.H.; Shin, O.S. Suppressor of cytokine signaling 3 expression induced by varicella-zoster virus infection results in the modulation of virus replication. Scand. J. Immunol. 2015, 82, 337–344. [Google Scholar] [CrossRef]
- Okumura, A.; Rasmussen, A.L.; Halfmann, P.; Feldmann, F.; Yoshimura, A.; Feldmann, H.; Kawaoka, Y.; Harty, R.N.; Katze, M.G. Suppressor of cytokine signaling 3 is an inducible host factor that regulates virus egress during ebola virus infection. J. Virol. 2015, 89, 10399–10406. [Google Scholar] [CrossRef]
- Pauli, E.K.; Schmolke, M.; Wolff, T.; Viemann, D.; Roth, J.; Bode, J.G.; Ludwig, S. Influenza A virus inhibits type I IFN signaling via NF-kappaB-dependent induction of SOCS-3 expression. PLoS Pathog. 2008, 4, e1000196. [Google Scholar] [CrossRef]
- Pothlichet, J.; Chignard, M.; Si-Tahar, M. Cutting edge: Innate immune response triggered by influenza A virus is negatively regulated by SOCS1 and SOCS3 through a RIG-I/IFNAR1-dependent pathway. J. Immunol. 2008, 180, 2034–2038. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Salmon, S.; Yajjala, V.K.; Bauer, C.; Metzger, D.W. Expression of suppressor of cytokine signaling 1 (SOCS1) impairs viral clearance and exacerbates lung injury during influenza infection. PLoS Pathog. 2014, 10, e1004560. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.A.; Seong, R.K.; Kumar, M.; Shin, O.S. Favipiravir and ribavirin inhibit replication of Asian and African strains of Zika virus in different cell models. Viruses 2018, 10, 72. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.J.; Lim, S.; Song, M.J.; Ahn, J.H.; Lee, C.H.; Shin, O.S. Whole transcriptome analyses reveal differential mRNA and microRNA expression profiles in primary human dermal fibroblasts infected with clinical or vaccine strains of Varicella Zoster virus. Pathogens 2019, 8, 183. [Google Scholar] [CrossRef]
- Sui, H.; Zhou, M.; Chen, Q.; Lane, H.C.; Imamichi, T. siRNA enhances DNA-mediated interferon lambda-1 response through crosstalk between RIG-I and IFI16 signalling pathway. Nucleic Acids Res. 2014, 42, 583–598. [Google Scholar] [CrossRef] [PubMed]
- Miner, J.J.; Diamond, M.S. Understanding how Zika virus enters and infects neural target cells. Cell Stem Cell 2016, 18, 559–560. [Google Scholar] [CrossRef] [PubMed]
- Nowakowski, T.J.; Pollen, A.A.; Di Lullo, E.; Sandoval-Espinosa, C.; Bershteyn, M.; Kriegstein, A.R. Expression analysis highlights AXL as a candidate Zika virus entry receptor in neural stem cells. Cell Stem Cell 2016, 18, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yang, Y.F.; Yang, Y.; Zou, P.; Chen, J.; He, Y.; Shui, S.L.; Cui, Y.R.; Bai, R.; Liang, Y.J.; et al. AXL promotes Zika virus infection in astrocytes by antagonizing type I interferon signalling. Nat. Microbiol. 2018, 3, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Bayer, A.; Lennemann, N.J.; Ouyang, Y.; Bramley, J.C.; Morosky, S.; Marques, E.T., Jr.; Cherry, S.; Sadovsky, Y.; Coyne, C.B. Type III interferons produced by human placental trophoblasts confer protection against Zika virus infection. Cell Host Microbe 2016, 19, 705–712. [Google Scholar] [CrossRef]
- Ye, S.; Lowther, S.; Stambas, J. Inhibition of reactive oxygen species production ameliorates inflammation induced by influenza A viruses via upregulation of SOCS1 and SOCS3. J. Virol. 2015, 89, 2672–2683. [Google Scholar] [CrossRef]
- Wei, H.; Wang, S.; Chen, Q.; Chen, Y.; Chi, X.; Zhang, L.; Huang, S.; Gao, G.F.; Chen, J.L. Suppression of interferon lambda signaling by SOCS-1 results in their excessive production during influenza virus infection. PLoS Pathog. 2014, 10, e1003845. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Martinez, G.; Cruz-Lagunas, A.; Jimenez-Alvarez, L.; Espinosa, E.; Ortiz-Quintero, B.; Santos-Mendoza, T.; Herrera, M.T.; Canche-Pool, E.; Mendoza, C.; Banales, J.L.; et al. Seasonal and pandemic influenza H1N1 viruses induce differential expression of SOCS-1 and RIG-I genes and cytokine/chemokine production in macrophages. Cytokine 2013, 62, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Reichard, A.C.; Cheemarla, N.R.; Bigley, N.J. SOCS1/3 expression levels in HSV-1-infected, cytokine-polarized and -unpolarized macrophages. J. Interferon Cytokine Res. 2015, 35, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K.; Ishibashi, K.; Ishioka, K.; Zhao, D.; Sato, M.; Ohara, S.; Abe, Y.; Kawasaki, Y.; Sato, Y.; Yokota, S.; et al. RSV replication is attenuated by counteracting expression of the suppressor of cytokine signaling (SOCS) molecules. Virology 2009, 391, 162–170. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.A.; Lin, W.; Tai, A.W.; Shao, R.X.; Weinberg, E.; De Sa Borges, C.B.; Bhan, A.K.; Zheng, H.; Kamegaya, Y.; Chung, R.T. Hepatic SOCS3 expression is strongly associated with non-response to therapy and race in HCV and HCV/HIV infection. J. Hepatol. 2009, 50, 705–711. [Google Scholar] [CrossRef][Green Version]
- Sood, V.; Lata, S.; Ramachandran, V.G.; Banerjea, A.C. Suppressor of cytokine signaling 3 (SOCS3) degrades p65 and regulate HIV-1 replication. Front. Microbiol. 2019, 10, 114. [Google Scholar] [CrossRef]
- Ubol, S.; Phuklia, W.; Kalayanarooj, S.; Modhiran, N. Mechanisms of immune evasion induced by a complex of dengue virus and preexisting enhancing antibodies. J. Infect. Dis. 2010, 201, 923–935. [Google Scholar] [CrossRef]
- Flores-Mendoza, L.K.; Estrada-Jimenez, T.; Sedeno-Monge, V.; Moreno, M.; Manjarrez, M.D.C.; Gonzalez-Ochoa, G.; Millan-Perez Pena, L.; Reyes-Leyva, J. IL-10 and socs3 are predictive biomarkers of dengue hemorrhagic fever. Mediat. Inflamm. 2017, 2017, 5197592. [Google Scholar] [CrossRef]
- Steffensen, M.A.; Fenger, C.; Christensen, J.E.; Jorgensen, C.K.; Bassi, M.R.; Christensen, J.P.; Finsen, B.; Thomsen, A.R. Suppressors of cytokine signaling 1 and 3 are upregulated in brain resident cells in response to virus-induced inflammation of the central nervous system via at least two distinctive pathways. J. Virol. 2014, 88, 14090–14104. [Google Scholar] [CrossRef]
- Sun, X.; Hua, S.; Chen, H.R.; Ouyang, Z.; Einkauf, K.; Tse, S.; Ard, K.; Ciaranello, A.; Yawetz, S.; Sax, P.; et al. Transcriptional changes during naturally acquired Zika virus infection render dendritic cells highly conducive to viral replication. Cell Rep. 2017, 21, 3471–3482. [Google Scholar] [CrossRef]
- Dhiman, G.; Abraham, R.; Griffin, D.E. Human schwann cells are susceptible to infection with Zika and yellow fever viruses, but not dengue virus. Sci. Rep. 2019, 9, 9951. [Google Scholar] [CrossRef] [PubMed]
- Dowall, S.D.; Graham, V.A.; Rayner, E.; Hunter, L.; Atkinson, B.; Pearson, G.; Dennis, M.; Hewson, R. Lineage-dependent differences in the disease progression of Zika virus infection in type-I interferon receptor knockout (A129) mice. PLoS Negl. Trop. Dis. 2017, 11, e0005704. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Sheng, C.; Gao, S.; Yao, C.; Li, J.; Jiang, W.; Chen, H.; Wu, J.; Pan, C.; Chen, S.; et al. SOCS3 drives proteasomal degradation of TBK1 and negatively regulates antiviral innate immunity. Mol. Cell Biol. 2015, 35, 2400–2413. [Google Scholar] [CrossRef] [PubMed]
- Blumer, T.; Coto-Llerena, M.; Duong, F.H.T.; Heim, M.H. SOCS1 is an inducible negative regulator of interferon lambda (IFN-lambda)-induced gene expression in vivo. J. Biol. Chem. 2017, 292, 17928–17938. [Google Scholar] [CrossRef]
- Jagger, B.W.; Miner, J.J.; Cao, B.; Arora, N.; Smith, A.M.; Kovacs, A.; Mysorekar, I.U.; Coyne, C.B.; Diamond, M.S. Gestational stage and IFN-lambda signaling regulate ZIKV infection in utero. Cell Host Microbe 2017, 22, 366–376. [Google Scholar] [CrossRef]
- Chen, J.; Liang, Y.; Yi, P.; Xu, L.; Hawkins, H.K.; Rossi, S.L.; Soong, L.; Cai, J.; Menon, R.; Sun, J. Outcomes of congenital Zika disease depend on timing of infection and maternal-fetal interferon action. Cell Rep. 2017, 21, 1588–1599. [Google Scholar] [CrossRef]
- Tang, H.; Hammack, C.; Ogden, S.C.; Wen, Z.; Qian, X.; Li, Y.; Yao, B.; Shin, J.; Zhang, F.; Lee, E.M.; et al. Zika virus infects human cortical neural progenitors and attenuates their growth. Cell Stem Cell 2016, 18, 587–590. [Google Scholar] [CrossRef]
- Zhang, P.; Li, F.; Li, N.; Zhu, Q.; Yang, C.; Han, Q.; Chen, J.; Lv, Y.; Yu, L.; Wei, P.; et al. Genetic variations of SOCS1 are associated with chronic hepatitis B virus infection. Hum. Immunol. 2014, 75, 709–714. [Google Scholar] [CrossRef]
- Aslam, R.; Raza, S.M.; Naeemi, H.; Mubarak, B.; Afzal, N.; Khaliq, S. SOCS3 mRNA expression and polymorphisms as pretreatment predictor of response to HCV genotype 3A IFN-based treatment. SpringerPlus 2016, 5, 1826. [Google Scholar] [CrossRef]
- Ahmed, C.M.; Dabelic, R.; Martin, J.P.; Jager, L.D.; Haider, S.M.; Johnson, H.M. Enhancement of antiviral immunity by small molecule antagonist of suppressor of cytokine signaling. J. Immunol. 2010, 185, 1103–1113. [Google Scholar] [CrossRef]
- Ahmed, C.M.; Dabelic, R.; Bedoya, S.K.; Larkin, J., III; Johnson, H.M. A SOCS1/3 antagonist peptide protects mice against lethal infection with influenza a virus. Front. Immunol. 2015, 6, 574. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seong, R.-K.; Lee, J.K.; Shin, O.S. Zika Virus-Induction of the Suppressor of Cytokine Signaling 1/3 Contributes to the Modulation of Viral Replication. Pathogens 2020, 9, 163. https://doi.org/10.3390/pathogens9030163
Seong R-K, Lee JK, Shin OS. Zika Virus-Induction of the Suppressor of Cytokine Signaling 1/3 Contributes to the Modulation of Viral Replication. Pathogens. 2020; 9(3):163. https://doi.org/10.3390/pathogens9030163
Chicago/Turabian StyleSeong, Rak-Kyun, Jae Kyung Lee, and Ok Sarah Shin. 2020. "Zika Virus-Induction of the Suppressor of Cytokine Signaling 1/3 Contributes to the Modulation of Viral Replication" Pathogens 9, no. 3: 163. https://doi.org/10.3390/pathogens9030163
APA StyleSeong, R.-K., Lee, J. K., & Shin, O. S. (2020). Zika Virus-Induction of the Suppressor of Cytokine Signaling 1/3 Contributes to the Modulation of Viral Replication. Pathogens, 9(3), 163. https://doi.org/10.3390/pathogens9030163