Decrease of miR-19b-3p in Brain Microvascular Endothelial Cells Attenuates Meningitic Escherichia coli-Induced Neuroinflammation via TNFAIP3-Mediated NF-κB Inhibition

 ,
, 
Abstract
1. Introduction
2. Results
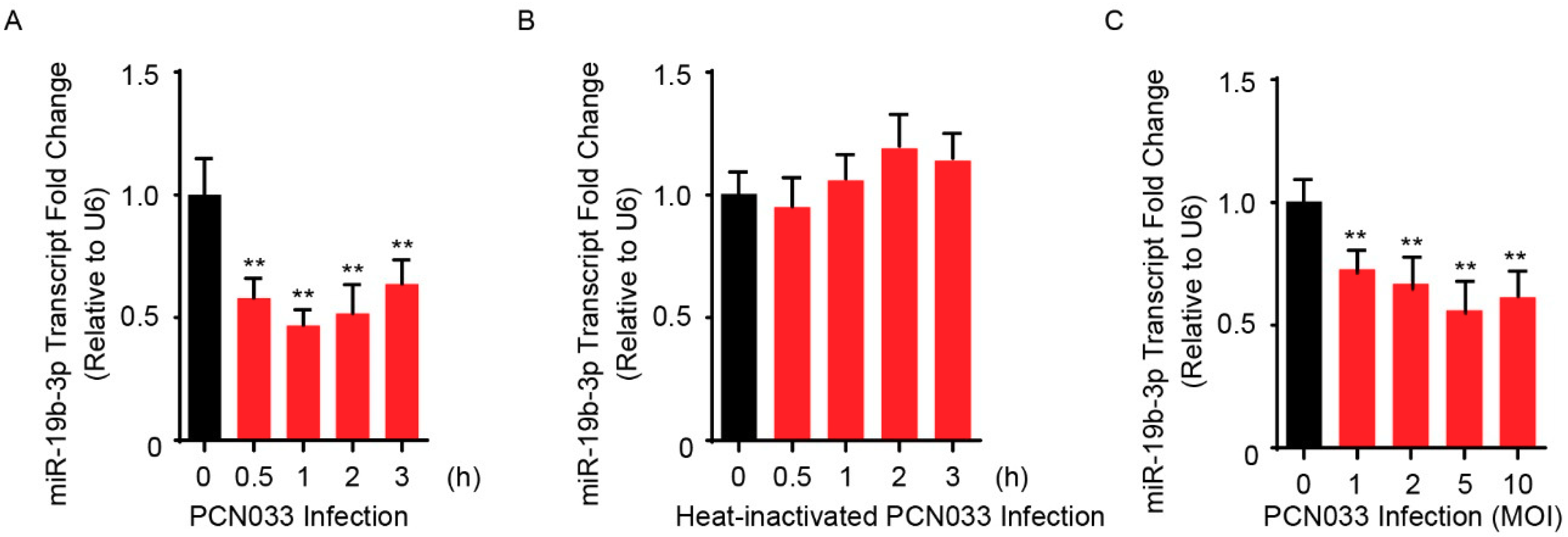
2.1. Downregulation of miR-19b-3p in Human BMECs (hBMECs) upon Meningitic E. coli PCN033 Infection
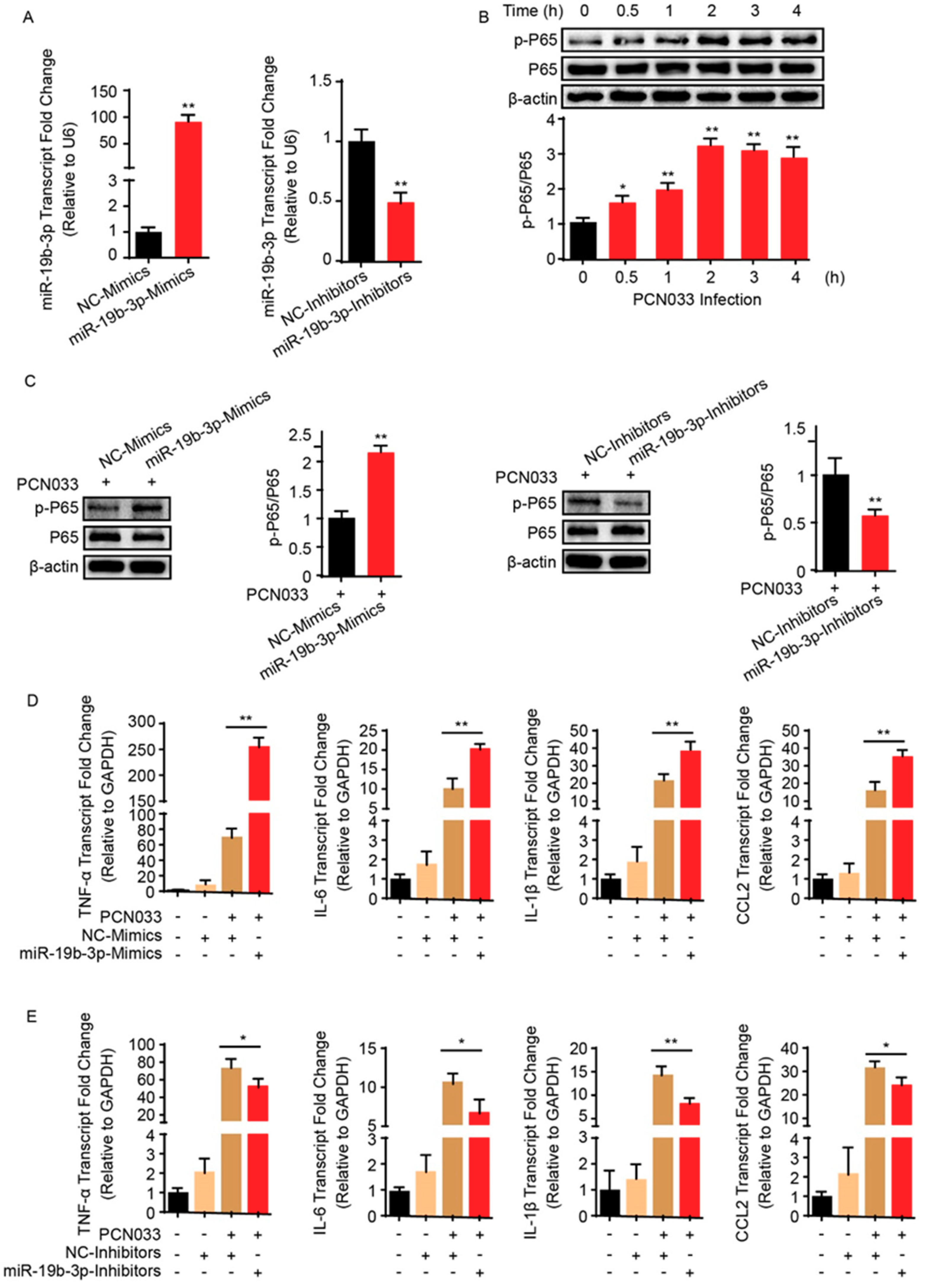
2.2. MiR-19b-3p Can Facilitate PCN033-Triggered Inflammatory Response
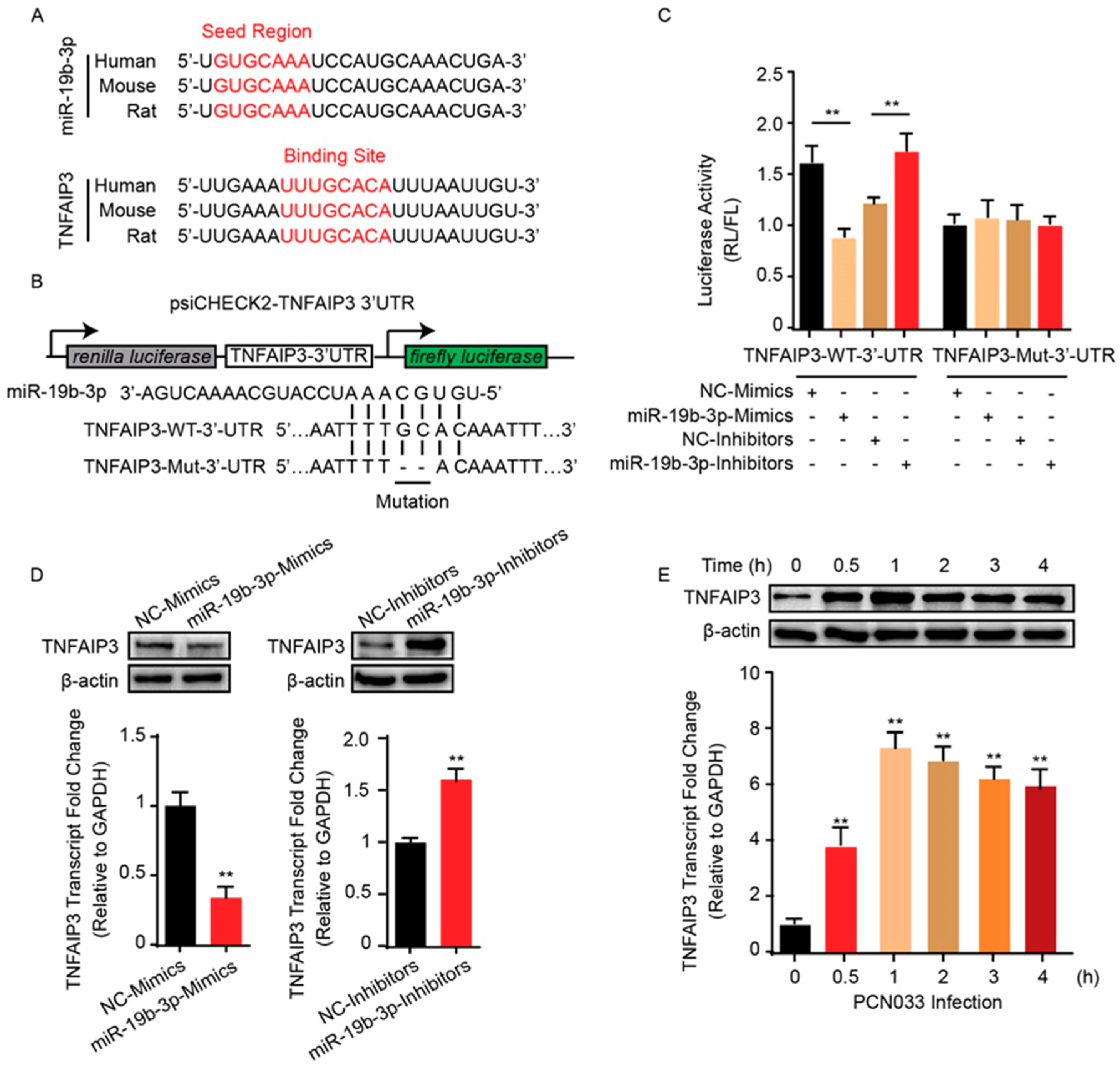
2.3. TNFAIP3 Is a Direct Target of miR-19b-3p in Meningitic E. coli Infection of hBMECs
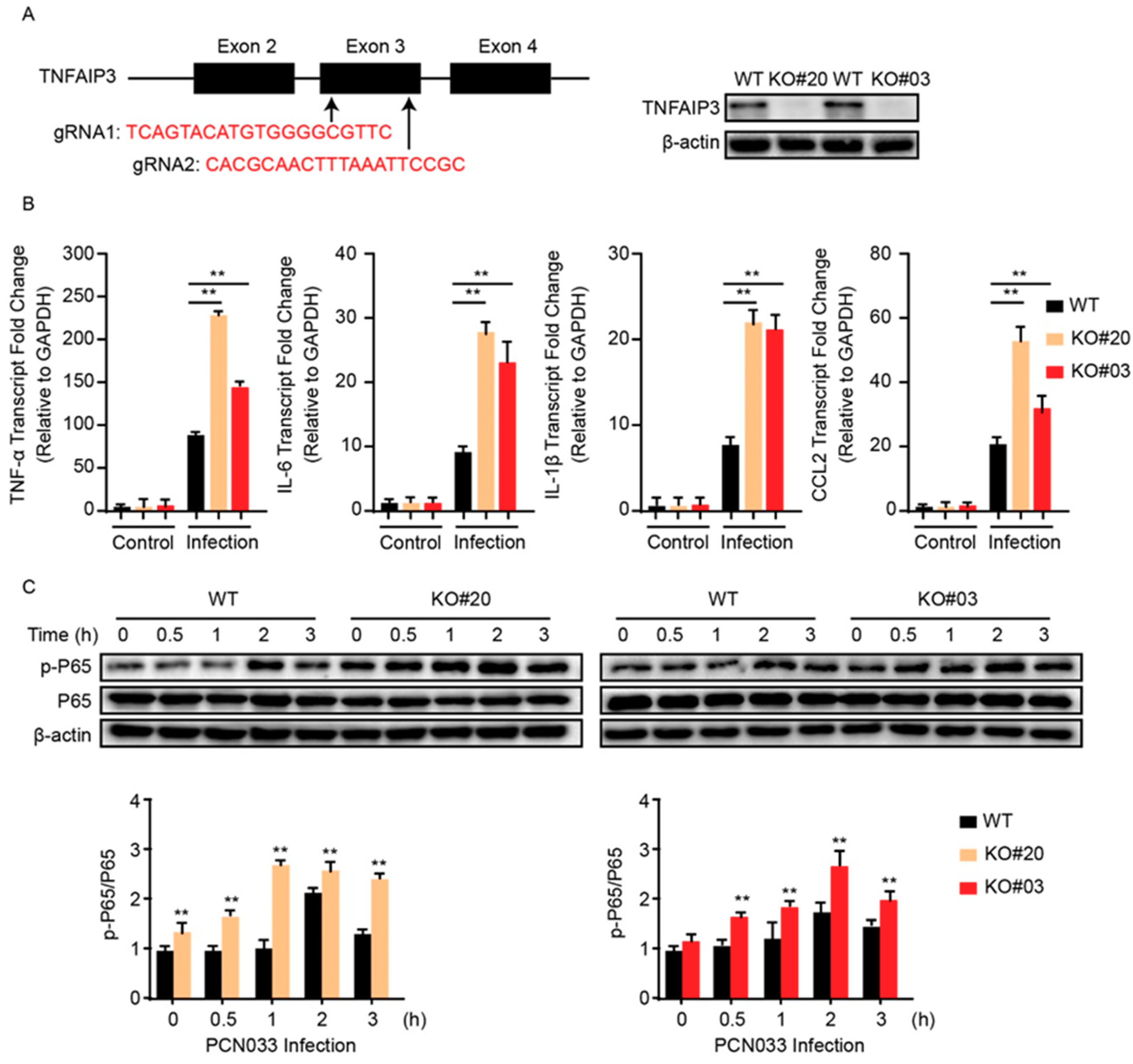
2.4. TNFAIP3 Negatively Participates the Inflammatory Responses in hBMECs
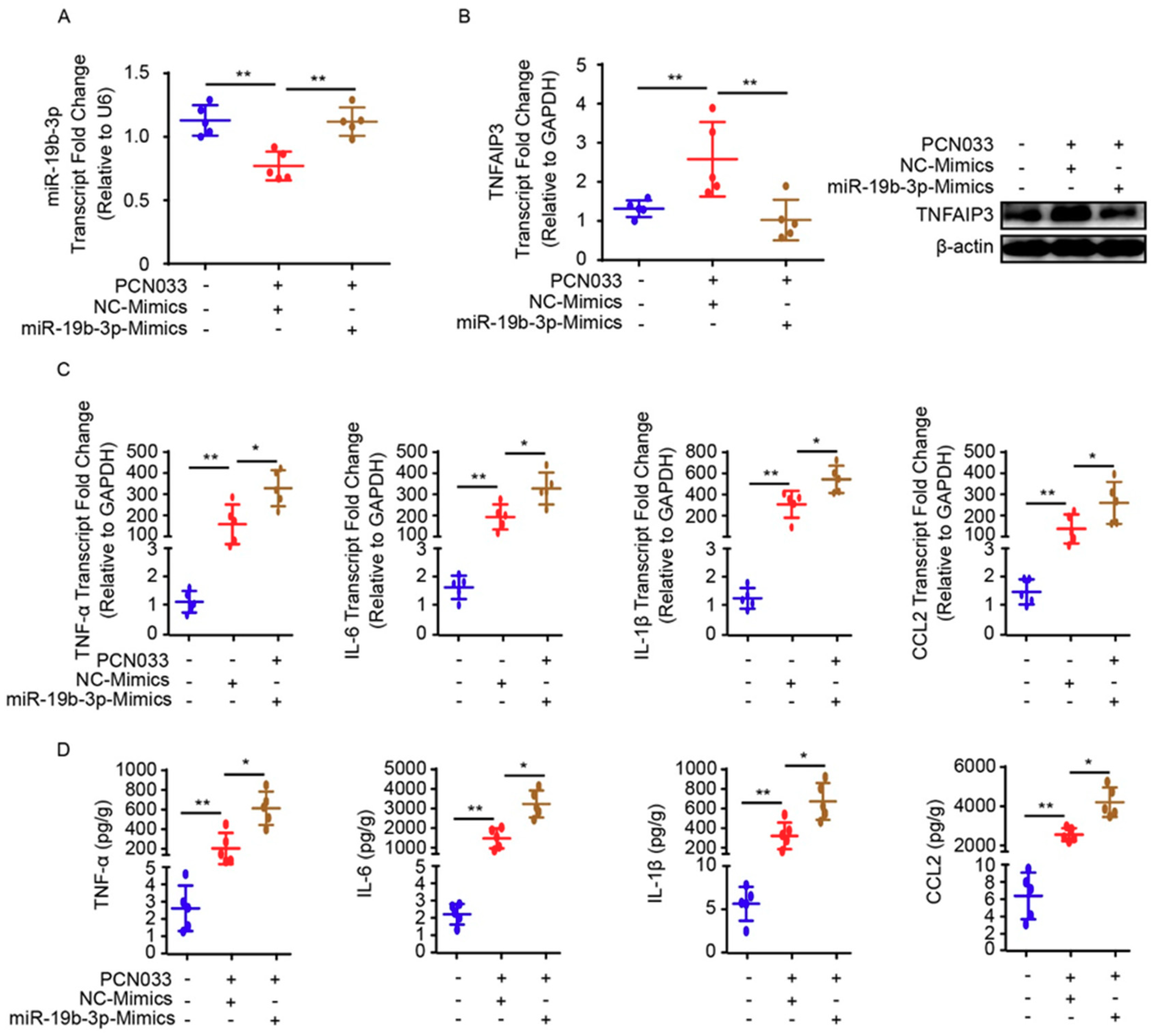
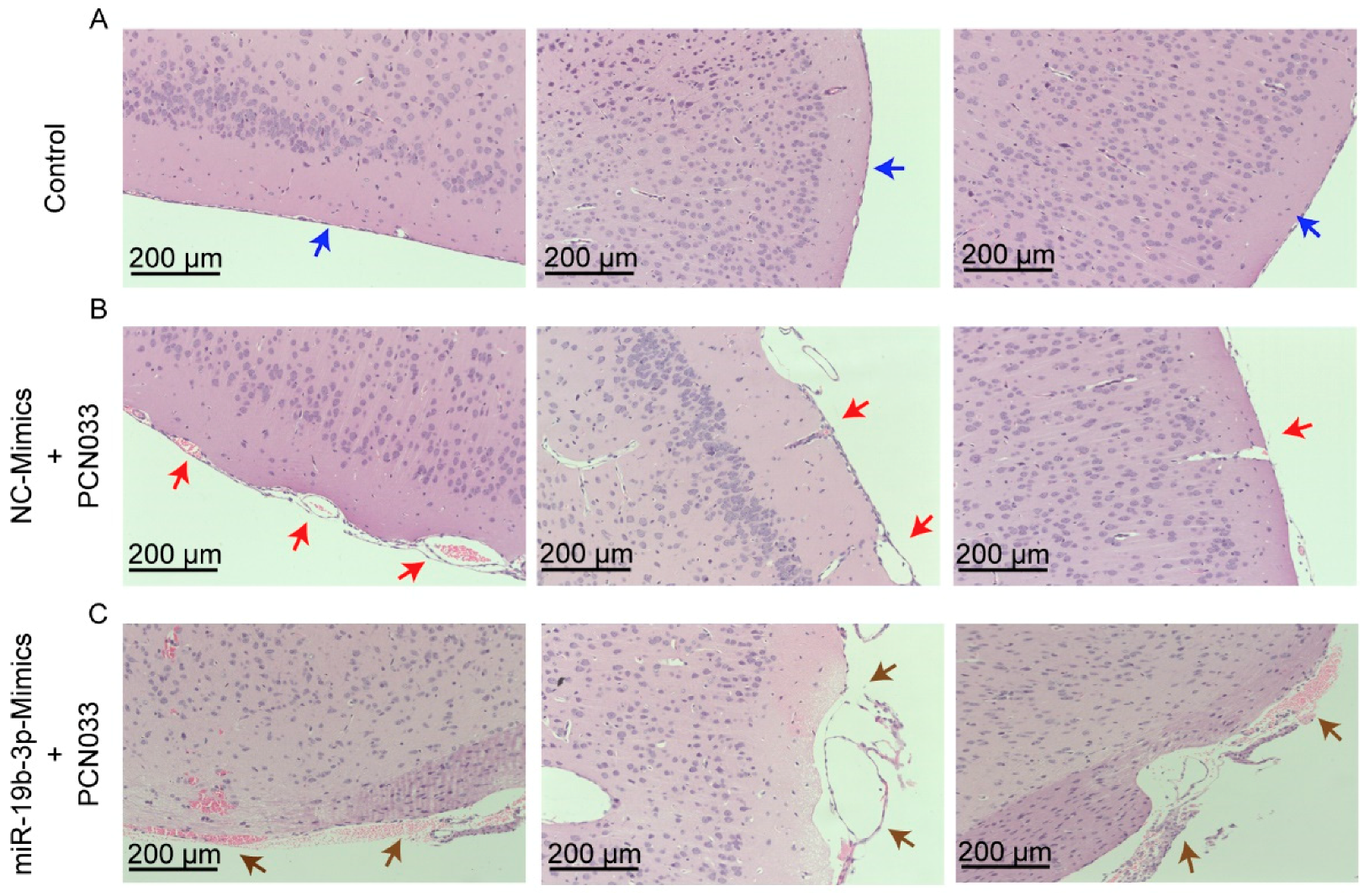
2.5. MiR-19b-3p Aggravates PCN033-Induced Inflammatory Responses in Mice
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains, Cell Culture, and Infection
4.2. Reagents and Antibodies
4.3. Plasmid Construction
4.4. Transfection
4.5. TNFAIP3 Knocking out via CRISPR/Cas9 Genomic Editing
4.6. Dual-Luciferase Reporter Assay
4.7. In Vivo Mice Infection Assays
4.8. RNA Extraction and Quantitative Real-Time PCR
4.9. Immunoblotting
4.10. Electrochemiluminescence (ECL) Assay
4.11. Histopathological Examination
4.12. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Kim, K.S. Acute bacterial meningitis in infants and children. Lancet Infect. Dis. 2010, 10, 32–42. [Google Scholar] [CrossRef]
- McGill, F.; Heyderman, R.S.; Panagiotou, S.; Tunkel, A.R.; Solomon, T. Acute bacterial meningitis in adults. Lancet 2016, 388, 3036–3047. [Google Scholar] [CrossRef]
- Kim, K.S. Pathogenesis of bacterial meningitis: From bacteraemia to neuronal injury. Nat. Rev. Neurosci. 2003, 4, 376–385. [Google Scholar] [CrossRef]
- Coureuil, M.; Lecuyer, H.; Bourdoulous, S.; Nassif, X. A journey into the brain: Insight into how bacterial pathogens cross blood-brain barriers. Nat. Rev. Microbiol. 2017, 15, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Blanchette, M.; Daneman, R. Formation and maintenance of the bbb. Mech. Dev. 2015, 138, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Carson-Walter, E.; Walter, K.A. Chemokine receptor cxcr7 is a functional receptor for cxcl12 in brain endothelial cells. PLoS ONE 2014, 9, e103938. [Google Scholar] [CrossRef]
- Yao, H.; Bethel-Brown, C.; Niu, F.; Yang, L.; Peng, F.; Buch, S. Yin and yang of pdgf-mediated signaling pathway in the context of hiv infection and drug abuse. J. Neuroimmune Pharm. 2014, 9, 161–167. [Google Scholar] [CrossRef]
- Coisne, C.; Engelhardt, B. Tight junctions in brain barriers during central nervous system inflammation. Antioxid Redox Signal. 2011, 15, 1285–1303. [Google Scholar] [CrossRef]
- Yang, X.P.; Fu, J.Y.; Yang, R.C.; Liu, W.T.; Zhang, T.; Yang, B.; Miao, L.; Dou, B.B.; Tan, C.; Chen, H.C.; et al. Egfr transactivation contributes to neuroinflammation in Streptococcus suis meningitis. J. Neuroinflammation 2016, 13, 274. [Google Scholar] [CrossRef]
- Yang, R.C.; Qu, X.Y.; Xiao, S.Y.; Li, L.; Xu, B.J.; Fu, J.Y.; Lv, Y.J.; Amjad, N.; Tan, C.; Kim, K.S.; et al. Meningitic escherichia coli-induced upregulation of pdgf-b and icam-1 aggravates blood-brain barrier disruption and neuroinflammatory response. J. Neuroinflammation 2019, 16, 101. [Google Scholar] [CrossRef]
- Bartel, D.P. Micrornas: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Swellam, M.; Ramadan, A.; El-Hussieny, E.A.; Bakr, N.M.; Hassan, N.M.; Sobeih, M.E.; EzzElArab, L.R. Clinical significance of blood-based mirnas as diagnostic and prognostic nucleic acid markers in breast cancer: Comparative to conventional tumor markers. J. Cell. Biochem. 2019, 120, 12321–12330. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Zhu, L.; Qiu, C.; Xu, X.; Zhang, L.; Ding, X.; Liao, Q.; Xu, J.; Zhang, X. Microrna mir-126-5p enhances the inflammatory responses of monocytes to lipopolysaccharide stimulation by suppressing cylindromatosis in chronic hiv-1 infection. J. Virol. 2017, 91, e20248-16. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Yu, T.; Shi, Y.; Ma, C.; Yang, W.; Fang, L.; Sun, M.; Wu, W.; Xiao, F.; Guo, F.; et al. Microrna 301a promotes intestinal inflammation and colitis-associated cancer development by inhibiting btg1. Gastroenterology 2017, 152, 1434–1448. [Google Scholar] [CrossRef] [PubMed]
- Zhan, C.Y.; Chen, D.; Luo, J.L.; Shi, Y.H.; Zhang, Y.P. Protective role of down-regulated microrna-31 on intestinal barrier dysfunction through inhibition of nf-kappab/hif-1alpha pathway by binding to hmox1 in rats with sepsis. Mol. Med. 2018, 24, 55. [Google Scholar] [CrossRef] [PubMed]
- Kalantari, P.; Harandi, O.F.; Agarwal, S.; Rus, F.; Kurt-Jones, E.A.; Fitzgerald, K.A.; Caffrey, D.R.; Golenbock, D.T. Mir-718 represses proinflammatory cytokine production through targeting phosphatase and tensin homolog (pten). J. Biol. Chem. 2017, 292, 5634–5644. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Li, X.; Ye, Y.; Zhao, K.; Zhuang, Y.; Li, Y.; Wei, Y.; Wu, M. Microrna-302b augments host defense to bacteria by regulating inflammatory responses via feedback to tlr/irak4 circuits. Nat. Commun. 2014, 5, 3619. [Google Scholar] [CrossRef]
- Kumar, M.; Sahu, S.K.; Kumar, R.; Subuddhi, A.; Maji, R.K.; Jana, K.; Gupta, P.; Raffetseder, J.; Lerm, M.; Ghosh, Z.; et al. Microrna let-7 modulates the immune response to mycobacterium tuberculosis infection via control of a20, an inhibitor of the nf-kappab pathway. Cell Host Microbe 2015, 17, 345–356. [Google Scholar] [CrossRef]
- Yang, R.; Huang, F.; Fu, J.; Dou, B.; Xu, B.; Miao, L.; Liu, W.; Yang, X.; Tan, C.; Chen, H.; et al. Differential transcription profiles of long non-coding rnas in primary human brain microvascular endothelial cells in response to meningitic escherichia coli. Sci. Rep. 2016, 6, 38903. [Google Scholar] [CrossRef]
- Gantier, M.P.; Stunden, H.J.; McCoy, C.E.; Behlke, M.A.; Wang, D.; Kaparakis-Liaskos, M.; Sarvestani, S.T.; Yang, Y.H.; Xu, D.; Corr, S.C.; et al. A mir-19 regulon that controls nf-kappab signaling. Nucleic Acids Res. 2012, 40, 8048–8058. [Google Scholar] [CrossRef]
- Yang, R.; Liu, W.; Miao, L.; Yang, X.; Fu, J.; Dou, B.; Cai, A.; Zong, X.; Tan, C.; Chen, H.; et al. Induction of vegfa and snail-1 by meningitic Escherichia coli mediates disruption of the blood-brain barrier. Oncotarget 2016, 7, 63839–63855. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, W.T.; Lv, Y.J.; Yang, R.C.; Fu, J.Y.; Liu, L.; Wang, H.; Cao, Q.; Tan, C.; Chen, H.C.; Wang, X.R. New insights into meningitic Escherichia coli infection of brain microvascular endothelial cells from quantitative proteomics analysis. J. Neuroinflammation 2018, 15, 291. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Yin, L.; Wu, J.; Gu, J.J.; Wu, J.Z.; Chen, D.; Yu, H.L.; Ding, K.; Zhang, N.; Du, M.Y.; et al. Microrna-19b-3p regulates nasopharyngeal carcinoma radiosensitivity by targeting tnfaip3/nf-kappab axis. J. Exp. Clin. Cancer Res. 2016, 35, 188. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, U.; Zhu, B.; Ye, J.; Wan, S.; Nie, Y.; Chen, Z.; Cui, M.; Wang, C.; Duan, X.; Zhang, H.; et al. Microrna-19b-3p modulates japanese encephalitis virus-mediated inflammation via targeting rnf11. J. Virol. 2016, 90, 4780–4795. [Google Scholar] [CrossRef]
- Kulczar, C.; Lubin, K.E.; Lefebvre, S.; Miller, D.W.; Knipp, G.T. Development of a direct contact astrocyte-human cerebral microvessel endothelial cells blood-brain barrier coculture model. J. Pharm. Pharmacol. 2017, 69, 1684–1696. [Google Scholar] [CrossRef]
- Stins, M.F.; Badger, J.; Sik Kim, K. Bacterial invasion and transcytosis in transfected human brain microvascular endothelial cells. Microb. Pathog. 2001, 30, 19–28. [Google Scholar] [CrossRef]
- He, L.; Thomson, J.M.; Hemann, M.T.; Hernando-Monge, E.; Mu, D.; Goodson, S.; Powers, S.; Cordon-Cardo, C.; Lowe, S.W.; Hannon, G.J.; et al. A microrna polycistron as a potential human oncogene. Nature 2005, 435, 828–833. [Google Scholar] [CrossRef]
- Lewis, A.; Mehta, S.; Hanna, L.N.; Rogalski, L.A.; Jeffery, R.; Nijhuis, A.; Kumagai, T.; Biancheri, P.; Bundy, J.G.; Bishop, C.L.; et al. Low serum levels of microrna-19 are associated with a stricturing crohn’s disease phenotype. Inflamm. Bowel Dis. 2015, 21, 1926–1934. [Google Scholar] [CrossRef]
- Zhang, J.; Song, Y.; Zhang, C.; Zhi, X.; Fu, H.; Ma, Y.; Chen, Y.; Pan, F.; Wang, K.; Ni, J.; et al. Circulating mir-16-5p and mir-19b-3p as two novel potential biomarkers to indicate progression of gastric cancer. Theranostics 2015, 5, 733–745. [Google Scholar] [CrossRef]
- Wang, K.J.; Zhao, X.; Liu, Y.Z.; Zeng, Q.T.; Mao, X.B.; Li, S.N.; Zhang, M.; Jiang, C.; Zhou, Y.; Qian, C.; et al. Circulating mir-19b-3p, mir-134-5p and mir-186-5p are promising novel biomarkers for early diagnosis of acute myocardial infarction. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2016, 38, 1015–1029. [Google Scholar] [CrossRef]
- Wertz, I.E.; O’Rourke, K.M.; Zhou, H.; Eby, M.; Aravind, L.; Seshagiri, S.; Wu, P.; Wiesmann, C.; Baker, R.; Boone, D.L.; et al. De-ubiquitination and ubiquitin ligase domains of a20 downregulate nf-kappab signalling. Nature 2004, 430, 694–699. [Google Scholar] [CrossRef] [PubMed]
- Ma, A.; Malynn, B.A. A20: Linking a complex regulator of ubiquitylation to immunity and human disease. Nat. Rev. Immunol. 2012, 12, 774–785. [Google Scholar] [CrossRef] [PubMed]
- Catrysse, L.; Vereecke, L.; Beyaert, R.; van Loo, G. A20 in inflammation and autoimmunity. Trends Immunol. 2014, 35, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Coornaert, B.; Baens, M.; Heyninck, K.; Bekaert, T.; Haegman, M.; Staal, J.; Sun, L.; Chen, Z.J.; Marynen, P.; Beyaert, R. T cell antigen receptor stimulation induces malt1 paracaspase-mediated cleavage of the nf-kappab inhibitor a20. Nat. Immunol. 2008, 9, 263–271. [Google Scholar] [CrossRef]
- Shembade, N.; Harhaj, E.W. Regulation of nf-kappab signaling by the a20 deubiquitinase. Cell. Mol. Immunol. 2012, 9, 123–130. [Google Scholar] [CrossRef]
- Bikker, R.; Christmann, M.; Preuss, K.; Welz, B.; Friesenhagen, J.; Dittrich-Breiholz, O.; Huber, R.; Brand, K. Tnf phase iii signalling in tolerant cells is tightly controlled by a20 and cyld. Cell. Signal. 2017, 37, 123–135. [Google Scholar] [CrossRef]
- Yokota, S.; Okabayashi, T.; Yokosawa, N.; Fujii, N. Measles virus p protein suppresses toll-like receptor signal through up-regulation of ubiquitin-modifying enzyme a20. FASEB J. 2008, 22, 74–83. [Google Scholar] [CrossRef]
- Maelfait, J.; Roose, K.; Bogaert, P.; Sze, M.; Saelens, X.; Pasparakis, M.; Carpentier, I.; van Loo, G.; Beyaert, R. A20 (tnfaip3) deficiency in myeloid cells protects against influenza a virus infection. PLoS Pathog. 2012, 8, e1002570. [Google Scholar] [CrossRef]
- Li, Y.; Mooney, E.C.; Holden, S.E.; Xia, X.J.; Cohen, D.J.; Walsh, S.W.; Ma, A.; Sahingur, S.E. A20 orchestrates inflammatory response in the oral mucosa through restraining nf-kappab activity. J. Immunol. 2019, 202, 2044–2056. [Google Scholar] [CrossRef]
- Turer, E.E.; Tavares, R.M.; Mortier, E.; Hitotsumatsu, O.; Advincula, R.; Lee, B.; Shifrin, N.; Malynn, B.A.; Ma, A. Homeostatic myd88-dependent signals cause lethal inflammation in the absence of a20. J. Exp. Med. 2008, 205, 451–464. [Google Scholar] [CrossRef]
- Liu, C.; Zheng, H.; Yang, M.; Xu, Z.; Wang, X.; Wei, L.; Tang, B.; Liu, F.; Zhang, Y.; Ding, Y.; et al. Genome analysis and in vivo virulence of porcine extraintestinal pathogenic escherichia coli strain pcn033. BMC Genom. 2015, 16, 717. [Google Scholar] [CrossRef] [PubMed]
- Stins, M.F.; Gilles, F.; Kim, K.S. Selective expression of adhesion molecules on human brain microvascular endothelial cells. J. Neuroimmunol. 1997, 76, 81–90. [Google Scholar] [CrossRef]
- Hoffmann, I.; Eugene, E.; Nassif, X.; Couraud, P.O.; Bourdoulous, S. Activation of erbb2 receptor tyrosine kinase supports invasion of endothelial cells by neisseria meningitidis. J. Cell Biol. 2001, 155, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Prosniak, M.; Curtis, M.T.; Weiss, M.L.; Faber, M.; Dietzschold, B.; Fu, Z.F. Silver-haired bat rabies virus variant does not induce apoptosis in the brain of experimentally infected mice. J. Neurovirol. 2001, 7, 518–527. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Sequence (5’ to 3’) |
|---|---|
| Human TNFAIP3 (Forward) | TCCTCAGGCTTTGTATTTGAGC |
| Human TNFAIP3 (reverse) | TGTGTATCGGTGCATGGTTTTA |
| Mouse TNFAIP3 (Forward) | ACAGTGGACCTGGTAAGAAAACA |
| Mouse TNFAIP3 (Reverse) | CCTCCGTGACTGATGACAAGAT |
| Human TNF-α (Forward) | CCTCTCTCTAATCAGCCCTCTG |
| Human TNF-α (Reverse) | GAGGACCTGGGAGTAGATGAG |
| Mouse TNF-α (Forward) | CCCTCACACTCAGATCATCTTCT |
| Mouse TNF-α (Reverse) | GCTACGACGTGGGCTACAG |
| Human IL-6 (Forward) | ACTCACCTCTTCAGAACGAATTG |
| Human IL-6 (Reverse) | CCATCTTTGGAAGGTTCAGGTTG |
| Mouse IL-6 (Forward) | TAGTCCTTCCTACCCCAATTTCC |
| Mouse IL-6 (Reverse) | TTGGTCCTTAGCCACTCCTTC |
| Human IL-1β (Forward) | ATGATGGCTTATTACAGTGGCAA |
| Human IL-1β (Reverse) | GTCGGAGATTCGTAGCTGGA |
| Mouse IL-1β (Forward) | GCAACTGTTCCTGAACTCAACT |
| Mouse IL-1β (Reverse) | ATCTTTTGGGGTCCGTCAACT |
| Human CCL2(Forward) | CAGCCAGATGCAATCAATGCC |
| Human CCL2 (Reverse) | TGGAATCCTGAACCCACTTCT |
| Mouse CCL2 (Forward) | TTAAAAACCTGGATCGGAACCAA |
| Mouse CCL2 (Reverse) | GCATTAGCTTCAGATTTACGGGT |
| Human GAPDH(Forward) | CAACAGCCTCAAGATCATCAG |
| Human GAPDH (Reverse) | GAGTCCTTCCACGATACCA |
| Mouse GAPDH (Forward) | AGGTCGGTGTGAACGGATTTG |
| Mouse GAPDH (Reverse) | TGTAGACCATGTAGTTGAGGTCA |
| Human U6 (Forward) | CTCGCTTCGGCAGCACA |
| Human U6 (Reverse) | AACGCTTCACGAATTTGCGT |
| Mouse U6 (Forward) | CTCGCTTCGGCAGCACA |
| Mouse U6 (Reverse) | AACGCTTCACGAATTTGCGT |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amjad, N.; Yang, R.; Li, L.; Fu, J.; Yang, B.; Xu, B.; Tan, C.; Chen, H.; Wang, X. Decrease of miR-19b-3p in Brain Microvascular Endothelial Cells Attenuates Meningitic Escherichia coli-Induced Neuroinflammation via TNFAIP3-Mediated NF-κB Inhibition. Pathogens 2019, 8, 268. https://doi.org/10.3390/pathogens8040268
Amjad N, Yang R, Li L, Fu J, Yang B, Xu B, Tan C, Chen H, Wang X. Decrease of miR-19b-3p in Brain Microvascular Endothelial Cells Attenuates Meningitic Escherichia coli-Induced Neuroinflammation via TNFAIP3-Mediated NF-κB Inhibition. Pathogens. 2019; 8(4):268. https://doi.org/10.3390/pathogens8040268
Chicago/Turabian StyleAmjad, Nouman, Ruicheng Yang, Liang Li, Jiyang Fu, Bo Yang, Bojie Xu, Chen Tan, Huanchun Chen, and Xiangru Wang. 2019. "Decrease of miR-19b-3p in Brain Microvascular Endothelial Cells Attenuates Meningitic Escherichia coli-Induced Neuroinflammation via TNFAIP3-Mediated NF-κB Inhibition" Pathogens 8, no. 4: 268. https://doi.org/10.3390/pathogens8040268
APA StyleAmjad, N., Yang, R., Li, L., Fu, J., Yang, B., Xu, B., Tan, C., Chen, H., & Wang, X. (2019). Decrease of miR-19b-3p in Brain Microvascular Endothelial Cells Attenuates Meningitic Escherichia coli-Induced Neuroinflammation via TNFAIP3-Mediated NF-κB Inhibition. Pathogens, 8(4), 268. https://doi.org/10.3390/pathogens8040268