Organization of the Influenza A Virus Genomic RNA in the Viral Replication Cycle—Structure, Interactions, and Implications for the Emergence of New Strains



Abstract
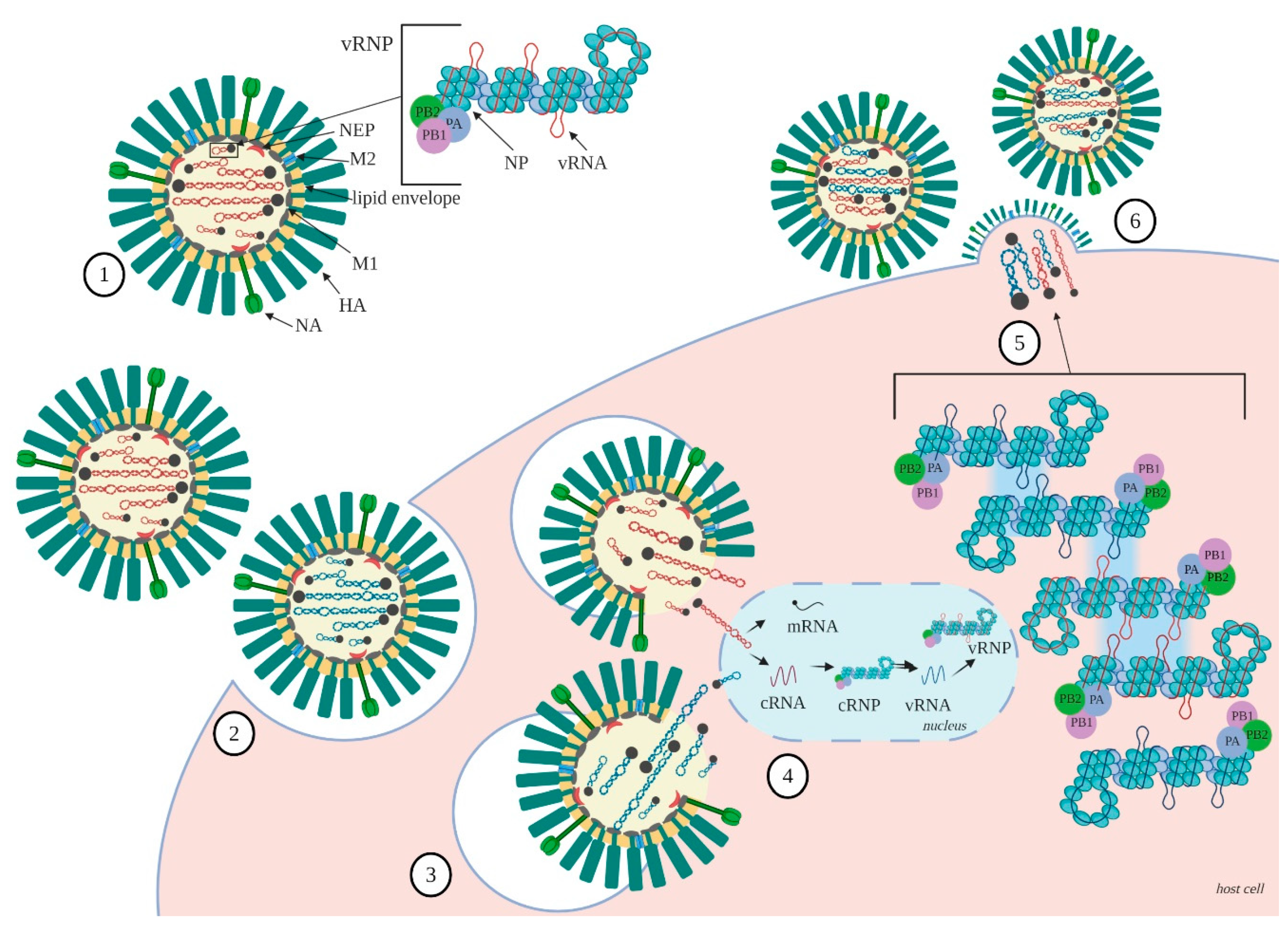
1. Introduction
2. Organization of the Influenza A Virus RNA
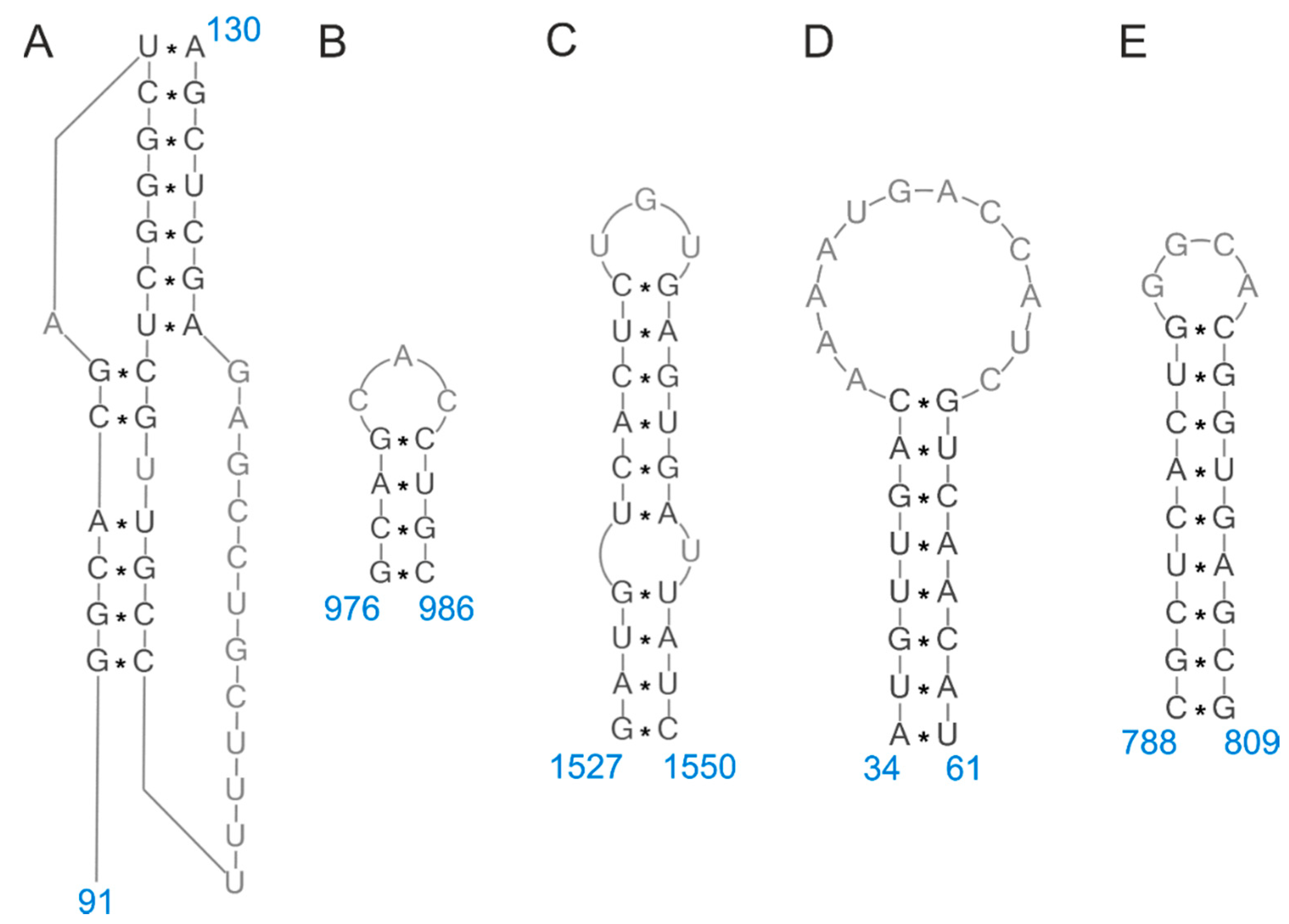
2.1. RNA Structure
2.2. NP—RNA Association
3. RNA Interactions
4. Implications for the Emergence of New Strains
4.1. Co-Segregation of PB1 in H3N2 and H1N1 Strains
4.2. Other Examples of Gene Co-Selection
4.3. Seasonal H3N2 Reassortment Potential
4.4. Role of the NP In Virion Production
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Palese, P.; Shaw, M.L. Fields of Virology, 5th ed.; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007. [Google Scholar]
- Hutchinson, E.C.; Charles, P.D.; Hester, S.S.; Thomas, B.; Trudgian, D.; Martínez-Alonso, M.; Fodor, E. Conserved and host-specific features of influenza virion architecture. Nat. Commun. 2014, 5, 4816. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-W.; Saif, Y.M. Avian influenza virus. Comp. Immunol. Microbiol. Infect. Dis. 2009, 32, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Arranz, R.; Coloma, R.; Chichon, F.J.; Conesa, J.J.; Carrascosa, J.L.; Valpuesta, J.M.; Ortin, J.; Martin-Benito, J. The structure of native influenza virion ribonucleoproteins. Science 2012, 338, 1634–1637. [Google Scholar] [CrossRef] [PubMed]
- Coloma, R.; Valpuesta, J.M.; Arranz, R.; Carrascosa, J.L.; Ortín, J.; Martín-Benito, J. The structure of a biologically active influenza virus ribonucleoprotein complex. PLoS Pathog. 2009, 5, e1000491. [Google Scholar] [CrossRef] [PubMed]
- Wandzik, J.M.; Kouba, T.; Karuppasamy, M.; Pflug, A.; Drncova, P.; Provaznik, J.; Azevedo, N.; Cusack, S. A structure-based model for the complete transcription cycle of influenza polymerase. Cell 2020, 181, 877–893.e21. [Google Scholar] [CrossRef] [PubMed]
- Pflug, A.; Guilligay, D.; Reich, S.; Cusack, S. Structure of influenza A polymerase bound to the viral RNA promoter. Nature 2014, 516, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Ferhadian, D.; Contrant, M.; Printz-Schweigert, A.; Smyth, R.P.; Paillart, J.C.; Marquet, R. Structural and functional motifs in influenza virus RNAs. Front. Microbiol. 2018, 9, 1–11. [Google Scholar] [CrossRef]
- Dadonaite, B.; Gilbertson, B.; Knight, M.L.; Trifkovic, S.; Rockman, S.; Laederach, A.; Brown, L.E.; Fodor, E.; Bauer, D.L.V. The structure of the influenza A virus genome. Nat. Microbiol. 2019, 4, 1781–1789. [Google Scholar] [CrossRef]
- Gultyaev, A.P.; Fouchier, R.A.M.; Olsthoorn, R.C.L. Influenza virus RNA structure: Unique and common features. Int. Rev. Immunol. 2010, 29, 533–556. [Google Scholar] [CrossRef]
- Ruigrok, R.W.; Crépin, T.; Hart, D.J.; Cusack, S. Towards an atomic resolution understanding of the influenza virus replication machinery. Curr. Opin. Struct. Biol. 2010, 20, 104–113. [Google Scholar] [CrossRef]
- Hutchinson, E.C.; von Kirchbach, J.C.; Gog, J.R.; Digard, P. Genome packaging in influenza A virus. J. Gen. Virol. 2010, 91, 313–328. [Google Scholar] [CrossRef] [PubMed]
- Fujii, Y.; Goto, H.; Watanabe, T.; Yoshida, T.; Kawaoka, Y. Selective incorporation of influenza virus RNA segments into virions. Proc. Natl. Acad. Sci. USA 2003, 100, 2002–2007. [Google Scholar] [CrossRef] [PubMed]
- Muramoto, Y.; Takada, A.; Fujii, K.; Noda, T.; Iwatsuki-Horimoto, K.; Watanabe, S.; Horimoto, T.; Kida, H.; Kawaoka, Y. Hierarchy among viral RNA (vRNA) segments in their role in vRNA incorporation into influenza A virions. J. Virol. 2006, 80, 2318–2325. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Hong, Y.; Parslow, T.G. Cis-acting packaging signals in the influenza virus PB1, PB2, and PA genomic RNA segments. J. Virol. 2005, 79, 10348–10355. [Google Scholar] [CrossRef]
- Hutchinson, E.C.; Curran, M.D.; Read, E.K.; Gog, J.R.; Digard, P. Mutational analysis of cis-acting RNA signals in segment 7 of influenza A virus. J. Virol. 2008, 82, 11869–11879. [Google Scholar] [CrossRef]
- Marsh, G.A.; Hatami, R.; Palese, P. Specific residues of the influenza A virus hemagglutinin viral RNA are important for efficient packaging into budding virions. J. Virol. 2007, 81, 9727–9736. [Google Scholar] [CrossRef]
- Marsh, G.A.; Rabadán, R.; Levine, A.J.; Palese, P. Highly conserved regions of influenza A virus polymerase gene segments are critical for efficient viral RNA packaging. J. Virol. 2008, 82, 2295–2304. [Google Scholar] [CrossRef]
- Gavazzi, C.; Yver, M.; Isel, C.; Smyth, R.P.; Rosa-Calatrava, M.; Lina, B.; Moulès, V.; Marquet, R. A functional sequence-specific interaction between influenza A virus genomic RNA segments. Proc. Natl. Acad. Sci. USA 2013, 110, 16604–16609. [Google Scholar] [CrossRef]
- Le Sage, V.; Kanarek, J.P.; Snyder, D.J.; Cooper, V.S.; Lakdawala, S.S.; Lee, N. Mapping of influenza virus RNA-RNA interactions reveals a flexible network. Cell Rep. 2020, 31, 107823. [Google Scholar] [CrossRef]
- Fournier, E.; Moules, V.; Essere, B.; Paillart, J.C.; Sirbat, J.D.; Cavalier, A.; Rolland, J.P.; Thomas, D.; Lina, B.; Isel, C.; et al. Interaction network linking the human H3N2 influenza A virus genomic RNA segments. Vaccine 2012, 30, 7359–7367. [Google Scholar] [CrossRef]
- Bolte, H.; Rosu, M.E.; Hagelauer, E.; García-Sastre, A.; Schwemmle, M. Packaging of the influenza virus genome is governed by a plastic network of RNA- and nucleoprotein-mediated interactions. J. Virol. 2018, 93, e01861-18. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Chou, Y.-Y.; Doganay, S.; Vafabakhsh, R.; Ha, T.; Palese, P. The influenza A virus PB2, PA, NP, and M segments play a pivotal role during genome packaging. J. Virol. 2012, 86, 7043–7051. [Google Scholar] [CrossRef] [PubMed]
- Lenartowicz, E.; Kesy, J.; Ruszkowska, A.; Soszynska-Jozwiak, M.; Michalak, P.; Moss, W.N.; Turner, D.H.; Kierzek, R.; Kierzek, E. Self-folding of naked segment 8 genomic RNA of influenza a virus. PLoS ONE 2016, 11, e0148281. [Google Scholar] [CrossRef] [PubMed]
- Michalak, P.; Soszynska-Jozwiak, M.; Biala, E.; Moss, W.N.; Kesy, J.; Szutkowska, B.; Lenartowicz, E.; Kierzek, R.; Kierzek, E. Secondary structure of the segment 5 genomic RNA of influenza A virus and its application for designing antisense oligonucleotides. Sci. Rep. 2019, 9, 3801. [Google Scholar] [CrossRef]
- Ruszkowska, A.; Lenartowicz, E.; Moss, W.N.; Kierzek, R.; Kierzek, E. Secondary structure model of the naked segment 7 influenza A virus genomic RNA. Biochem. J. 2016, 473, 4327–4348. [Google Scholar] [CrossRef]
- Noble, E.; Mathews, D.H.; Chen, J.L.; Turner, D.H.; Takimoto, T.; Kim, B. Biophysical analysis of influenza A virus RNA promoter at physiological temperatures. J. Biol. Chem. 2011, 286, 22965–22970. [Google Scholar] [CrossRef]
- Pflug, A.; Lukarska, M.; Resa-Infante, P.; Reich, S.; Cusack, S. Structural insights into RNA synthesis by the influenza virus transcription-replication machine. Virus Res. 2017, 234, 103–117. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.; Zhao, L.; Cao, M.; Deng, T. Dual roles of the hemagglutinin segment-specific noncoding nucleotides in the extended duplex region of the influenza A virus RNA promoter. J. Virol. 2017, 91, e01931-16. [Google Scholar] [CrossRef]
- Anchisi, S.; Guerra, J.; Mottet-Osman, G.; Garcin, D. Mismatches in the influenza A virus RNA panhandle prevent retinoic acid-inducible gene I (RIG-I) sensing by impairing RNA/RIG-I complex formation. J. Virol. 2016, 90, 586–590. [Google Scholar] [CrossRef]
- Soszynska-Jozwiak, M.; Michalak, P.; Moss, W.N.; Kierzek, R.; Kierzek, E. A conserved secondary structural element in the coding region of the influenza a virus nucleoprotein (NP) mRNA is important for the regulation of viral proliferation. PLoS ONE 2015, 10, e0141132. [Google Scholar] [CrossRef]
- Soszynska-Jozwiak, M.; Michalak, P.; Moss, W.N.; Kierzek, R.; Kesy, J.; Kierzek, E. Influenza virus segment 5 (+)RNA-secondary structure and new targets for antiviral strategies. Sci. Rep. 2017, 7, 15041. [Google Scholar] [CrossRef] [PubMed]
- Gultyaev, A.P.; Tsyganov-Bodounov, A.; Spronken, M.I.J.; Van Der Kooij, S.; Fouchier, R.A.M.; Olsthoorn, R.C.L. RNA structural constraints in the evolution of the influenza A virus genome NP segment. RNA Biol. 2014, 11, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, M.; Fujii, K.; Muramoto, Y.; Yamada, S.; Yamayoshi, S.; Takada, A.; Goto, H.; Horimoto, T.; Kawaoka, Y. Contributions of two nuclear localization signals of influenza A virus nucleoprotein to viral replication. J. Virol. 2007, 81, 30–41. [Google Scholar] [CrossRef]
- Ozawa, M.; Maeda, J.; Iwatsuki-Horimoto, K.; Watanabe, S.; Goto, H.; Horimoto, T.; Kawaoka, Y. Nucleotide sequence requirements at the 5′ end of the influenza A virus M RNA segment for efficient virus replication. J. Virol. 2009, 83, 3384–3388. [Google Scholar] [CrossRef][Green Version]
- Gog, J.R.; Dos Santos Afonso, E.; Dalton, R.M.; Leclercq, I.; Tiley, L.; Elton, D.; von Kirchbach, J.C.; Naffakh, N.; Escriou, N.; Digard, P. Codon conservation in the influenza A virus genome defines RNA packaging signals. Nucleic Acids Res. 2007, 35, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Lenartowicz, E.; Nogales, A.; Kierzek, E.; Kierzek, R.; Martínez-Sobrido, L.; Turner, D.H. Antisense oligonucleotides targeting influenza A segment 8 genomic RNA inhibit viral replication. Nucleic Acid Ther. 2016, 26, 277–285. [Google Scholar] [CrossRef]
- Fujii, K.; Fujii, Y.; Noda, T.; Muramoto, Y.; Watanabe, T.; Takada, A.; Goto, H.; Horimoto, T.; Kawaoka, Y. Importance of both the coding and the segment-specific noncoding regions of the influenza A virus NS segment for its efficient incorporation into virions. J. Virol. 2005, 79, 3766–3774. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Dadonaite, B.; van Doremalen, N.; Suzuki, Y.; Barclay, W.S.; Pybus, O.G. Computational and molecular analysis of conserved influenza A virus RNA secondary structures involved in infectious virion production. RNA Biol. 2016, 13, 883–894. [Google Scholar] [CrossRef]
- Kesy, J.; Patil, K.M.; Kumar, S.R.; Shu, Z.; Yong, H.Y.; Zimmermann, L.; Ong, A.A.L.; Toh, D.-F.K.; Krishna, M.S.; Yang, L.; et al. A short chemically modified dsRNA-binding PNA (dbPNA) inhibits influenza viral replication by targeting viral RNA panhandle structure. Bioconjug. Chem. 2019, 30, 931–943. [Google Scholar] [CrossRef]
- Williams, G.D.; Townsend, D.; Wylie, K.M.; Kim, P.J.; Amarasinghe, G.K.; Kutluay, S.B.; Boon, A.C.M. Nucleotide resolution mapping of influenza A virus nucleoprotein-RNA interactions reveals RNA features required for replication. Nat. Commun. 2018, 9, 465. [Google Scholar] [CrossRef]
- Takizawa, N.; Higashi, K.; Kawaguchi, R.K.; Gotoh, Y.; Suzuki, Y.; Hayashi, T.; Kurokawa, K. A functional RNA structure in the influenza A virus ribonucleoprotein complex for segment bundling. bioRxiv 2020. [Google Scholar] [CrossRef]
- Takizawa, N.; Ogura, Y.; Fujita, Y.; Noda, T.; Shigematsu, H.; Hayashi, T.; Kurokawa, K. Local structural changes of the influenza A virus ribonucleoprotein complex by single mutations in the specific residues involved in efficient genome packaging. Virology 2019, 531, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Spronken, M.I.; van de Sandt, C.E.; de Jongh, E.P.; Vuong, O.; van der Vliet, S.; Bestebroer, T.M.; Olsthoorn, R.C.L.; Rimmelzwaan, G.F.; Fouchier, R.A.M.; Gultyaev, A.P. A compensatory mutagenesis study of a conserved hairpin in the M gene segment of influenza A virus shows its role in virus replication. RNA Biol. 2017, 14, 1606–1616. [Google Scholar] [CrossRef] [PubMed]
- Lee, N.; Le Sage, V.; Nanni, A.V.; Snyder, D.J.; Cooper, V.S.; Lakdawala, S.S. Genome-wide analysis of influenza viral RNA and nucleoprotein association. Nucleic Acids Res. 2017, 45, 8968–8977. [Google Scholar] [CrossRef]
- Coloma, R.; Arranz, R.; de la Rosa-Trevín, J.M.; Sorzano, C.O.S.; Munier, S.; Carlero, D.; Naffakh, N.; Ortín, J.; Martín-Benito, J. Structural insights into influenza A virus ribonucleoproteins reveal a processive helical track as transcription mechanism. Nat. Microbiol. 2020, 5, 727–734. [Google Scholar] [CrossRef]
- Noda, T.; Sagara, H.; Yen, A.; Takada, A.; Kida, H.; Cheng, R.H.; Kawaoka, Y. Architecture of ribonucleoprotein complexes in influenza A virus particles. Nature 2006, 439, 490–492. [Google Scholar] [CrossRef]
- Fournier, E.; Moules, V.; Essere, B.; Paillart, J.C.; Sirbat, J.D.; Isel, C.; Cavalier, A.; Rolland, J.P.; Thomas, D.; Lina, B.; et al. A supramolecular assembly formed by influenza A virus genomic RNA segments. Nucleic Acids Res. 2012, 40, 2197–2209. [Google Scholar] [CrossRef]
- Noda, T.; Sugita, Y.; Aoyama, K.; Hirase, A.; Kawakami, E.; Miyazawa, A.; Sagara, H.; Kawaoka, Y. Three-dimensional analysis of ribonucleoprotein complexes in influenza A virus. Nat. Commun. 2012, 3, 639. [Google Scholar] [CrossRef]
- Harris, A.; Cardone, G.; Winkler, D.C.; Heymann, J.B.; Brecher, M.; White, J.M.; Steven, A.C. Influenza virus pleiomorphy characterized by cryoelectron tomography. Proc. Natl. Acad. Sci. USA 2006, 103, 19123–19127. [Google Scholar] [CrossRef]
- Noda, T.; Murakami, S.; Nakatsu, S.; Imai, H.; Muramoto, Y.; Shindo, K.; Sagara, H.; Kawaoka, Y. Importance of the 1+7 configuration of ribonucleoprotein complexes for influenza A virus genome packaging. Nat. Commun. 2018, 9, 1–10. [Google Scholar] [CrossRef]
- Liang, Y.; Huang, T.; Ly, H.; Parslow, T.G.; Liang, Y. Mutational analyses of packaging signals in influenza virus PA, PB1, and PB2 genomic RNA segments. J. Virol. 2008, 82, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, S.; Noda, T. In vitro vRNA–vRNA interactions in the H1N1 influenza A virus genome. Microbiol. Immunol. 2020, 64, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.F.; Nogales, A.; Finch, C.; Tuffy, K.M.; Domm, W.; Perez, D.R.; Topham, D.J.; Martinez-Sobrido, L. Influenza A and B virus intertypic reassortment through compatible viral packaging signals. J. Virol. 2014, 88, 10778–10791. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos Afonso, E.; Escriou, N.; Leclercq, I.; Van Der Werf, S.; Naffakh, N. The generation of recombinant influenza A viruses expressing a PB2 fusion protein requires the conservation of a packaging signal overlapping the coding and noncoding regions at the 5′ end of the PB2 segment. Virology 2005, 341, 34–46. [Google Scholar] [CrossRef] [PubMed]
- Gavazzi, C.; Isel, C.; Fournier, E.; Moules, V.; Cavalier, A.; Thomas, D.; Lina, B.; Marquet, R. An in vitro network of intermolecular interactions between viral RNA segments of an avian H5N2 influenza A virus: Comparison with a human H3N2 virus. Nucleic Acids Res. 2013, 41, 1241–1254. [Google Scholar] [CrossRef]
- Reperant, L.A.; Grenfell, B.T.; Osterhaus, A.D.M.E. Quantifying the risk of pandemic influenza virus evolution by mutation and re-assortment. Vaccine 2015, 33, 6955–6966. [Google Scholar] [CrossRef]
- Fonville, J.M.; Marshall, N.; Tao, H.; Steel, J.; Lowen, A.C. Influenza virus reassortment is enhanced by semi-infectious particles but can be suppressed by defective interfering particles. PLoS Pathog. 2015, 11, e1005204. [Google Scholar] [CrossRef]
- Simonsen, L.; Viboud, C.; Grenfell, B.T.; Dushoff, J.; Jennings, L.; Smit, M.; Macken, C.; Hata, M.; Gog, J.; Miller, M.A.; et al. The genesis and spread of reassortment human influenza A/H3N2 viruses conferring adamantane resistance. Mol. Biol. Evol. 2007, 24, 1811–1820. [Google Scholar] [CrossRef]
- Yang, J.R.; Lin, Y.C.; Huang, Y.P.; Su, C.H.; Lo, J.; Ho, Y.L.; Yao, C.Y.; Hsu, L.C.; Wu, H.S.; Liu, M.T. Reassortment and mutations associated with emergence and spread of oseltamivir-resistant seasonal influenza A/H1N1 viruses in 2005-2009. PLoS ONE 2011, 6, e18177. [Google Scholar] [CrossRef][Green Version]
- Gilbertson, B.; Zheng, T.; Gerber, M.; Printz-Schweigert, A.; Ong, C.; Marquet, R.; Isel, C.; Rockman, S.; Brown, L. Influenza NA and PB1 gene segments interact during the formation of viral progeny: Localization of the binding region within the PB1 gene. Viruses 2016, 8, 238. [Google Scholar] [CrossRef]
- Greenbaum, B.D.; Li, O.T.W.; Poon, L.L.M.; Levine, A.J.; Rabadan, R. Viral reassortment as an information exchange between viral segments. Proc. Natl. Acad. Sci. USA 2012, 109, 3341–3346. [Google Scholar] [CrossRef] [PubMed]
- Essere, B.; Yver, M.; Gavazzi, C.; Terrier, O.; Isel, C.; Fournier, E.; Giroux, F.; Textoris, J.; Julien, T.; Socratous, C.; et al. Critical role of segment-specific packaging signals in genetic reassortment of influenza A viruses. Proc. Natl. Acad. Sci. USA 2013, 110, E3840–E3848. [Google Scholar] [CrossRef] [PubMed]
- Marshall, N.; Priyamvada, L.; Ende, Z.; Steel, J.; Lowen, A.C. Influenza virus reassortment occurs with high frequency in the absence of segment mismatch. PLoS Pathog. 2013, 9, e1003421. [Google Scholar] [CrossRef] [PubMed]
- White, M.C.; Steel, J.; Lowen, A.C. Heterologous packaging signals on segment 4, but not segment 6 or segment 8, limit influenza A virus reassortment. J. Virol. 2017, 91, 1–16. [Google Scholar] [CrossRef]
- Cobbin, J.C.A.; Verity, E.E.; Gilbertson, B.P.; Rockman, S.P.; Brown, L.E. The source of the PB1 gene in influenza vaccine reassortants selectively alters the hemagglutinin content of the resulting seed virus. J. Virol. 2013, 87, 5577–5585. [Google Scholar] [CrossRef]
- Cobbin, J.C.A.; Ong, C.; Verity, E.; Gilbertson, B.P.; Rockman, S.P.; Brown, L.E. Influenza virus PB1 and neuraminidase gene segments can cosegregate during vaccine reassortment driven by interactions in the PB1 coding region. J. Virol. 2014, 88, 8971–8980. [Google Scholar] [CrossRef]
- Bergeron, C.; Valette, M.; Lina, B.; Ottmann, M. Genetic content of influenza H3N2 vaccine seeds. PLoS Curr. 2010, 1, 2–9. [Google Scholar] [CrossRef]
- Watanabe, T.; Watanabe, S.; Noda, T.; Fujii, Y.; Kawaoka, Y. Exploitation of nucleic acid packaging signals to generate a novel influenza virus-based vector stably expressing two foreign genes. J. Virol. 2003, 77, 10575–10583. [Google Scholar] [CrossRef]
- Brooke, C.B.; Ince, W.L.; Wrammert, J.; Ahmed, R.; Wilson, P.C.; Bennink, J.R.; Yewdell, J.W. Most influenza A virions fail to express at least one essential viral protein. J. Virol. 2013, 87, 3155–3162. [Google Scholar] [CrossRef]
- Jacobs, N.T.; Onuoha, N.O.; Antia, A.; Steel, J.; Antia, R.; Lowen, A.C. Incomplete influenza A virus genomes occur frequently but are readily complemented during localized viral spread. Nat. Commun. 2019, 10, 3526. [Google Scholar] [CrossRef]
- White, M.C.; Tao, H.; Steel, J.; Lowen, A.C. H5N8 and H7N9 packaging signals constrain HA reassortment with a seasonal H3N2 influenza A virus. Proc. Natl. Acad. Sci. USA 2019, 116, 4611–4618. [Google Scholar] [CrossRef] [PubMed]
- Gao, Q.; Brydon, E.W.A.; Palese, P. A seven-segmented influenza A virus expressing the influenza C virus glycoprotein HEF. J. Virol. 2008, 82, 6419–6426. [Google Scholar] [CrossRef] [PubMed]
- Eisfeld, A.J.; Neumann, G.; Kawaoka, Y. At the centre: Influenza A virus ribonucleoproteins. Nat. Rev. Microbiol. 2015, 13, 28–41. [Google Scholar] [CrossRef] [PubMed]
- Kakisaka, M.; Yamada, K.; Yamaji-Hasegawa, A.; Kobayashi, T.; Aida, Y. Intrinsically disordered region of influenza A NP regulates viral genome packaging via interactions with viral RNA and host PI(4,5)P2. Virology 2016, 496, 116–126. [Google Scholar] [CrossRef]
- Moreira, É.A.; Weber, A.; Bolte, H.; Kolesnikova, L.; Giese, S.; Lakdawala, S.; Beer, M.; Zimmer, G.; García-Sastre, A.; Schwemmle, M.; et al. A conserved influenza A virus nucleoprotein code controls specific viral genome packaging. Nat. Commun. 2016, 7, 12861. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| vRNA Segment | Nucleotide Region | Predicted Structure | Publication | |
|---|---|---|---|---|
| (+) Sense Numbering | (−) Sense Numbering | |||
| 1 | 1823–1944 | 398–519 | motif with long helical regions and two hairpins | [41] |
| 2 | 289–309 | 2033–2053 | hairpin | [19] |
| 497–561 | 1781–1845 | hairpin | [41] | |
| 5 | 16–39 | 1527–1550 | hairpin | [9,25,33] |
| 22–68 | 1498–1544 | two hairpins | [41] | |
| 70–82 | 1484–1496 | hairpin | [9,19,25] | |
| 89–105 | 1461–1477 | hairpin | [33] | |
| 191–203 | 1363–1375 | hairpin | [9,25] | |
| 580–590 | 976–986 | hairpin | [9,25,33] | |
| 922–938 | 628–644 | hairpin | [33] | |
| 1090–1106 | 460–476 | hairpin | [9,25] | |
| 1144–1160 | 406–422 | hairpin | [9,25] | |
| 1431–1479 | 87–135 | hairpin/pseudoknot | [9,19,25,33,41,42] | |
| 1476–1530 | 36–90 | hairpin | [33] | |
| 7 | 21–63 | 965–1007 | dynamic | [16,26] |
| 219–240 | 788–809 | hairpin | [9,26,39] | |
| 249–260 | 768–779 | hairpin | [9,26] | |
| 318–333 | 695–710 | hairpin | [9,26] | |
| 443–450 | 578–585 | hairpin | [9,26] | |
| 671–691 | 337–357 | hairpin | [9,26] | |
| 857–890 | 138–171 | hairpin | [9,26] | |
| 967–994 | 34–61 | hairpin | [9,16,26,34,36,39,43,44] | |
| 8 | 22–86 | 790–854 | hairpin | [41] |
| 96–101/175–180 | 696–701/775–780 | helix | [24,38] | |
| 109–118/163–172 | 704–713/758–767 | helix | [24,38] | |
| 128–140 | 736–748 | hairpin | [24,38] | |
| 257–277 | 599–619 | hairpin | [19] | |
| 529–534/578–583 | 293–298/342–347 | helix | [9,24] | |
| 549–564 | 312–327 | hairpin | [9,24] | |
| 588–615 | 261–288 | hairpin | [9,24] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piasecka, J.; Jarmolowicz, A.; Kierzek, E. Organization of the Influenza A Virus Genomic RNA in the Viral Replication Cycle—Structure, Interactions, and Implications for the Emergence of New Strains. Pathogens 2020, 9, 951. https://doi.org/10.3390/pathogens9110951
Piasecka J, Jarmolowicz A, Kierzek E. Organization of the Influenza A Virus Genomic RNA in the Viral Replication Cycle—Structure, Interactions, and Implications for the Emergence of New Strains. Pathogens. 2020; 9(11):951. https://doi.org/10.3390/pathogens9110951
Chicago/Turabian StylePiasecka, Julita, Aleksandra Jarmolowicz, and Elzbieta Kierzek. 2020. "Organization of the Influenza A Virus Genomic RNA in the Viral Replication Cycle—Structure, Interactions, and Implications for the Emergence of New Strains" Pathogens 9, no. 11: 951. https://doi.org/10.3390/pathogens9110951
APA StylePiasecka, J., Jarmolowicz, A., & Kierzek, E. (2020). Organization of the Influenza A Virus Genomic RNA in the Viral Replication Cycle—Structure, Interactions, and Implications for the Emergence of New Strains. Pathogens, 9(11), 951. https://doi.org/10.3390/pathogens9110951