The Key Roles of Interferon Lambda in Human Molecular Defense against Respiratory Viral Infections

 ,
,  ,
, 
Abstract
1. Introduction
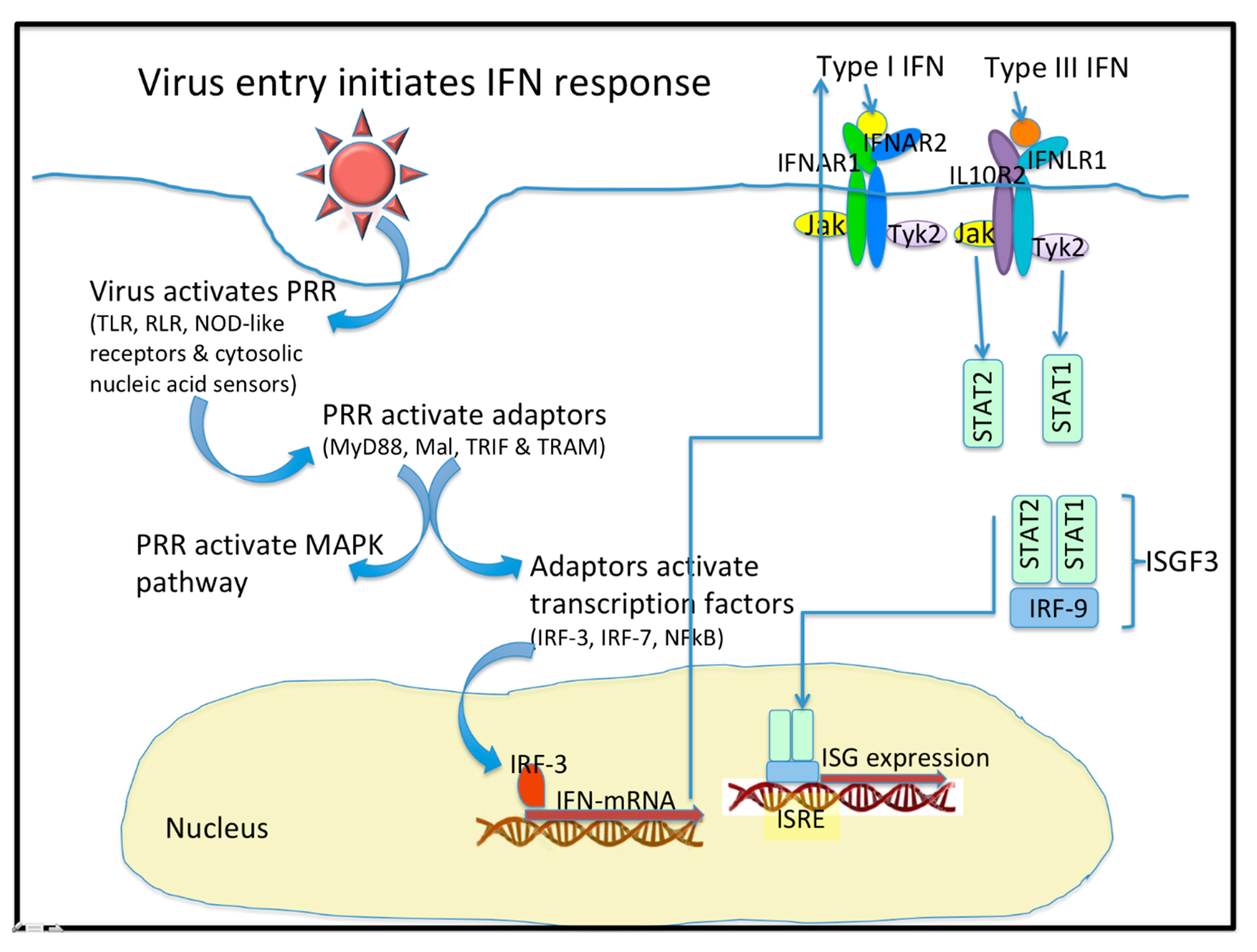
1.1. Virus Entry Triggers Host Signaling Responses
1.2. IFN Are Class II Cytokines
2. IFN-λ Play a Distinct Anti-Viral Role in Collaboration with Other IFN
2.1. IFN-λ Structure
2.2. Expression of IFN-λ
2.3. Molecular Mechanism of IFN-λ Induction
2.4. The IFN-λ Receptor (IFNλR)
2.5. The Effects of IFN-λ on Cells
2.6. Immuno-Modulatory Activity of IFN-λ
3. Antiviral Effects
3.1. IFN-λ Are Universal Antivirals
3.2. INF-λ Exhibit Antiviral Activity against Coronaviruses
3.3. Antiviral Activity against Other Respiratory Viruses
4. The Role of IFN-λ Specifically during Influenza Virus Infection
4.1. Influenza Virus Infection and Respiratory Airway Epithelium
4.2. Knockout Mouse Models
4.3. Induction of IFN-λ in Influenza Virus Infection
4.4. The Antiviral Effect of IFN-λ in Influenza Virus Infection
4.5. Immuno-Modulatory Effects
4.6. Use of Recombinant IFN-λ: A Two-Faced Janus
5. Conclusions
Funding
Conflicts of Interest
Abbreviations
| BALF | bronchoalveolar fluid |
| HA | hemagglutinin |
| HBV | hepatitis B virus |
| HCV | hepatitis C virus |
| HSV | herpes simplex virus |
| IAV | influenza A virus |
| IBV | influenza B virus |
| IFN | interferon(s) |
| IFNαR | type I (α/β) interferon receptor |
| IFNαR1 | interferon-α/β receptor subunit 1 |
| ISG | interferon stimulated gene(s) |
| mDC | myeloid dendritic cell(s) |
| MTEC | murine tracheal epithelial cell(s) |
| pDC | plasmacytoid dendritic cell(s) |
| RLR | RIG-I-like receptor(s) |
| PRR | pattern recognition receptor(s) |
| SAR | sialic acid receptor(s) |
| SARS-CoV | severe acute respiratory syndrome coronavirus |
| TLR | Toll-like receptor(s) |
| URT | upper respiratory tract |
References
- Thompson, M.R.; Kaminski, J.J.; Kurt-Jones, E.A.; Fitzgerald, K.A. Pattern Recognition Receptors and the Innate Immune Response to Viral Infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef] [PubMed]
- Kaisho, T.; Akira, S. Toll-like receptor function and signaling. J. Allergy Clin. Immunol. 2006, 117, 979–987. [Google Scholar] [CrossRef] [PubMed]
- Randall, R.E.; Goodbourn, S. Interferons and viruses: An interplay between induction, signalling, antiviral responses and virus countermeasures. J. Gen. Virol. 2008, 89, 1–47. [Google Scholar] [CrossRef] [PubMed]
- Hamming, O.J.; Terczyńska-Dyla, E.; Vieyres, G.; Dijkman, R.; Jørgensen, S.E.; Akhtar, H.; Siupka, P.; Pietschmann, T.; Thiel, V.; Hartmann, R. Interferon lambda 4 signals via the IFNλ receptor to regulate antiviral activity against HCV and coronaviruses. EMBO J. 2013, 32, 3055–3065. [Google Scholar] [CrossRef] [PubMed]
- Baños-Lara, M.D.R.; Harvey, L.; Mendoza, A.; Simms, D.; Chouljenko, V.N.; Wakamatsu, N.; Kousoulas, K.G.; Guerrero-Plata, A. Impact and Regulation of Lambda Interferon Response in Human Metapneumovirus Infection. J. Virol. 2015, 89, 730–742. [Google Scholar] [CrossRef] [PubMed]
- Killip, M.J.; Fodor, E.; Randall, R.E. Influenza virus activation of the interferon system. Virus Res. 2015, 209, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Syedbasha, M.; Egli, A. Interferon Lambda: Modulating immunity in infectious diseases. Front. Immunol. 2017, 8, 119. [Google Scholar] [CrossRef]
- Capobianchi, M.R.; Uleri, E.; Caglioti, C.; Dolei, A. Type I IFN family members: Similarity, differences and interaction. Cytokine Growth Factor Rev. 2015, 26, 103–111. [Google Scholar] [CrossRef]
- François-Newton, V.; de Freitas Almeida, G.M.; Payelle-Brogard, B.; Monneron, D.; Pichard-Garcia, L.; Piehler, J.; Pellegrini, S.; Uzé, G. USP18-based negative feedback control is induced by type I and type III interferons and specifically inactivates interferon α response. PLoS ONE 2011, 6, e22200. [Google Scholar] [CrossRef]
- Jaitin, D.A.; Roisman, L.C.; Jaks, E.; Gavutis, M.; Piehler, J.; Van Der Heyden, J.; Uze, G.; Schreiber, G. Inquiring into the Differential Action of Interferons (IFNs): An IFN-2 Mutant with Enhanced Affinity to IFNAR1 Is Functionally Similar to IFN. Mol. Cell. Biol. 2006, 26, 1888–1897. [Google Scholar] [CrossRef]
- Sommereyns, C.; Paul, S.; Staeheli, P.; Michiels, T. IFN-lambda (IFN-λ) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 2008, 4, e1000017. [Google Scholar] [CrossRef] [PubMed]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.Y.; Ma, Y.; Wang, J.; Fan, Y.; Feng, S.; Lu, Q.; Yu, Q.; Sui, D.; Rothbart, M.K.; Fan, M.; et al. Short-term meditation training improves attention and self-regulation. Proc. Natl. Acad. Sci. USA 2007, 104, 17152–17156. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Hamming, O.J.; Ank, N.; Paludan, S.R.; Nielsen, A.L.; Hartmann, R. Type III Interferon (IFN) Induces a Type I IFN-Like Response in a Restricted Subset of Cells through Signaling Pathways Involving both the Jak-STAT Pathway and the Mitogen-Activated Protein Kinases. J. Virol. 2007, 81, 7749–7758. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.C.; Tavares, L.P.; Dias, A.C.F.; Kehdy, F.; Alvarado-Arnez, L.E.; Queiroz-Junior, C.M.; Galvão, I.; Lima, B.H.; Matos, A.R.; Gonçalves, A.P.F.; et al. Phosphatidyl inositol 3 kinase-gamma balances antiviral and inflammatory responses during influenza A H1N1 infection: From murine model to genetic association in patients. Front. Immunol. 2018, 9, 975. [Google Scholar] [CrossRef]
- Katsoulidis, E.; Li, Y.; Mears, H.; Platanias, L.C. The p38 mitogen-activated protein kinase pathway in interferon signal transduction. J. Int. Cytokine Res. 2005, 25, 749–756. [Google Scholar] [CrossRef]
- Jewell, N.A.; Vaghefi, N.; Mertz, S.E.; Akter, P.; Peebles, R.S.; Bakaletz, L.O.; Durbin, R.K.; Flaño, E.; Durbin, J.E. Differential Type I Interferon Induction by Respiratory Syncytial Virus and Influenza A Virus In Vivo. J. Virol. 2007, 81, 9790–9800. [Google Scholar] [CrossRef]
- Honda, K.; Ohba, Y.; Yanai, H.; Hegishi, H.; Mizutani, T.; Takaoka, A.; Taya, C.; Taniguchi, T. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature 2005, 434, 1035–1040. [Google Scholar] [CrossRef]
- Stetson, D.B.; Medzhitov, R. Type I Interferons in Host Defense. Immunity 2006, 25, 373–381. [Google Scholar] [CrossRef]
- Sarasin-Filipowicz, M.; Wang, X.; Yan, M.; Duong, F.H.T.; Poli, V.; Hilton, D.J.; Zhang, D.E.; Heim, M.H. Alpha Interferon Induces Long-Lasting Refractoriness of JAK-STAT Signaling in the Mouse Liver through Induction of USP18/UBP43. Mol. Cell. Biol. 2009, 29, 4841–4851. [Google Scholar] [CrossRef]
- Delgado-Ortega, M.; Marc, D.; Dupont, J.; Trapp, S.; Berri, M.; Meurens, F. SOCS proteins in infectious diseases of mammals. Vet. Immunol. Immunopathol. 2013, 151, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Malakhov, M.P.; Malakhova, O.A.; Il Kim, K.; Ritchie, K.J.; Zhang, D.E. UBP43 (USP18) specifically removes ISG15 from conjugated proteins. J. Biol. Chem. 2002, 277, 9976–9981. [Google Scholar] [CrossRef] [PubMed]
- Malakhova, O.A.; Kim, K., II; Luo, J.K.; Zou, W.; Kumar, K.G.S.; Fuchs, S.Y.; Shuai, K.; Zhang, D.E. UBP43 is a novel regulator of interferon signaling independent of its ISG15 isopeptidase activity. EMBO J. 2006, 25, 2358–2367. [Google Scholar] [CrossRef] [PubMed]
- Dellgren, C.; Gad, H.; Hamming, O.; Melchjorsen, J.; Hartmann, R. Human interferon-λ3 is a potent member of the type III interferon family. Genes Immun. 2009, 10, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Miknis, Z.; Magracheva, E.; Li, W.; Zdanov, A.; Kotenko, S.V.; Wlodawer, A. Crystal structure of human interferon-λ1 in complex with its high-affinity receptor interferon-λR1. J. Mol. Biol. 2010, 404, 650–664. [Google Scholar] [CrossRef]
- Lazear, H.M.; Nice, T.J.; Diamond, M.S. Interferon-λ: Immune Functions at Barrier Surfaces and Beyond. Immunity 2015, 43, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Lasfar, A.; Lewis-Antes, A.; Smirnov, S.V.; Anantha, S.; Abushahba, W.; Tian, B.; Reuhl, K.; Dickensheets, H.; Sheikh, F.; Donnelly, R.P.; et al. Characterization of the mouse IFN-λ ligand-receptor system: IFN-λs exhibit antitumor activity against B16 melanoma. Cancer Res. 2006, 66, 4468–4477. [Google Scholar] [CrossRef]
- Prokunina-Olsson, L.; Muchmore, B.; Tang, W.; Pfeiffer, R.M.; Park, H.; Dickensheets, H.; Hergott, D.; Porter-Gill, P.; Mumy, A.; Kohaar, I.; et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat. Genet. 2013, 45, 164–171. [Google Scholar] [CrossRef]
- Egli, A.; Santer, D.M.; O’Shea, D.; Tyrrell, D.L.; Houghton, M. The impact of the interferon-lambda family on the innate and adaptive immune response to viral infections. Emerg. Microbes Infect. 2014, 3, e51. [Google Scholar] [CrossRef]
- Hermant, P.; Michiels, T. Interferon-λ in the context of viral infections: Production, response and therapeutic implications. J. Innate Immun. 2014, 6, 563–574. [Google Scholar] [CrossRef]
- Pott, J.; Mahlakõiv, T.; Mordstein, M.; Duerr, C.U.; Michiels, T.; Stockinger, S.; Staeheli, P.; Hornef, M.W. IFN-λ determines the intestinal epithelial antiviral host defense. Proc. Natl. Acad. Sci. USA 2011, 108, 7944–7949. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Nie, P.; Secombes, C.J.; Zou, J. Intron-Containing Type I and Type III IFN Coexist in Amphibians: Refuting the Concept That a Retroposition Event Gave Rise to Type I IFNs. J. Immunol. 2010, 184, 5038–5046. [Google Scholar] [CrossRef] [PubMed]
- Hamming, O.J.; Lutfalla, G.; Levraud, J.P.; Hartmann, R. Crystal Structure of Zebrafish Interferons I and II Reveals Conservation of Type I Interferon Structure in Vertebrates. J. Virol. 2011, 85, 8181–8187. [Google Scholar] [CrossRef] [PubMed]
- Reuter, A.; Soubies, S.; Hartle, S.; Schusser, B.; Kaspers, B.; Staeheli, P.; Rubbenstroth, D.; Garcia-Sastre, A. Antiviral Activity of Lambda Interferon in Chickens. J. Virol. 2014, 88, 2835–2843. [Google Scholar] [CrossRef]
- Hong, M.A.; Schwerk, J.; Lim, C.; Kell, A.; Jarret, A.; Pangallo, J.; Loo, Y.; Liu, S.; Hagedorn, C.H.; Gale, M.; et al. Interferon lambda 4 expression is suppressed by the host during viral infection. J. Exp. Med. 2016, 213, 2539–2552. [Google Scholar] [CrossRef]
- Terczyńska-Dyla, E.; Bibert, S.; Duong, F.H.T.; Krol, I.; Jørgensen, S.; Collinet, E.; Kutalik, Z.; Aubert, V.; Cerny, A.; Kaiser, L.; et al. Reduced IFNλ4 activity is associated with improved HCV clearance and reduced expression of interferon-stimulated genes. Nat. Commun. 2014, 5, 5699. [Google Scholar] [CrossRef]
- Bamford, C.G.G.; Aranday-Cortes, E.; Filipe, I.C.; Sukumar, S.; Mair, D.; da Silva Filipe, A.; Mendoza, J.L.; Garcia, K.C.; Fan, S.; Tishkoff, S.A.; et al. A polymorphic residue that attenuates the antiviral potential of interferon lambda 4 in hominid lineages. PLoS Pathog. 2018, 14, e1007307. [Google Scholar] [CrossRef]
- Ioannidis, I.; Ye, F.; McNally, B.; Willette, M.; Flano, E. Toll-Like Receptor Expression and Induction of Type I and Type III Interferons in Primary Airway Epithelial Cells. J. Virol. 2013, 87, 3261–3270. [Google Scholar] [CrossRef]
- Wang, J.; Oberley-Deegan, R.; Wang, S.; Nikrad, M.; Funk, C.J.; Hartshorn, K.L.; Mason, R.J. Differentiated Human Alveolar Type II Cells Secrete Antiviral IL-29 (IFN-λ1) in Response to Influenza A Infection. J. Immunol. 2009, 182, 1296–1304. [Google Scholar] [CrossRef]
- Delgado-Ortega, M.; Melo, S.; Punyadarsaniya, D.; Ramé, C.; Olivier, M.; Soubieux, D.; Marc, D.; Simon, G.; Herrler, G.; Berri, M.; et al. Innate immune response to a H3N2 subtype swine influenza virus in newborn porcine trachea cells, alveolar macrophages, and precision-cut lung slices. Vet. Res. 2014, 45. [Google Scholar] [CrossRef]
- Contoli, M.; Message, S.D.; Laza-Stanca, V.; Edwards, M.R.; Wark, P.A.B.; Bartlett, N.W.; Kebadze, T.; Mallia, P.; Stanciu, L.A.; Parker, H.L.; et al. Role of deficient type III interferon-λ production in asthma exacerbations. Nat. Med. 2006, 12, 1023–1026. [Google Scholar] [CrossRef] [PubMed]
- Koltsida, O.; Hausding, M.; Stavropoulos, A.; Koch, S.; Tzelepis, G.; Übel, C.; Kotenko, S.V.; Sideras, P.; Lehr, H.A.; Tepe, M.; et al. IL-28A (IFN-λ2) modulates lung DC function to promote Th1 immune skewing and suppress allergic airway disease. EMBO Mol. Med. 2011, 3, 348–361. [Google Scholar] [CrossRef] [PubMed]
- Hillyer, P.; Mane, V.P.; Schramm, L.M.; Puig, M.; Verthelyi, D.; Chen, A.; Zhao, Z.; Navarro, M.B.; Kirschman, K.D.; Bykadi, S.; et al. Expression profiles of human interferon-alpha and interferon-lambda subtypes are ligand- and cell-dependent. Immunol. Cell Biol. 2012, 90, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Banos-Lara, M.D.R.; Ghosh, A.; Guerrero-Plata, A. Critical Role of MDA5 in the Interferon Response Induced by Human Metapneumovirus Infection in Dendritic Cells and In Vivo. J. Virol. 2013, 87, 1242–1251. [Google Scholar] [CrossRef] [PubMed]
- Onoguchi, K.; Yoneyama, M.; Takemura, A.; Akira, S.; Taniguchi, T.; Namiki, H.; Fujita, T. Viral infections activate types I and III interferon genes through a common mechanism. J. Biol. Chem. 2007, 282, 7576–7581. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Brann, T.W.; Zhou, M.; Yang, J.; Oguariri, R.M.; Lidie, K.B.; Imamichi, H.; Huang, D.W.; Lempicki, R.A.; Baseler, M.W.; et al. Cutting Edge: Ku70 Is a Novel Cytosolic DNA Sensor That Induces Type III Rather Than Type I IFN. J. Immunol. 2011, 186, 4541–4545. [Google Scholar] [CrossRef] [PubMed]
- Österlund, P.I.; Pietilä, T.E.; Veckman, V.; Kotenko, S.V.; Julkunen, I. IFN Regulatory Factor Family Members Differentially Regulate the Expression of Type III IFN (IFN-λ) Genes. J. Immunol. 2007, 179, 3434–3442. [Google Scholar] [CrossRef]
- Griffiths, S.J.; Koegl, M.; Boutell, C.; Zenner, H.L.; Crump, C.M.; Pica, F.; Gonzalez, O.; Friedel, C.C.; Barry, G.; Martin, K.; et al. A Systematic Analysis of Host Factors Reveals a Med23-Interferon-λ Regulatory Axis against Herpes Simplex Virus Type 1 Replication. PLoS Pathog. 2013, 9, e1003514. [Google Scholar] [CrossRef]
- Egli, A.; Levin, A.; Santer, D.M.; Joyce, M.; O’Shea, D.; Thomas, B.S.; Lisboa, L.F.; Barakat, K.; Bhat, R.; Fischer, K.P.; et al. Immunomodulatory function of interleukin 28B during primary infection with cytomegalovirus. J. Infect. Dis. 2014, 210, 717–727. [Google Scholar] [CrossRef]
- Stanifer, M.L.; Pervolaraki, K.; Boulant, S. Differential regulation of type I and type III interferon signaling. Int. J. Mol. Sci. 2019, 20, 1445. [Google Scholar] [CrossRef]
- Liu, B.; Chen, S.; Guan, Y.; Chen, L. Type III interferon induces distinct SOCS1 expression pattern that contributes to delayed but prolonged activation of Jak/STAT signaling pathway: Implications for treatment non-response in HCV patients. PLoS ONE 2015, 10, e0133800. [Google Scholar] [CrossRef] [PubMed]
- Jordan, W.; Eskdale, J.; Srinivas, S.; Pekarek, V.; Kelner, D.; Rodia, M.; Gallagher, G. Human interferon lambda-1 (IFN-λ 1/IL-29) modulates the Th1/Th2 response. Genes Immun. 2007, 8, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Wolk, K.; Witte, E.; Wallace, E.; Döcke, W.D.; Kunz, S.; Asadullah, K.; Volk, H.D.; Sterry, W.; Sabat, R. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: A potential role in psoriasis. Eur. J. Immunol. 2006, 36, 1309–1323. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, J.; Schneider, W.M.; Hoffmann, H.; Vercauteren, K.; Jude, K.M.; Xiong, A.; Moraga, I.; Horton, T.M.; Glenn, J.S.; de Jong, Y.P.; et al. The IFN-λ-IFN-λR1-IL-10Rβ complex reveals structural features underlying type III IFN functional plasticity. Immunity 2017, 46, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.; Wavreil, F.; Schepens, B.; Gad, H.H.; Hartmann, R.; Rocha-Pereira, J.; Neyts, J.; Saelens, X.; Michiels, T. Species specificity of type III interferon activity and development of a sensitive luciferase-based bioassay for quantitation of mouse interferon-λ. J. Int. Cytokine Res. 2018, 38, 469–479. [Google Scholar] [CrossRef]
- Zahn, S.; Rehkämper, C.; Kümmerer, B.M.; Ferring-Schmidt, S.; Bieber, T.; Tüting, T.; Wenzel, J. Evidence for a pathophysiological role of keratinocyte-derived type III interferon (IFNλ) in cutaneous lupus erythematosus. J. Investig. Dermatol. 2011, 131, 133–140. [Google Scholar] [CrossRef]
- Yin, Z.; Dai, J.; Deng, J.; Sheikh, F.; Natalia, M.; Shih, T.; Lewis-Antes, A.; Amrute, S.B.; Garrigues, U.; Doyle, S.; et al. Type III IFNs Are Produced by and Stimulate Human Plasmacytoid Dendritic Cells. J. Immunol. 2012, 189, 2735–2745. [Google Scholar] [CrossRef]
- Zhang, S.; Kodys, K.; Li, K.; Szabo, G. Human type 2 myeloid dendritic cells produce interferon-λ and amplify interferon-α in response to hepatitis C virus infection. Gastroenterology 2013, 144, 414–425. [Google Scholar] [CrossRef]
- Dickensheets, H.; Sheikh, F.; Park, O.; Gao, B.; Donnelly, R.P. Interferon-lambda (IFN-) induces signal transduction and gene expression in human hepatocytes, but not in lymphocytes or monocytes. J. Leukoc. Biol. 2013, 93, 377–385. [Google Scholar] [CrossRef]
- Mennechet, F.J.D.; Uzé, G. Interferon-λ-treated dendritic cells specifically induce proliferation of FOXP3-expressing suppressor T cells. Blood 2006, 107, 4417–4423. [Google Scholar] [CrossRef]
- Sun, Y.; Jiang, J.; Tien, P.; Liu, W.; Li, J. IFN-λ: A new spotlight in innate immunity against influenza virus infection. Protein Cell 2018, 9, 832–837. [Google Scholar] [CrossRef] [PubMed]
- Major, J.; Crotta, S.; Llorian, M.; McCabe, T.M.; Gad, H.H.; Priestnall, S.L.; Hartmann, R.; Wack, A. Type I and III interferons disrupt lung epithelial repair during recovery from viral infection. Science 2020, 369, 712–717. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Yanagi, Y.; Ohno, S. Both type I and type III interferons are required to restrict measles virus growth in lung epithelial cells. Arch. Virol. 2019, 164, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Lukacikova, L.; Oveckova, I.; Betakova, T.; Laposova, K.; Polcicova, K.; Pastorekova, S.; Pastorek, J.; Tomaskova, J. Antiviral Effect of Interferon Lambda Against Lymphocytic Choriomeningitis Virus. J. Int. Cytokine Res. 2015, 35, 540–553. [Google Scholar] [CrossRef]
- Ge, D.; Fellay, J.; Thompson, A.J.; Simon, J.S.; Shianna, K.V.; Urban, T.J.; Heinzen, E.L.; Qiu, P.; Bertelsen, A.H.; Muir, A.J.; et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 2009, 461, 399–401. [Google Scholar] [CrossRef]
- Hsu, Y.L.; Wang, M.Y.; Ho, L.J.; Lai, J.H. Dengue virus infection induces interferon-lambda1 to facilitate cell migration. Sci. Rep. 2016, 6, 24530. [Google Scholar] [CrossRef]
- Palma-Ocampo, H.K.; Flores-Alonso, J.C.; Vallejo-Ruiz, V.; Reyes-Leyva, J.; Flores-Mendoza, L.; Herrera-Camacho, I.; Rosas-Murrieta, N.H.; Santos-López, G. Interferon lambda inhibits dengue virus replication in epithelial cells. Virol. J. 2015, 12, 150. [Google Scholar] [CrossRef]
- Nice, T.J.; Baldridge, M.T.; McCune, B.T.; Norman, J.M.; Lazear, H.M.; Artyomov, M.; Diamond, M.S.; Virgin, H.W. Interferon-λ cures persistent murine norovirus infection in the absence of adaptive immunity. Science 2015, 347, 269–273. [Google Scholar] [CrossRef]
- Lind, K.; Svedin, E.; Utorova, R.; Stone, V.M.; Flodström-Tullberg, M. Type III interferons are expressed by Coxsackievirus-infected human primary hepatocytes and regulate hepatocyte permissiveness to infection. Clin. Exp. Immunol. 2014, 177, 687–695. [Google Scholar] [CrossRef]
- Dinnon, K.H.; Leist, S.R.; Schäfer, A.; Edwards, C.E.; Martinez, D.R.; Montgomery, S.A.; West, A.; Yount, B.L.; Hou, Y.J.; Adams, L.E.; et al. A mouse-adapted SARS-CoV-2 model for the evaluation of COVID-19 medical countermeasures. bioRxiv Prepr. Serv. Biol. 2020. [Google Scholar] [CrossRef]
- Doyle, S.E.; Schreckhise, H.; Khuu-Duong, K.; Henderson, K.; Rosler, R.; Storey, H.; Yao, L.; Liu, H.; Barahmand-Pour, F.; Sivakumar, P.; et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology 2006, 44, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Jewell, N.A.; Cline, T.; Mertz, S.E.; Smirnov, S.V.; Flaño, E.; Schindler, C.; Grieves, J.L.; Durbin, R.K.; Kotenko, S.V.; Durbin, J.E. Lambda Interferon Is the Predominant Interferon Induced by Influenza A Virus Infection In Vivo. J. Virol. 2010, 84, 11515–11522. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Wang, S.; Chen, Q.; Chen, Y.; Chi, X.; Zhang, L.; Huang, S.; Gao, G.F.; Chen, J.L. Suppression of Interferon Lambda Signaling by SOCS-1 Results in Their Excessive Production during Influenza Virus Infection. PLoS Pathog. 2014, 10, e1003845. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Finotto, S. Role of Interferon-λ in allergic asthma. J. Innate Immun. 2015, 7, 224–230. [Google Scholar] [CrossRef]
- Odendall, C.; Dixit, E.; Stavru, F.; Bierne, H.; Franz, K.M.; Durbin, A.F.; Boulant, S.; Gehrke, L.; Cossart, P.; Kagan, J.C. Diverse intracellular pathogens activate type III interferon expression from peroxisomes. Nat. Immunol. 2014, 15, 717. [Google Scholar] [CrossRef]
- Jordan, W.; Eskdale, J.; Boniotto, M.; Rodia, M.; Kellner, D.; Gallagher, G. Modulation of the human cytokine response by interferon lambda-1 (IFN-λ 1/IL-29). Genes Immun. 2007, 8, 13–20. [Google Scholar] [CrossRef]
- Hernández, P.P.; Mahlakõiv, T.; Yang, I.; Schwierzeck, V.; Nguyen, N.; Guendel, F.; Gronke, K.; Ryffel, B.; Hölscher, C.; Dumoutier, L.; et al. Interferon-γ and interleukin 22 act synergistically for the induction of interferon-stimulated genes and control of rotavirus infection. Nat. Immunol. 2015, 16, 698–707. [Google Scholar] [CrossRef]
- Bierne, H.; Travier, L.; Mahlakõiv, T.; Tailleux, L.; Subtil, A.; Lebreton, A.; Paliwal, A.; Gicquel, B.; Staeheli, P.; Lecuit, M.; et al. Activation of type III interferon genes by pathogenic bacteria in infected epithelial cells and mouse placenta. PLoS ONE 2012, 7, e39080. [Google Scholar] [CrossRef]
- Cohen, T.S.; Prince, A.S. Activation of inflammasome signaling mediates pathology of acute P. aeruginosa pneumonia. J. Clin. Investig. 2013, 123, 1630–1637. [Google Scholar] [CrossRef]
- Zhou, L.; Li, J.L.; Zhou, Y.; Liu, J.B.; Zhuang, K.; Gao, J.F.; Liu, S.; Sang, M.; Wu, J.G.; Ho, W.Z. Induction of interferon-λ contributes to TLR3 and RIG-I activation-mediated inhibition of herpes simplex virus type 2 replication in human cervical epithelial cells. Mol. Hum. Reprod. 2015, 21, 917–929. [Google Scholar] [CrossRef]
- Diegelmann, J.; Beigel, F.; Zitzmann, K.; Kaul, A.; Göke, B.; Auernhammer, C.J.; Bartenschlager, R.; Diepolder, H.M.; Brand, S. Comparative analysis of the lambda-interferons IL-28A and IL-29 regarding their transcriptome and their antiviral properties against hepatitis C virus. PLoS ONE. 2010, 5, e15200. [Google Scholar] [CrossRef] [PubMed]
- Marcello, T.; Grakoui, A.; Barba-Spaeth, G.; Machlin, E.S.; Kotenko, S.V.; Macdonald, M.R.; Rice, C.M. Interferons α and λ Inhibit Hepatitis C Virus Replication with Distinct Signal Transduction and Gene Regulation Kinetics. Gastroenterology 2006, 131, 1887–1898. [Google Scholar] [CrossRef] [PubMed]
- Langhans, B.; Kupfer, B.; Braunschweiger, I.; Arndt, S.; Schulte, W.; Nischalke, H.D.; Nattermann, J.; Oldenburg, J.; Sauerbruch, T.; Spengler, U. Interferon-lambda serum levels in hepatitis C. J. Hepatol. 2011, 54, 859–865. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Pan, Y.; Wang, M.; Wang, D.; Li, W.; Jiang, T.; Zhang, P.; Chi, X.; Jiang, Y.; Gao, Y.; et al. IL28B genetic variation is associated with spontaneous clearance of hepatitis C virus, treatment response, serum IL-28B levels in Chinese population. PLoS ONE 2012, 7, e37054. [Google Scholar] [CrossRef]
- Mordstein, M.; Neugebauer, E.; Ditt, V.; Jessen, B.; Rieger, T.; Falcone, V.; Sorgeloos, F.; Ehl, S.; Mayer, D.; Kochs, G.; et al. Lambda Interferon Renders Epithelial Cells of the Respiratory and Gastrointestinal Tracts Resistant to Viral Infections. J. Virol. 2010, 84, 5670–5677. [Google Scholar] [CrossRef]
- Chan, R.W.Y.; Chan, M.C.W.; Agnihothram, S.; Chan, L.L.Y.; Kuok, D.I.T.; Fong, J.H.M.; Guan, Y.; Poon, L.L.M.; Baric, R.S.; Nicholls, J.M.; et al. Tropism of and Innate Immune Responses to the Novel Human Betacoronavirus Lineage C Virus in Human Ex Vivo Respiratory Organ Cultures. J. Virol. 2013, 87, 6604–6614. [Google Scholar] [CrossRef]
- Zhou, J.; Chu, H.; Li, C.; Wong, B.H.Y.; Cheng, Z.S.; Poon, V.K.M.; Sun, T.; Lau, C.C.Y.; Wong, K.K.Y.; Chan, J.Y.W.; et al. Active replication of middle east respiratory syndrome coronavirus and aberrant induction of inflammatory cytokines and chemokines in human macrophages: Implications for pathogenesis. J. Infect. Dis. 2014, 209, 1331–1342. [Google Scholar] [CrossRef]
- Qian, Z.; Travanty, E.A.; Oko, L.; Edeen, K.; Berglund, A.; Wang, J.; Ito, Y.; Holmes, K.V.; Mason, R.J. Innate Immune Response of Human Alveolar Type II Cells Infected with Severe Acute Respiratory Syndrome-Coronavirus. Am. J. Respir. Cell Mol. Biol. 2013, 48, 742–748. [Google Scholar] [CrossRef]
- Chu, H.; Chan, J.F.W.; Wang, Y.; Yuen, T.T.T.; Chai, Y.; Hou, Y.; Shuai, H.; Yang, D.; Hu, B.; Huang, X.; et al. Comparative replication and immune activation profiles of SARS-CoV-2 and SARS-CoV in human lungs: An ex vivo study with implications for the pathogenesis of COVID-19. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- O’Brien, T.R.; Thomas, D.L.; Jackson, S.S.; Prokunina-Olsson, L.; Donnelly, R.P.; Hartmann, R. Weak Induction of Interferon Expression by SARS-CoV-2 Supports Clinical Trials of Interferon Lambda to Treat Early COVID-19. Clin. Infect. Dis. 2020. [Google Scholar] [CrossRef]
- Felgenhauer, U.; Schoen, A.; Gad, H.H.; Hartmann, R.; Schaubmar, A.R.; Failing, K.; Drosten, C.; Weber, F. Inhibition of SARS-CoV-2 by type I and type III interferons. J. Biol. Chem. 2020. [Google Scholar] [CrossRef] [PubMed]
- Stanifer, M.L.; Kee, C.; Cortese, M.; Zumaran, C.M.; Triana, S.; Mukenhirn, M.; Kraeusslich, H.G.; Alexandrov, T.; Bartenschlager, R.; Boulant, S. Critical Role of Type III Interferon in Controlling SARS-CoV-2 Infection in Human Intestinal Epithelial Cells. Cell Rep. 2020, 32, 107863. [Google Scholar] [CrossRef] [PubMed]
- Villenave, R.; Broadbent, L.; Douglas, I.; Lyons, J.D.; Coyle, P.V.; Teng, M.N.; Tripp, R.A.; Heaney, L.G.; Shields, M.D.; Power, U.F. Induction and Antagonism of Antiviral Responses in Respiratory Syncytial Virus-Infected Pediatric Airway Epithelium. J. Virol. 2015, 89, 12309–12318. [Google Scholar] [CrossRef] [PubMed]
- Essaidi-Laziosi, M.; Geiser, J.; Huang, S.; Constant, S.; Kaiser, L.; Tapparel, C. Interferon-Dependent and Respiratory Virus-Specific Interference in Dual Infections of Airway Epithelia. Sci. Rep. 2020, 10, 10246. [Google Scholar] [CrossRef]
- Srinivas, S.; Dai, J.; Eskdale, J.; Gallagher, G.E.; Megjugorac, N.J.; Gallagher, G. Interferon-λ1 (interleukin-29) preferentially down-regulates interleukin-13 over other T helper type 2 cytokine responses in vitro. Immunology 2008, 125, 492–502. [Google Scholar] [CrossRef]
- Li, Y.; Gao, Q.; Yuan, X.; Zhou, M.; Peng, X.; Liu, X.; Zheng, X.; Xu, D.; Li, M. Adenovirus expressing IFN-λ1 (IL-29) attenuates allergic airway inflammation and airway hyperreactivity in experimental asthma. Int. Immunopharmacol. 2014, 21, 156–162. [Google Scholar] [CrossRef]
- Yu, R.Y.; Dai, J.; Megjugorac, N.J.; Gallagher, G.E.; Yu, R.Y.L.; Gallagher, G. IFN-1 (IL-29) inhibits GATA3 expression and suppresses Th2 responses in human naive and memory T cells. Blood J. Am. Soc. Hematol. 2009. [Google Scholar] [CrossRef]
- Wareing, M.D.; Shea, A.L.; Inglis, C.A.; Dias, P.B.; Sarawar, S.R. CXCR2 is required for neutrophil recruitment to the lung during influenza virus infection, but is not essential for viral clearance. Viral Immunol. 2007, 20, 369–378. [Google Scholar] [CrossRef]
- Suzuki, Y.; Ito, T.; Suzuki, T.; Holland, R.E.; Chambers, T.M.; Kiso, M.; Ishida, H.; Kawaoka, Y. Sialic Acid Species as a Determinant of the Host Range of Influenza A Viruses. J. Virol. 2000, 74, 11825–11831. [Google Scholar] [CrossRef]
- Tate, M.D.; Pickett, D.L.; van Rooijen, N.; Brooks, A.G.; Reading, P.C. Critical Role of Airway Macrophages in Modulating Disease Severity during Influenza Virus Infection of Mice. J. Virol. 2010, 84, 7569–7580. [Google Scholar] [CrossRef]
- Mason, R.J. Biology of alveolar type II cells. Respirology 2006, 11, S12–S15. [Google Scholar] [CrossRef]
- Iwasaki, A.; Pillai, P.S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 2014, 14, 315–328. [Google Scholar] [CrossRef]
- Cole, S.L.; Ho, L.P. Contribution of innate immune cells to pathogenesis of severe influenza virus infection. Clin. Sci. 2017, 131, 269–283. [Google Scholar] [CrossRef]
- Manicassamy, B.; Manicassamy, S.; Belicha-Villanueva, A.; Pisanelli, G.; Pulendran, B.; García-Sastre, A. Analysis of in vivo dynamics of influenza virus infection in mice using a GFP reporter virus. Proc. Natl. Acad. Sci. USA 2010, 107, 11531–11536. [Google Scholar] [CrossRef]
- Chan, M.C.W.; Cheung, C.Y.; Chui, W.H.; Tsao, G.S.W.; Nicholls, J.M.; Chan, Y.O.; Chan, R.W.Y.; Long, H.T.; Poon, L.L.M.; Guan, Y.; et al. Proinflammatory cytokine responses induced by influenza A (H5N1) viruses in primary human alveolar and bronchial epithelial cells. Respir. Res. 2005, 6, 135. [Google Scholar] [CrossRef]
- Schmidt, N.; Domingues, P.; Golebiowski, F.; Patzina, C.; Tatham, M.H.; Hay, R.T.; Hale, B.G. An influenza virus-triggered SUMO switch orchestrates co-opted endogenous retroviruses to stimulate host antiviral immunity. Proc. Natl. Acad. Sci. USA 2019, 116, 17399–17408. [Google Scholar] [CrossRef]
- Crotta, S.; Davidson, S.; Mahlakoiv, T.; Desmet, C.J.; Buckwalter, M.R.; Albert, M.L.; Staeheli, P.; Wack, A. Type I and Type III Interferons Drive Redundant Amplification Loops to Induce a Transcriptional Signature in Influenza-Infected Airway Epithelia. PLoS Pathog. 2013, 9, e1003773. [Google Scholar] [CrossRef]
- Davidson, S.; McCabe, T.M.; Crotta, S.; Gad, H.H.; Hessel, E.M.; Beinke, S.; Hartmann, R.; Wack, A. IFN λ is a potent anti-influenza therapeutic without the inflammatory side effects of IFN α treatment. EMBO Mol. Med. 2016, 8, 1099–1112. [Google Scholar] [CrossRef]
- Mordstein, M.; Kochs, G.; Dumoutier, L.; Renauld, J.C.; Paludan, S.R.; Klucher, K.; Staeheli, P. Interferon-λ contributes to innate immunity of mice against influenza A virus but not against hepatotropic viruses. PLoS Pathog. 2008, 4, e1000151. [Google Scholar] [CrossRef]
- Peterson, S.T.; Kennedy, E.A.; Brigleb, P.H.; Taylor, G.M.; Urbanek, K.; Bricker, T.L.; Lee, S.; Shin, H.; Dermody, T.S.; Boon, A.C.M.; et al. Disruption of Type III Interferon (IFN) Genes Ifnl2 and Ifnl3 Recapitulates Loss of the Type III IFN Receptor in the Mucosal Antiviral Response. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Galani, I.E.; Triantafyllia, V.; Eleminiadou, E.E.; Koltsida, O.; Stavropoulos, A.; Manioudaki, M.; Thanos, D.; Doyle, S.E.; Kotenko, S.V.; Thanopoulou, K.; et al. Interferon-λ Mediates Non-redundant Front-Line Antiviral Protection against Influenza Virus Infection without Compromising Host Fitness. Immunity 2017, 46, 875–890. [Google Scholar] [CrossRef]
- Guo, X.; Zhi, J.; Thomas, P.G. New fronts emerge in the influenza cytokine storm. Semin. Immunopathol. 2017, 39, 541–550. [Google Scholar] [CrossRef]
- Klinkhammer, J.; Schnepf, D.; Ye, L.; Schwaderlapp, M.; Gad, H.H.; Hartmann, R.; Garcin, D.; Mahlakõiv, T.; Staeheli, P. IFN-λ prevents influenza virus spread from the upper airways to the lungs and limits virus transmission. Elife 2018, 7, e33354. [Google Scholar] [CrossRef]
- Ramos, I.; Smith, G.; Ruf-Zamojski, F.; Martínez-Romero, C.; Fribourg, M.; Carbajal, E.A.; Hartmann, B.M.; Nair, V.D.; Marjanovic, N.; Monteagudo, P.L.; et al. Innate Immune Response to Influenza Virus at Single-Cell Resolution in Human Epithelial Cells Revealed Paracrine Induction of Interferon Lambda 1. J. Virol. 2019, 93, e00559-19. [Google Scholar] [CrossRef]
- Wu, W.; Zhang, W.; Duggan, E.S.; Booth, J.L.; Zou, M.H.; Metcalf, J.P. RIG-I and TLR3 are both required for maximum interferon induction by influenza virus in human lung alveolar epithelial cells. Virology 2015, 482, 181–188. [Google Scholar] [CrossRef]
- Švancarová, P.; Svetlíková, D.; Betáková, T. Induction of interferon lambda in influenza a virus infected cells treated with shRNAs against M1 transcript. Acta Virol. 2015, 59, 148. [Google Scholar] [CrossRef][Green Version]
- Niwa, M.; Fujisawa, T.; Mori, K.; Yamanaka, K.; Yasui, H.; Suzuki, Y.; Karayama, M.; Hozumi, H.; Furuhashi, K.; Enomoto, N.; et al. IL-17A Attenuates IFN-λ Expression by Inducing Suppressor of Cytokine Signaling Expression in Airway Epithelium. J. Immunol. 2018, 201, 2392–2402. [Google Scholar] [CrossRef]
- Saade, G.; Deblanc, C.; Bougon, J.; Marois-Créhan, C.; Fablet, C.; Auray, G.; Belloc, C.; Leblanc-Maridor, M.; Gagnon, C.A.; Zhu, J.; et al. Coinfections and their molecular consequences in the porcine respiratory tract. Vet. Res. 2020, 51, 80. [Google Scholar] [CrossRef]
- Kim, H.J.; Jo, A.; Jeon, Y.J.; An, S.; Lee, K.M.; Yoon, S.S.; Choi, J.Y. Nasal commensal Staphylococcus epidermidis enhances interferon-λ-dependent immunity against influenza virus. Microbiome 2019, 7, 80. [Google Scholar] [CrossRef]
- Knosp, C.A.; Johnston, J.A. Regulation of CD4+ T-cell polarization by suppressor of cytokine signalling proteins. Immunology 2012, 135, 101–111. [Google Scholar] [CrossRef]
- Skorvanova, L.; Svancarova, P.; Svetlikova, D.; Betakova, T. Protective efficacy of IFN-ω AND IFN-λs against influenza viruses in induced A549 cells. Acta Virol. 2015, 59, 4137. [Google Scholar] [CrossRef]
- Lachová, V.; Škorvanová, L.; Svetlíková, D.; Turianová, L.; Kostrábová, A.; Betáková, T. Comparison of transcriptional profiles of interferons, CXCL10 and RIG-1 in influenza infected A549 cells stimulated wiThexogenous interferons. Acta Virol. 2017, 61, 183. [Google Scholar] [CrossRef][Green Version]
- Cao, Y.; Huang, Y.; Xu, K.; Liu, Y.; Li, X.; Xu, Y.; Zhong, W.; Hao, P. Differential responses of innate immunity triggered by different subtypes of influenza a viruses in human and avian hosts. BMC Med. Genom. 2017, 10, 70. [Google Scholar] [CrossRef]
- de Groen, R.A.; Groothuismink, Z.M.A.; Liu, B.S.; Boonstra, A. IFN- is able to augment TLR-mediated activation and subsequent function of primary human B cells. J. Leukoc. Biol. 2015, 98, 623–630. [Google Scholar] [CrossRef]
- Ye, L.; Schnepf, D.; Becker, J.; Ebert, K.; Tanriver, Y.; Bernasconi, V.; Gad, H.H.; Hartmann, R.; Lycke, N.; Staeheli, P. Interferon-λ enhances adaptive mucosal immunity by boosting release of thymic stromal lymphopoietin. Nat. Immunol. 2019, 20, 593–601. [Google Scholar] [CrossRef]
- Hemann, E.A.; Green, R.; Turnbull, J.B.; Langlois, R.A.; Savan, R.; Gale, M. Interferon-λ modulates dendritic cells to facilitate T cell immunity during infection with influenza A virus. Nat. Immunol. 2019, 20, 1035–1045. [Google Scholar] [CrossRef]
- Fox, J.M.; Crabtree, J.M.; Sage, L.K.; Tompkins, S.M.; Tripp, R.A. Interferon Lambda Upregulates IDO1 Expression in Respiratory Epithelial Cells after Influenza Virus Infection. J. Int. Cytokine Res. 2015, 35, 554–562. [Google Scholar] [CrossRef]
- Wang, Y.; Li, T.; Chen, Y.; Wei, H.; Sun, R.; Tian, Z. Involvement of NK Cells in IL-28B–Mediated Immunity against Influenza Virus Infection. J. Immunol. 2017, 199, 1012–1020. [Google Scholar] [CrossRef]
- Davidson, S.; Crotta, S.; McCabe, T.M.; Wack, A. Pathogenic potential of interferon αβ in acute influenza infection. Nat. Commun. 2014, 5, 3864. [Google Scholar] [CrossRef]
- Jeon, Y.J.; Lim, J.H.; An, S.; Jo, A.; Han, D.H.; Won, T.B.; Kim, D.Y.; Rhee, C.S.; Kim, H.J. Type III interferons are critical host factors that determine susceptibility to Influenza A viral infection in allergic nasal mucosa. Clin. Exp. Allergy 2018, 48, 253–265. [Google Scholar] [CrossRef]
- Planet, P.J.; Parker, D.; Cohen, T.S.; Smith, H.; Leon, J.D.; Ryan, C.; Hammer, T.J.; Fierer, N.; Chen, E.I.; Prince, A.S. Lambda interferon restructures the nasal microbiome and increases susceptibility to Staphylococcus aureus superinfection. MBio 2016, 7. [Google Scholar] [CrossRef]
- Rich, H.E.; McCourt, C.C.; Zheng, W.Q.; McHugh, K.J.; Robinson, K.M.; Wang, J.; Alcorna, J.F. Interferon lambda inhibits bacterial uptake during influenza superinfection. Infect. Immun. 2019, 87. [Google Scholar] [CrossRef]
- Robertson, C.M.; Perrone, E.E.; McConnell, K.W.; Dunne, W.M.; Boody, B.; Brahmbhatt, T.; Diacovo, M.J.; Van Rooijen, N.; Hogue, L.A.; Cannon, C.L.; et al. Neutrophil Depletion Causes a Fatal Defect in Murine Pulmonary Staphylococcus aureus clearance. J. Surg. Res. 2008, 150, 278–285. [Google Scholar] [CrossRef]


{kind=link}
| Virus Family Common Names | Virus Genome | Infected Cells Expressing IFN-λ and IFN λR | Effects of IFN-λ |
|---|---|---|---|
| Myxoviridae Influenza A Influenza B | − strand ssRNA | Respiratory epithelia, keratinocytes, mDC and pDC, hepatocytes and primary neuronal cells; NOT macrophage | IFN-λ decreases influenza virus replication in a dose-dependent manner in respiratory and gastrointestinal epithelial cells by up-regulating ISG (MX1, OAS, IFITM1) [61]. IFN-λ is more anti-proliferative and anti-inflammatory than IFN α/β [62]. Anti-proliferative effects due to up-regulation of p53 can increase susceptibility to bacterial pathogens [62]. |
| Paramyxoviridae Resp. syncitial virus (RSV); Metapneumovirus Measles virus | − strand ssRNA | Respiratory epithelia | Mice treated with IFN-λ2 and -λ3 had decreased viral titers, less pulmonary inflammation, and higher survival rates [5]. Metapneumovirus replication is attenuated in DC through MDA-5-mediated IFN response [44]. IFN-λ restricts measles replication in lung epithelial cells [63]. |
| Arenaviridae Lymphocytic choriomeningitis virus (LCMV) | − strand ssRNA | Respiratory epithelia, DC | IFN-λ2 and -λ3 elicit an antiviral effect against LCMV in lung cell culture [64]. |
| Flaviviridae Hepatitis C Dengue virus | + strand ssRNA | Primary hepatocytes DC and Lung epithelial cells | Successful Heptitis C treatment is associated with human genetic SNPs in IFN-λ3 promoter [65] and in IFN-λ4 [28]. IFN-λ1 reduced DC migration by reducing CCR-7 expr [66]. IFN-λ1 and -λ2 increase antiviral ISG (OAS and Mx1) and thus decrease virus loads [67]. |
| Caliciviridae Norovirus (NoV) | + strand ssRNA | Intestinal epithelia | IFN-λ clears persistent NoV, affects gut microbiota, and prevents transmission of acute NoV [68]. |
| Picornaviridae Rhinovirus | + strand ssRNA | Respiratory epithelia, DC | IFN-λ decreases rhinovirus replication and the asthmatic effects of rhinovirus. In a murine model for asthma, treatment with IFN-λ2 reduces Th2, eosinophils and neutrophils in bronchial fluid [30,41,42]. |
| Picornaviridae Coxsackie virus | + strand ssRNA | Primary human hepatocytes | Coxsackie titers were 10–100X lower in IFN-λ-treated cells [69]. |
| Coronaviridae MERS-CoV SARS-CoV-1 and -2 | + strand ssRNA | Respiratory epithelia | The coronaviruses induce little type I or type III IFN, but treatment with PEGylated IFN-λ1 decreased SARS-CoV-2 titers [70]. |
| Herpesviridae Cytomegalo- virus (CMV) Herpes (HSV-1 HSV-2) | dsDNA | CMV infects human foreskin fibroblasts. HSV infects buccal or genital mucosa | IFN-λ3 lowers infection with CMV [49]. IFN-λ lowers infection with HSV-1 [48]. |
| Hepadnaviridae Hepatitis B (HBV) | + strand ssDNA | HBV infects primary hepatocytes. | HBV infection up-regulates expression of IFN-λ [26]. IFN-λ1 significantly reduced viral load during infection with HBV [71]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lozhkov, A.A.; Klotchenko, S.A.; Ramsay, E.S.; Moshkoff, H.D.; Moshkoff, D.A.; Vasin, A.V.; Salvato, M.S. The Key Roles of Interferon Lambda in Human Molecular Defense against Respiratory Viral Infections. Pathogens 2020, 9, 989. https://doi.org/10.3390/pathogens9120989
Lozhkov AA, Klotchenko SA, Ramsay ES, Moshkoff HD, Moshkoff DA, Vasin AV, Salvato MS. The Key Roles of Interferon Lambda in Human Molecular Defense against Respiratory Viral Infections. Pathogens. 2020; 9(12):989. https://doi.org/10.3390/pathogens9120989
Chicago/Turabian StyleLozhkov, Alexey A., Sergey A. Klotchenko, Edward S. Ramsay, Herman D. Moshkoff, Dmitry A. Moshkoff, Andrey V. Vasin, and Maria S. Salvato. 2020. "The Key Roles of Interferon Lambda in Human Molecular Defense against Respiratory Viral Infections" Pathogens 9, no. 12: 989. https://doi.org/10.3390/pathogens9120989
APA StyleLozhkov, A. A., Klotchenko, S. A., Ramsay, E. S., Moshkoff, H. D., Moshkoff, D. A., Vasin, A. V., & Salvato, M. S. (2020). The Key Roles of Interferon Lambda in Human Molecular Defense against Respiratory Viral Infections. Pathogens, 9(12), 989. https://doi.org/10.3390/pathogens9120989