Host Single Nucleotide Polymorphisms Modulating Influenza A Virus Disease in Humans



Abstract
1. Introduction
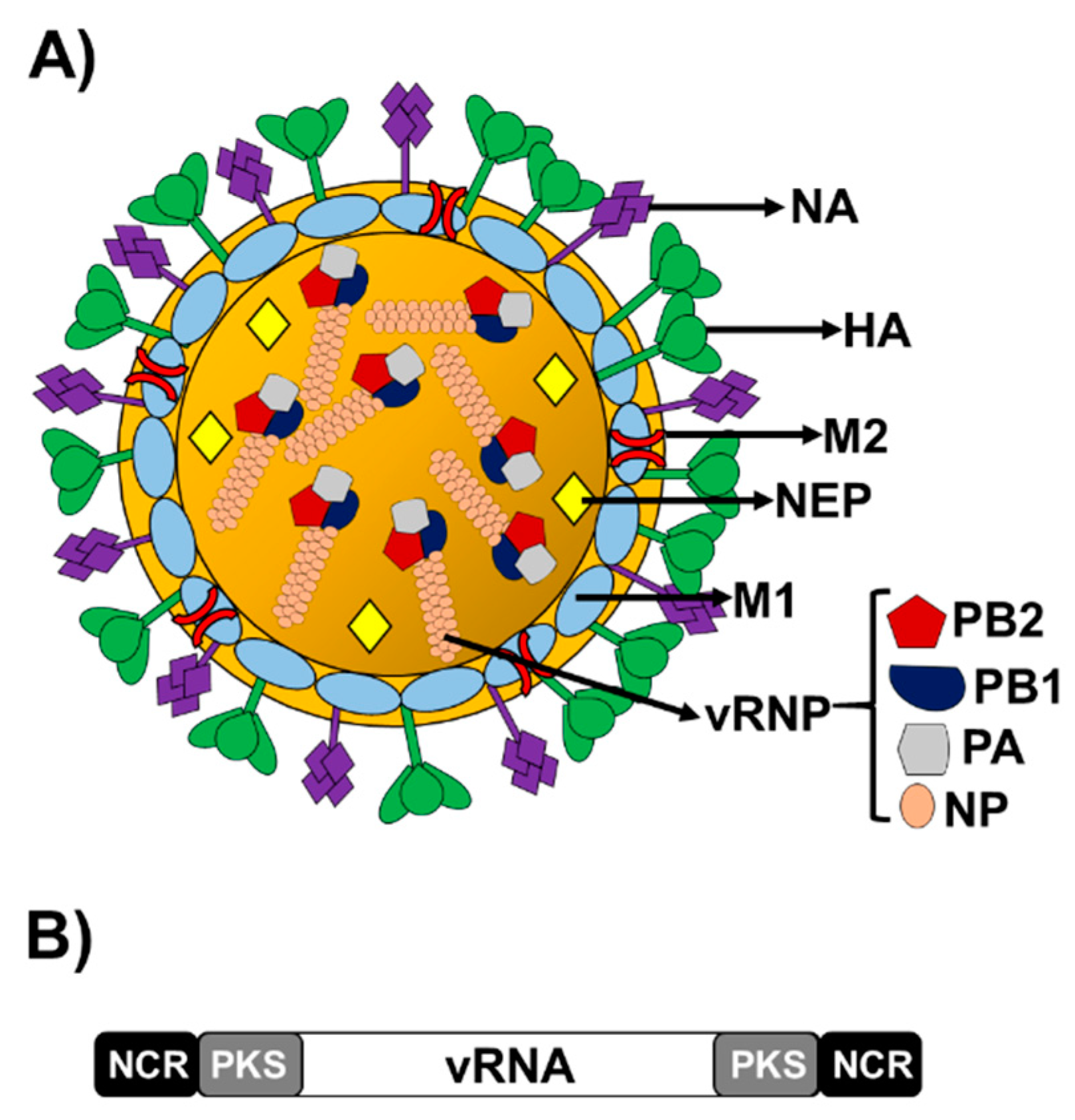
1.1. Influenza A Virus (IAV)
1.2. Influenza Virus Importance in Human Health
1.3. Innate Immunity in IAV Infections
1.4. Single Nucleotide Polymorphisms (SNPs)
2. SNPs in Host Genes Affecting IAV Disease
3. SNPs in Genes that Influence the IAV Vaccine Response
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Heldt, F.S.; Frensing, T.; Reichl, U. Modeling the intracellular dynamics of influenza virus replication to understand the control of viral RNA synthesis. J. Virol. 2012, 86, 7806–7817. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Sobrido, L.; Peersen, O.; Nogales, A. Temperature Sensitive Mutations in Influenza A Viral Ribonucleoprotein Complex Responsible for the Attenuation of the Live Attenuated Influenza Vaccine. Viruses 2018, 10, 560. [Google Scholar] [CrossRef] [PubMed]
- Arranz, R.; Coloma, R.; Chichon, F.J.; Conesa, J.J.; Carrascosa, J.L.; Valpuesta, J.M.; Ortin, J.; Martin-Benito, J. The structure of native influenza virion ribonucleoproteins. Science 2012, 338, 1634–1637. [Google Scholar] [CrossRef]
- Pflug, A.; Guilligay, D.; Reich, S.; Cusack, S. Structure of influenza A polymerase bound to the viral RNA promoter. Nature 2014, 516, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Pflug, A.; Lukarska, M.; Resa-Infante, P.; Reich, S.; Cusack, S. Structural insights into RNA synthesis by the influenza virus transcription-replication machine. Virus Res. 2017, 234, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Resa-Infante, P.; Jorba, N.; Coloma, R.; Ortin, J. The influenza virus RNA synthesis machine: Advances in its structure and function. RNA Biol. 2011, 8, 207–215. [Google Scholar] [CrossRef]
- Baker, S.F.; Nogales, A.; Martinez-Sobrido, L. Downregulating viral gene expression: Codon usage bias manipulation for the generation of novel influenza A virus vaccines. Future Virol. 2015, 10, 715–730. [Google Scholar] [CrossRef]
- Wise, H.M.; Foeglein, A.; Sun, J.; Dalton, R.M.; Patel, S.; Howard, W.; Anderson, E.C.; Barclay, W.S.; Digard, P. A complicated message: Identification of a novel PB1-related protein translated from influenza A virus segment 2 mRNA. J. Virol. 2009, 83, 8021–8031. [Google Scholar] [CrossRef]
- Jagger, B.W.; Wise, H.M.; Kash, J.C.; Walters, K.A.; Wills, N.M.; Xiao, Y.L.; Dunfee, R.L.; Schwartzman, L.M.; Ozinsky, A.; Bell, G.L.; et al. An overlapping protein-coding region in influenza A virus segment 3 modulates the host response. Science 2012, 337, 199–204. [Google Scholar] [CrossRef]
- Bouvier, N.M.; Palese, P. The biology of influenza viruses. Vaccine 2008, 26, D49–D53. [Google Scholar] [CrossRef]
- Byrd-Leotis, L.; Cummings, R.D.; Steinhauer, D.A. The Interplay between the Host Receptor and Influenza Virus Hemagglutinin and Neuraminidase. Int. J. Mol. Sci. 2017, 18, 1541. [Google Scholar] [CrossRef] [PubMed]
- De, A. Molecular evolution of hemagglutinin gene of Influenza A virus. Front. Biosci. 2018, 10, 101–118. [Google Scholar] [CrossRef]
- Imai, M.; Kawaoka, Y. The role of receptor binding specificity in interspecies transmission of influenza viruses. Curr. Opin. Virol. 2012, 2, 160–167. [Google Scholar] [CrossRef] [PubMed]
- McAuley, J.L.; Gilbertson, B.P.; Trifkovic, S.; Brown, L.E.; McKimm-Breschkin, J.L. Influenza Virus Neuraminidase Structure and Functions. Front. Microbiol. 2019, 10, 39. [Google Scholar] [CrossRef] [PubMed]
- Gaymard, A.; Le Briand, N.; Frobert, E.; Lina, B.; Escuret, V. Functional balance between neuraminidase and haemagglutinin in influenza viruses. Clin. Microbiol. Infect. 2016, 22, 975–983. [Google Scholar] [CrossRef] [PubMed]
- Jagadesh, A.; Salam, A.A.; Mudgal, P.P.; Arunkumar, G. Influenza virus neuraminidase (NA): A target for antivirals and vaccines. Arch. Virol. 2016, 161, 2087–2094. [Google Scholar] [CrossRef]
- Chen, B.J.; Leser, G.P.; Jackson, D.; Lamb, R.A. The influenza virus M2 protein cytoplasmic tail interacts with the M1 protein and influences virus assembly at the site of virus budding. J. Virol. 2008, 82, 10059–10070. [Google Scholar] [CrossRef] [PubMed]
- Rossman, J.S.; Lamb, R.A. Influenza virus assembly and budding. Virology 2011, 411, 229–236. [Google Scholar] [CrossRef]
- Chen, B.J.; Leser, G.P.; Morita, E.; Lamb, R.A. Influenza virus hemagglutinin and neuraminidase, but not the matrix protein, are required for assembly and budding of plasmid-derived virus-like particles. J. Virol. 2007, 81, 7111–7123. [Google Scholar] [CrossRef]
- Nayak, D.P.; Hui, E.K.; Barman, S. Assembly and budding of influenza virus. Virus Res. 2004, 106, 147–165. [Google Scholar] [CrossRef]
- Ward, A.C.; Castelli, L.A.; Lucantoni, A.C.; White, J.F.; Azad, A.A.; Macreadie, I.G. Expression and analysis of the NS2 protein of influenza A virus. Arch. Virol. 1995, 140, 2067–2073. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, J.; Nakada, S.; Kato, A.; Toyoda, T.; Ishihama, A. Molecular assembly of influenza virus: Association of the NS2 protein with virion matrix. Virology 1993, 196, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Richardson, J.C.; Akkina, R.K. NS2 protein of influenza virus is found in purified virus and phosphorylated in infected cells. Arch. Virol. 1991, 116, 69–80. [Google Scholar] [CrossRef] [PubMed]
- Paterson, D.; Fodor, E. Emerging roles for the influenza A virus nuclear export protein (NEP). PLoS Pathog. 2012, 8, e1003019. [Google Scholar] [CrossRef] [PubMed]
- Nogales, A.; Martinez-Sobrido, L.; Topham, D.J.; DeDiego, M.L. Modulation of Innate Immune Responses by the Influenza A NS1 and PA-X Proteins. Viruses 2018, 10, 708. [Google Scholar] [CrossRef]
- Nogales, A.; Rodriguez, L.; DeDiego, M.L.; Topham, D.J.; Martinez-Sobrido, L. Interplay of PA-X and NS1 Proteins in Replication and Pathogenesis of a Temperature-Sensitive 2009 Pandemic H1N1 Influenza A Virus. J. Virol. 2017, 91, e00720-17. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Sun, H.; Hu, J.; Qi, L.; Wang, J.; Xiong, X.; Wang, Y.; He, Q.; Lin, Y.; Kong, W.; et al. Twenty amino acids at the C-terminus of PA-X are associated with increased influenza A virus replication and pathogenicity. J. Gen. Virol. 2015, 96, 2036–2049. [Google Scholar] [CrossRef]
- Hu, J.; Mo, Y.; Gao, Z.; Wang, X.; Gu, M.; Liang, Y.; Cheng, X.; Hu, S.; Liu, W.; Liu, H.; et al. PA-X-associated early alleviation of the acute lung injury contributes to the attenuation of a highly pathogenic H5N1 avian influenza virus in mice. Med. Microbiol. Immunol. 2016, 205, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.; Mo, Y.; Wang, X.; Gu, M.; Hu, Z.; Zhong, L.; Wu, Q.; Hao, X.; Hu, S.; Liu, W.; et al. PA-X decreases the pathogenicity of highly pathogenic H5N1 influenza A virus in avian species by inhibiting virus replication and host response. J. Virol. 2015, 89, 4126–4142. [Google Scholar] [CrossRef]
- Khaperskyy, D.A.; Schmaling, S.; Larkins-Ford, J.; McCormick, C.; Gaglia, M.M. Selective Degradation of Host RNA Polymerase II Transcripts by Influenza A Virus PA-X Host Shutoff Protein. PLoS Pathog. 2016, 12, e1005427. [Google Scholar] [CrossRef]
- Barr, I.G.; McCauley, J.; Cox, N.; Daniels, R.; Engelhardt, O.G.; Fukuda, K.; Grohmann, G.; Hay, A.; Kelso, A.; Klimov, A.; et al. Epidemiological, antigenic and genetic characteristics of seasonal influenza A(H1N1), A(H3N2) and B influenza viruses: Basis for the WHO recommendation on the composition of influenza vaccines for use in the 2009-2010 northern hemisphere season. Vaccine 2010, 28, 1156–1167. [Google Scholar] [CrossRef] [PubMed]
- Grohskopf, L.A.; Sokolow, L.Z.; Broder, K.R.; Olsen, S.J.; Karron, R.A.; Jernigan, D.B.; Bresee, J.S. Prevention and Control of Seasonal Influenza with Vaccines. MMWR Recomm. Rep. 2016, 65, 1–54. [Google Scholar] [CrossRef] [PubMed]
- Molinari, N.A.; Ortega-Sanchez, I.R.; Messonnier, M.L.; Thompson, W.W.; Wortley, P.M.; Weintraub, E.; Bridges, C.B. The annual impact of seasonal influenza in the US: Measuring disease burden and costs. Vaccine 2007, 25, 5086–5096. [Google Scholar] [CrossRef] [PubMed]
- Bailey, E.S.; Choi, J.Y.; Fieldhouse, J.K.; Borkenhagen, L.K.; Zemke, J.; Zhang, D.; Gray, G.C. The continual threat of influenza virus infections at the human-animal interface: What is new from a one health perspective? Evol. Med. Public Health 2018, 2018, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Parrish, C.R.; Murcia, P.R.; Holmes, E.C. Influenza virus reservoirs and intermediate hosts: Dogs, horses, and new possibilities for influenza virus exposure of humans. J. Virol. 2015, 89, 2990–2994. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.; Zhu, X.; Li, Y.; Shi, M.; Zhang, J.; Bourgeois, M.; Yang, H.; Chen, X.; Recuenco, S.; Gomez, J.; et al. New world bats harbor diverse influenza A viruses. PLoS Pathog. 2013, 9, e1003657. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.W.; Webby, R.J.; Webster, R.G. Evolution and ecology of influenza A viruses. Curr. Top. Microbiol. Immunol. 2014, 385, 359–375. [Google Scholar] [CrossRef] [PubMed]
- Clark, A.M.; DeDiego, M.L.; Anderson, C.S.; Wang, J.; Yang, H.; Nogales, A.; Martinez-Sobrido, L.; Zand, M.S.; Sangster, M.Y.; Topham, D.J. Antigenicity of the 2015-2016 seasonal H1N1 human influenza virus HA and NA proteins. PLoS ONE 2017, 12, e0188267. [Google Scholar] [CrossRef]
- Grohskopf, L.A.; Sokolow, L.Z.; Broder, K.R.; Walter, E.B.; Bresee, J.S.; Fry, A.M.; Jernigan, D.B. Prevention and Control of Seasonal Influenza with Vaccines: Recommendations of the Advisory Committee on Immunization Practices—United States, 2017–2018 Influenza Season. MMWR Recomm. Rep. 2017, 66, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Garten, R.J.; Davis, C.T.; Russell, C.A.; Shu, B.; Lindstrom, S.; Balish, A.; Sessions, W.M.; Xu, X.; Skepner, E.; Deyde, V.; et al. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science 2009, 325, 197–201. [Google Scholar] [CrossRef]
- Smith, G.J.; Vijaykrishna, D.; Bahl, J.; Lycett, S.J.; Worobey, M.; Pybus, O.G.; Ma, S.K.; Cheung, C.L.; Raghwani, J.; Bhatt, S.; et al. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 2009, 459, 1122–1125. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.F.; Nogales, A.; Santiago, F.W.; Topham, D.J.; Martinez-Sobrido, L. Competitive detection of influenza neutralizing antibodies using a novel bivalent fluorescence-based microneutralization assay (BiFMA). Vaccine 2015, 33, 3562–3570. [Google Scholar] [CrossRef] [PubMed]
- Nogales, A.; Martinez-Sobrido, L. Reverse Genetics Approaches for the Development of Influenza Vaccines. Int. J. Mol. Sci. 2016, 18, 20. [Google Scholar] [CrossRef] [PubMed]
- Schotsaert, M.; Garcia-Sastre, A. Inactivated influenza virus vaccines: The future of TIV and QIV. Curr. Opin. Virol. 2017, 23, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.J.; Cooper, K.L.; Tappenden, P.; Rees, A.; Simpson, E.L.; Read, R.C.; Nicholson, K.G. Oseltamivir, zanamivir and amantadine in the prevention of influenza: A systematic review. J. Infect. 2011, 62, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Lobo, P.; Nogales, A.; Rodriguez, L.; Martinez-Sobrido, L. Novel Approaches for The Development of Live Attenuated Influenza Vaccines. Viruses 2019, 11, 190. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.Y.; Brass, A.L. Host genetic determinants of influenza pathogenicity. Curr. Opin. Virol. 2013, 3, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Bui, C.M.; Chughtai, A.A.; Adam, D.C.; MacIntyre, C.R. An overview of the epidemiology and emergence of influenza A infection in humans over time. Arch. Public Health 2017, 75, 15. [Google Scholar] [CrossRef] [PubMed]
- Perez, D.R.; Garcia-Sastre, A. H5N1, a wealth of knowledge to improve pandemic preparedness. Virus Res. 2013, 178, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, A.; Pillai, P.S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 2014, 14, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Goff, P.H.; Hayashi, T.; He, W.; Yao, S.; Cottam, H.B.; Tan, G.S.; Crain, B.; Krammer, F.; Messer, K.; Pu, M.; et al. Synthetic Toll-Like Receptor 4 (TLR4) and TLR7 Ligands Work Additively via MyD88 To Induce Protective Antiviral Immunity in Mice. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Weber-Gerlach, M.; Weber, F. Standing on three legs: Antiviral activities of RIG-I against influenza viruses. Curr. Opin. Immunol. 2016, 42, 71–75. [Google Scholar] [CrossRef] [PubMed]
- Sarvestani, S.T.; McAuley, J.L. The role of the NLRP3 inflammasome in regulation of antiviral responses to influenza A virus infection. Antivir. Res. 2017, 148, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Luo, J.; Alcorn, J.F.; Chen, K.; Fan, S.; Pilewski, J.; Liu, A.; Chen, W.; Kolls, J.K.; Wang, J. AIM2 Inflammasome Is Critical for Influenza-Induced Lung Injury and Mortality. J. Immunol. 2017, 198, 4383–4393. [Google Scholar] [CrossRef] [PubMed]
- Wolff, T.; Ludwig, S. Influenza viruses control the vertebrate type I interferon system: Factors, mechanisms, and consequences. J. Interferon Cytokine Res. 2009, 29, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Hermant, P.; Michiels, T. Interferon-lambda in the context of viral infections: Production, response and therapeutic implications. J. Innate Immun. 2014, 6, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, T.H.; Paludan, S.R. Molecular pathways in virus-induced cytokine production. Microbiol. Mol. Biol. Rev. 2001, 65, 131–150. [Google Scholar] [CrossRef]
- Crotta, S.; Davidson, S.; Mahlakoiv, T.; Desmet, C.J.; Buckwalter, M.R.; Albert, M.L.; Staeheli, P.; Wack, A. Type I and type III interferons drive redundant amplification loops to induce a transcriptional signature in influenza-infected airway epithelia. PLoS Pathog. 2013, 9, e1003773. [Google Scholar] [CrossRef] [PubMed]
- Desai, T.M.; Marin, M.; Chin, C.R.; Savidis, G.; Brass, A.L.; Melikyan, G.B. IFITM3 restricts influenza A virus entry by blocking the formation of fusion pores following virus-endosome hemifusion. PLoS Pathog. 2014, 10, e1004048. [Google Scholar] [CrossRef]
- Levy, D.E.; Marie, I.J.; Durbin, J.E. Induction and function of type I and III interferon in response to viral infection. Curr. Opin. Virol. 2011, 1, 476–486. [Google Scholar] [CrossRef]
- Schindler, C.; Levy, D.E.; Decker, T. JAK-STAT signaling: From interferons to cytokines. J. Biol. Chem. 2007, 282, 20059–20063. [Google Scholar] [CrossRef] [PubMed]
- Tenoever, B.R.; Ng, S.L.; Chua, M.A.; McWhirter, S.M.; Garcia-Sastre, A.; Maniatis, T. Multiple functions of the IKK-related kinase IKKepsilon in interferon-mediated antiviral immunity. Science 2007, 315, 1274–1278. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.L.; Friedman, B.A.; Schmid, S.; Gertz, J.; Myers, R.M.; tenOever, B.R.; Maniatis, T. I kappa B kinase epsilon (IKK epsilon) regulates the balance between type I and type II interferon responses. Proc. Natl. Acad. Sci. USA 2011, 108, 21170–21175. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, H.H.; Schneider, W.M.; Rice, C.M. Interferons and viruses: An evolutionary arms race of molecular interactions. Trends Immunol. 2015, 36, 124–138. [Google Scholar] [CrossRef] [PubMed]
- Kos, F.J.; Engleman, E.G. Role of natural killer cells in the generation of influenza virus-specific cytotoxic T cells. Cell. Immunol. 1996, 173, 1–6. [Google Scholar] [CrossRef] [PubMed]
- He, X.S.; Draghi, M.; Mahmood, K.; Holmes, T.H.; Kemble, G.W.; Dekker, C.L.; Arvin, A.M.; Parham, P.; Greenberg, H.B. T cell-dependent production of IFN-gamma by NK cells in response to influenza A virus. J. Clin. Investig. 2004, 114, 1812–1819. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Moki, T.; Takizawa, T.; Shiratsuchi, A.; Nakanishi, Y. Evidence for phagocytosis of influenza virus-infected, apoptotic cells by neutrophils and macrophages in mice. J. Immunol. 2007, 178, 2448–2457. [Google Scholar] [CrossRef]
- Legge, K.L.; Braciale, T.J. Accelerated migration of respiratory dendritic cells to the regional lymph nodes is limited to the early phase of pulmonary infection. Immunity 2003, 18, 265–277. [Google Scholar] [CrossRef]
- Legge, K.L.; Braciale, T.J. Lymph node dendritic cells control CD8+ T cell responses through regulated FasL expression. Immunity 2005, 23, 649–659. [Google Scholar] [CrossRef]
- Chiu, C.; Openshaw, P.J. Antiviral B cell and T cell immunity in the lungs. Nat. Immunol. 2015, 16, 18–26. [Google Scholar] [CrossRef]
- Guarda, G.; Zenger, M.; Yazdi, A.S.; Schroder, K.; Ferrero, I.; Menu, P.; Tardivel, A.; Mattmann, C.; Tschopp, J. Differential expression of NLRP3 among hematopoietic cells. J. Immunol. 2011, 186, 2529–2534. [Google Scholar] [CrossRef] [PubMed]
- Pothlichet, J.; Meunier, I.; Davis, B.K.; Ting, J.P.; Skamene, E.; von Messling, V.; Vidal, S.M. Type I IFN triggers RIG-I/TLR3/NLRP3-dependent inflammasome activation in influenza A virus infected cells. PLoS Pathog. 2013, 9, e1003256. [Google Scholar] [CrossRef] [PubMed]
- Lupfer, C.; Kanneganti, T.D. The expanding role of NLRs in antiviral immunity. Immunol. Rev. 2013, 255, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; McElvania-TeKippe, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 2009, 30, 556–565. [Google Scholar] [CrossRef] [PubMed]
- McAuley, J.L.; Tate, M.D.; MacKenzie-Kludas, C.J.; Pinar, A.; Zeng, W.; Stutz, A.; Latz, E.; Brown, L.E.; Mansell, A. Activation of the NLRP3 inflammasome by IAV virulence protein PB1-F2 contributes to severe pathophysiology and disease. PLoS Pathog. 2013, 9, e1003392. [Google Scholar] [CrossRef] [PubMed]
- Ichinohe, T.; Pang, I.K.; Iwasaki, A. Influenza virus activates inflammasomes via its intracellular M2 ion channel. Nat. Immunol. 2010, 11, 404–410. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, P.; Nawadkar, R.; Ojha, H.; Kumar, J.; Sahu, A. Complement Evasion Strategies of Viruses: An Overview. Front. Microbiol. 2017, 8, 1117. [Google Scholar] [CrossRef]
- Kishore, U.; Reid, K.B. C1q: Structure, function, and receptors. Immunopharmacology 2000, 49, 159–170. [Google Scholar] [CrossRef]
- Kim, D.D.; Song, W.C. Membrane complement regulatory proteins. Clin. Immunol. 2006, 118, 127–136. [Google Scholar] [CrossRef]
- Wang, R.; Xiao, H.; Guo, R.; Li, Y.; Shen, B. The role of C5a in acute lung injury induced by highly pathogenic viral infections. Emerg Microbes Infect 2015, 4, e28. [Google Scholar] [CrossRef]
- Kwok, P.Y.; Chen, X. Detection of single nucleotide polymorphisms. Curr. Issues Mol. Biol. 2003, 5, 43–60. [Google Scholar] [PubMed]
- Ramirez-Bello, J.; Jimenez-Morales, M. Functional implications of single nucleotide polymorphisms (SNPs) in protein-coding and non-coding RNA genes in multifactorial diseases. Gac. Med. Mex. 2017, 153, 238–250. [Google Scholar] [PubMed]
- Ramirez-Bello, J.; Vargas-Alarcon, G.; Tovilla-Zarate, C.; Fragoso, J.M. Single nucleotide polymorphisms (SNPs): Functional implications of regulatory-SNP (rSNP) and structural RNA (srSNPs) in complex diseases. Gac. Med. Mex. 2013, 149, 220–228. [Google Scholar] [PubMed]
- Skevaki, C.; Pararas, M.; Kostelidou, K.; Tsakris, A.; Routsias, J.G. Single nucleotide polymorphisms of Toll-like receptors and susceptibility to infectious diseases. Clin. Exp. Immunol. 2015, 180, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Hill, A.V. The genomics and genetics of human infectious disease susceptibility. Annu. Rev. Genom. Hum. Genet. 2001, 2, 373–400. [Google Scholar] [CrossRef]
- Dai, W.; Ye, Z.; Lu, H.; Su, Q.; Li, H.; Li, L. Meta-analysis of the relationship between single nucleotide polymorphism of IL-10-1082G/A and rheumatic heart disease. Oncotarget 2018, 9, 12343–12350. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Koberle, B.; Koch, B.; Fischer, B.M.; Hartwig, A. Single nucleotide polymorphisms in DNA repair genes and putative cancer risk. Arch Toxicol 2016, 90, 2369–2388. [Google Scholar] [CrossRef]
- Li, X.; Zhou, J.; Chen, H.; Wang, F.; Mei, Q.; Sun, H. The association between the UBQLN1 polymorphism and Alzheimer’s disease risk: A systematic review. Cell. Mol. Biol. 2017, 63, 94–96. [Google Scholar] [CrossRef]
- Luykx, J.J.; Broersen, J.L.; de Leeuw, M. The DRD2 rs1076560 polymorphism and schizophrenia-related intermediate phenotypes: A systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2017, 74, 214–224. [Google Scholar] [CrossRef]
- Shaw, V.; Bullock, K.; Greenhalf, W. Single-Nucleotide Polymorphism to Associate Cancer Risk. Methods Mol. Biol. 2016, 1381, 93–110. [Google Scholar] [CrossRef]
- Zhang, Y.; Bai, R.; Liu, C.; Ma, C.; Chen, X.; Yang, J.; Sun, D. MicroRNA single-nucleotide polymorphisms and diabetes mellitus: A comprehensive review. Clin. Genet. 2019, 95, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Imahara, S.D.; O’Keefe, G.E. Genetic determinants of the inflammatory response. Curr. Opin. Crit. Care 2004, 10, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, T.L.; Lima, R.E.; Goes, G.H.B.; Pereira, L.A.; Fernandes, M.S.S.; Moura, P.; Vasconcelos, L.R.S.; Correia, C.C. Cognitive Dysfunction and Single Nucleotide Polymorphisms in Hepatitis C Virus-Infected Persons: A Systematic Review. Viral Immunol. 2017, 30, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Jaeger, M.; Stappers, M.H.; Joosten, L.A.; Gyssens, I.C.; Netea, M.G. Genetic variation in pattern recognition receptors: Functional consequences and susceptibility to infectious disease. Future Med. 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Kenney, A.D.; Dowdle, J.A.; Bozzacco, L.; McMichael, T.M.; St Gelais, C.; Panfil, A.R.; Sun, Y.; Schlesinger, L.S.; Anderson, M.Z.; Green, P.L.; et al. Human Genetic Determinants of Viral Diseases. Annu. Rev. Genet. 2017, 51, 241–263. [Google Scholar] [CrossRef] [PubMed]
- Mathew, S.; Abdel-Hafiz, H.; Raza, A.; Fatima, K.; Qadri, I. Host nucleotide polymorphism in hepatitis B virus-associated hepatocellular carcinoma. World J. Hepatol. 2016, 8, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.U.; Rauf, M.; Abbas, Z.; Hamed, M.H.; Qadri, I. Role of Some Predominant Host Immunomodulators’ Single Nucleotide Polymorphisms in Severity of Hepatitis B Virus and Hepatitis C Virus Infection. Viral Immunol. 2016, 29, 536–545. [Google Scholar] [CrossRef] [PubMed]
- Hidaka, F.; Matsuo, S.; Muta, T.; Takeshige, K.; Mizukami, T.; Nunoi, H. A missense mutation of the Toll-like receptor 3 gene in a patient with influenza-associated encephalopathy. Clin. Immunol. 2006, 119, 188–194. [Google Scholar] [CrossRef]
- Esposito, S.; Molteni, C.G.; Giliani, S.; Mazza, C.; Scala, A.; Tagliaferri, L.; Pelucchi, C.; Fossali, E.; Plebani, A.; Principi, N. Toll-like receptor 3 gene polymorphisms and severity of pandemic A/H1N1/2009 influenza in otherwise healthy children. Virol J. 2012, 9, 270. [Google Scholar] [CrossRef]
- Lee, N.; Cao, B.; Ke, C.; Lu, H.; Hu, Y.; Tam, C.H.T.; Ma, R.C.W.; Guan, D.; Zhu, Z.; Li, H.; et al. IFITM3, TLR3, and CD55 Gene SNPs and Cumulative Genetic Risks for Severe Outcomes in Chinese Patients With H7N9/H1N1pdm09 Influenza. J. Infect. Dis. 2017, 216, 97–104. [Google Scholar] [CrossRef]
- Her, Z.; Teng, T.S.; Tan, J.J.; Teo, T.H.; Kam, Y.W.; Lum, F.M.; Lee, W.W.; Gabriel, C.; Melchiotti, R.; Andiappan, A.K.; et al. Loss of TLR3 aggravates CHIKV replication and pathology due to an altered virus-specific neutralizing antibody response. EMBO Mol. Med. 2015, 7, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Y.; Jouanguy, E.; Ugolini, S.; Smahi, A.; Elain, G.; Romero, P.; Segal, D.; Sancho-Shimizu, V.; Lorenzo, L.; Puel, A.; et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science 2007, 317, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Lafaille, F.G.; Pessach, I.M.; Zhang, S.Y.; Ciancanelli, M.J.; Herman, M.; Abhyankar, A.; Ying, S.W.; Keros, S.; Goldstein, P.A.; Mostoslavsky, G.; et al. Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature 2012, 491, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Perez de Diego, R.; Sancho-Shimizu, V.; Lorenzo, L.; Puel, A.; Plancoulaine, S.; Picard, C.; Herman, M.; Cardon, A.; Durandy, A.; Bustamante, J.; et al. Human TRAF3 adaptor molecule deficiency leads to impaired Toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity 2010, 33, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Sancho-Shimizu, V.; Perez de Diego, R.; Lorenzo, L.; Halwani, R.; Alangari, A.; Israelsson, E.; Fabrega, S.; Cardon, A.; Maluenda, J.; Tatematsu, M.; et al. Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency. J. Clin. Investig. 2011, 121, 4889–4902. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.; Ciancanelli, M.; Ou, Y.H.; Lorenzo, L.; Klaudel-Dreszler, M.; Pauwels, E.; Sancho-Shimizu, V.; Perez de Diego, R.; Abhyankar, A.; Israelsson, E.; et al. Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood. J. Exp. Med. 2012, 209, 1567–1582. [Google Scholar] [CrossRef]
- Al-Anazi, M.R.; Matou-Nasri, S.; Abdo, A.A.; Sanai, F.M.; Alkahtani, S.; Alarifi, S.; Alkahtane, A.A.; Al-Yahya, H.; Ali, D.; Alessia, M.S.; et al. Association of Toll-Like Receptor 3 Single-Nucleotide Polymorphisms and Hepatitis C Virus Infection. J. Immunol. Res. 2017, 2017, 1590653. [Google Scholar] [CrossRef]
- Al-Qahtani, A.; Al-Ahdal, M.; Abdo, A.; Sanai, F.; Al-Anazi, M.; Khalaf, N.; Viswan, N.A.; Al-Ashgar, H.; Al-Humaidan, H.; Al-Suwayeh, R.; et al. Toll-like receptor 3 polymorphism and its association with hepatitis B virus infection in Saudi Arabian patients. J. Med. Virol. 2012, 84, 1353–1359. [Google Scholar] [CrossRef]
- Jorgensen, S.E.; Christiansen, M.; Ryo, L.B.; Gad, H.H.; Gjedsted, J.; Staeheli, P.; Mikkelsen, J.G.; Storgaard, M.; Hartmann, R.; Mogensen, T.H. Defective RNA sensing by RIG-I in severe influenza virus infection. Clin. Exp. Immunol. 2018, 192, 366–376. [Google Scholar] [CrossRef]
- Honda, K.; Yanai, H.; Negishi, H.; Asagiri, M.; Sato, M.; Mizutani, T.; Shimada, N.; Ohba, Y.; Takaoka, A.; Yoshida, N.; et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature 2005, 434, 772–777. [Google Scholar] [CrossRef]
- Marie, I.; Durbin, J.E.; Levy, D.E. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. EMBO J. 1998, 17, 6660–6669. [Google Scholar] [CrossRef] [PubMed]
- Osterlund, P.I.; Pietila, T.E.; Veckman, V.; Kotenko, S.V.; Julkunen, I. IFN regulatory factor family members differentially regulate the expression of type III IFN (IFN-lambda) genes. J. Immunol. 2007, 179, 3434–3442. [Google Scholar] [CrossRef] [PubMed]
- Ciancanelli, M.J.; Huang, S.X.; Luthra, P.; Garner, H.; Itan, Y.; Volpi, S.; Lafaille, F.G.; Trouillet, C.; Schmolke, M.; Albrecht, R.A.; et al. Infectious disease. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science 2015, 348, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Lindsay, R.J.; Kulkarni, S.; Lifson, J.D.; Carrington, M.; Altfeld, M. Polymorphisms in interferon regulatory factor 7 reduce interferon-alpha responses of plasmacytoid dendritic cells to HIV-1. AIDS 2011, 25, 715–717. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, N.; Melki, I.; Jing, H.; Habib, T.; Huang, S.S.Y.; Danielson, J.; Kula, T.; Drutman, S.; Belkaya, S.; Rattina, V.; et al. Life-threatening influenza pneumonitis in a child with inherited IRF9 deficiency. J. Exp. Med. 2018, 215, 2567–2585. [Google Scholar] [CrossRef]
- Bravo Garcia-Morato, M.; Calvo Apalategi, A.; Bravo-Gallego, L.Y.; Blazquez Moreno, A.; Simon-Fuentes, M.; Garmendia, J.V.; Mendez Echevarria, A.; Del Rosal Rabes, T.; Dominguez-Soto, A.; Lopez-Granados, E.; et al. Impaired control of multiple viral infections in a family with complete IRF9 deficiency. J. Allergy Clin. Immunol. 2019, 144, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Wakim, L.M.; Gupta, N.; Mintern, J.D.; Villadangos, J.A. Enhanced survival of lung tissue-resident memory CD8(+) T cells during infection with influenza virus due to selective expression of IFITM3. Nat. Immunol. 2013, 14, 238–245. [Google Scholar] [CrossRef]
- Everitt, A.R.; Clare, S.; Pertel, T.; John, S.P.; Wash, R.S.; Smith, S.E.; Chin, C.R.; Feeley, E.M.; Sims, J.S.; Adams, D.J.; et al. IFITM3 restricts the morbidity and mortality associated with influenza. Nature 2012, 484, 519–523. [Google Scholar] [CrossRef]
- Weidner, J.M.; Jiang, D.; Pan, X.B.; Chang, J.; Block, T.M.; Guo, J.T. Interferon-induced cell membrane proteins, IFITM3 and tetherin, inhibit vesicular stomatitis virus infection via distinct mechanisms. J. Virol. 2010, 84, 12646–12657. [Google Scholar] [CrossRef]
- Zhang, Y.H.; Zhao, Y.; Li, N.; Peng, Y.C.; Giannoulatou, E.; Jin, R.H.; Yan, H.P.; Wu, H.; Liu, J.H.; Liu, N.; et al. Interferon-induced transmembrane protein-3 genetic variant rs12252-C is associated with severe influenza in Chinese individuals. Nat. Commun. 2013, 4, 1418. [Google Scholar] [CrossRef]
- Yang, X.; Tan, B.; Zhou, X.; Xue, J.; Zhang, X.; Wang, P.; Shao, C.; Li, Y.; Li, C.; Xia, H.; et al. Interferon-Inducible Transmembrane Protein 3 Genetic Variant rs12252 and Influenza Susceptibility and Severity: A Meta-Analysis. PLoS ONE 2015, 10, e0124985. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, S.S.; Chakraborty, T.T.; Kumar, N.; Banerjee, I. Association between IFITM3 rs12252 polymorphism and influenza susceptibility and severity: A meta-analysis. Gene 2018, 674, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.K.; Randolph, A.G.; Bhangale, T.; Dogra, P.; Ohlson, M.; Oshansky, C.M.; Zamora, A.E.; Shannon, J.P.; Finkelstein, D.; Dressen, A.; et al. SNP-mediated disruption of CTCF binding at the IFITM3 promoter is associated with risk of severe influenza in humans. Nat. Med. 2017, 23, 975–983. [Google Scholar] [CrossRef]
- Eisfeld, A.J.; Kawaoka, Y. Calculated risk: A new single-nucleotide polymorphism linked to severe influenza disease. Nat. Med. 2017, 23, 911–912. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. IL-1: Discoveries, controversies and future directions. Eur. J. Immunol. 2010, 40, 599–606. [Google Scholar] [CrossRef]
- Liu, Y.; Li, S.; Zhang, G.; Nie, G.; Meng, Z.; Mao, D.; Chen, C.; Chen, X.; Zhou, B.; Zeng, G. Genetic variants in IL1A and IL1B contribute to the susceptibility to 2009 pandemic H1N1 influenza A virus. BMC Immunol. 2013, 14, 37. [Google Scholar] [CrossRef]
- Zhang, G.; Zhou, B.; Li, S.; Yue, J.; Yang, H.; Wen, Y.; Zhan, S.; Wang, W.; Liao, M.; Zhang, M.; et al. Allele-specific induction of IL-1beta expression by C/EBPbeta and PU.1 contributes to increased tuberculosis susceptibility. PLoS Pathog. 2014, 10, e1004426. [Google Scholar] [CrossRef]
- Lind, H.; Haugen, A.; Zienolddiny, S. Differential binding of proteins to the IL1B -31 T/C polymorphism in lung epithelial cells. Cytokine 2007, 38, 43–48. [Google Scholar] [CrossRef]
- Luft, T.; Jefford, M.; Luetjens, P.; Hochrein, H.; Masterman, K.A.; Maliszewski, C.; Shortman, K.; Cebon, J.; Maraskovsky, E. IL-1 beta enhances CD40 ligand-mediated cytokine secretion by human dendritic cells (DC): A mechanism for T cell-independent DC activation. J. Immunol. 2002, 168, 713–722. [Google Scholar] [CrossRef]
- Antonopoulou, A.; Baziaka, F.; Tsaganos, T.; Raftogiannis, M.; Koutoukas, P.; Spyridaki, A.; Mouktaroudi, M.; Kotsaki, A.; Savva, A.; Georgitsi, M.; et al. Role of tumor necrosis factor gene single nucleotide polymorphisms in the natural course of 2009 influenza A H1N1 virus infection. Int. J. Infect. Dis. 2012, 16, e204–e208. [Google Scholar] [CrossRef]
- Garcia-Ramirez, R.A.; Ramirez-Venegas, A.; Quintana-Carrillo, R.; Camarena, A.E.; Falfan-Valencia, R.; Mejia-Arangure, J.M. TNF, IL6, and IL1B Polymorphisms Are Associated with Severe Influenza A (H1N1) Virus Infection in the Mexican Population. PLoS ONE 2015, 10, e0144832. [Google Scholar] [CrossRef] [PubMed]
- Deval, H.; Alagarasu, K.; Mittal, M.; Srivastava, N.; Bachal, R.; Gondhalekar, A.; Chaudhary, U.; Chowdhary, D.; Bondre, V.P. Association of single nucleotide polymorphisms in TNFA and CCR5 genes with Japanese Encephalitis: A study from an endemic region of North India. J. Neuroimmunol. 2019, 336, 577043. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, J.; Zheng, Y.; Zhou, S.; Zheng, J.; Wang, F.; Ma, X.; Zeng, Z.; Consortium, H.B.V.S. A study of TNF-alpha-238 and -308 polymorphisms with different outcomes of persistent hepatitis B virus infection in China. Pathology 2010, 42, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Glass, W.G.; McDermott, D.H.; Lim, J.K.; Lekhong, S.; Yu, S.F.; Frank, W.A.; Pape, J.; Cheshier, R.C.; Murphy, P.M. CCR5 deficiency increases risk of symptomatic West Nile virus infection. J. Exp. Med. 2006, 203, 35–40. [Google Scholar] [CrossRef] [PubMed]
- Keynan, Y.; Juno, J.; Meyers, A.; Ball, T.B.; Kumar, A.; Rubinstein, E.; Fowke, K.R. Chemokine receptor 5 big up tri, open32 allele in patients with severe pandemic (H1N1) 2009. Emerg. Infect. Dis. 2010, 16, 1621–1622. [Google Scholar] [CrossRef] [PubMed]
- Samson, M.; Libert, F.; Doranz, B.J.; Rucker, J.; Liesnard, C.; Farber, C.M.; Saragosti, S.; Lapoumeroulie, C.; Cognaux, J.; Forceille, C.; et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 1996, 382, 722–725. [Google Scholar] [CrossRef]
- Zhou, J.; To, K.K.; Dong, H.; Cheng, Z.S.; Lau, C.C.; Poon, V.K.; Fan, Y.H.; Song, Y.Q.; Tse, H.; Chan, K.H.; et al. A functional variation in CD55 increases the severity of 2009 pandemic H1N1 influenza A virus infection. J. Infect. Dis. 2012, 206, 495–503. [Google Scholar] [CrossRef] [PubMed]
- Chatzopoulou, F.; Gioula, G.; Kioumis, I.; Chatzidimitriou, D.; Exindari, M. Identification of complement-related host genetic risk factors associated with influenza A(H1N1)pdm09 outcome: Challenges ahead. Med. Microbiol. Immunol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ewulonu, U.K.; Ravi, L.; Medof, M.E. Characterization of the decay-accelerating factor gene promoter region. Proc. Natl. Acad. Sci. USA 1991, 88, 4675–4679. [Google Scholar] [CrossRef]
- Zuniga, J.; Buendia-Roldan, I.; Zhao, Y.; Jimenez, L.; Torres, D.; Romo, J.; Ramirez, G.; Cruz, A.; Vargas-Alarcon, G.; Sheu, C.C.; et al. Genetic variants associated with severe pneumonia in A/H1N1 influenza infection. Eur. Respir J 2012, 39, 604–610. [Google Scholar] [CrossRef] [PubMed]
- Ng, W.C.; Tate, M.D.; Brooks, A.G.; Reading, P.C. Soluble host defense lectins in innate immunity to influenza virus. J Biomed Biotechnol 2012, 2012, 732191. [Google Scholar] [CrossRef]
- LeVine, A.M.; Hartshorn, K.; Elliott, J.; Whitsett, J.; Korfhagen, T. Absence of SP-A modulates innate and adaptive defense responses to pulmonary influenza infection. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L563–L572. [Google Scholar] [CrossRef]
- Li, G.; Siddiqui, J.; Hendry, M.; Akiyama, J.; Edmondson, J.; Brown, C.; Allen, L.; Levitt, S.; Poulain, F.; Hawgood, S. Surfactant protein-A--deficient mice display an exaggerated early inflammatory response to a beta-resistant strain of influenza A virus. Am. J. Respir. Cell Mol. Biol. 2002, 26, 277–282. [Google Scholar] [CrossRef]
- Hartshorn, K.L.; White, M.R.; Shepherd, V.; Reid, K.; Jensenius, J.C.; Crouch, E.C. Mechanisms of anti-influenza activity of surfactant proteins A and D: Comparison with serum collectins. Am. J. Physiol. 1997, 273, L1156–L1166. [Google Scholar] [CrossRef]
- Herrera-Ramos, E.; Lopez-Rodriguez, M.; Ruiz-Hernandez, J.J.; Horcajada, J.P.; Borderias, L.; Lerma, E.; Blanquer, J.; Perez-Gonzalez, M.C.; Garcia-Laorden, M.I.; Florido, Y.; et al. Surfactant protein A genetic variants associate with severe respiratory insufficiency in pandemic influenza A virus infection. Crit. Care 2014, 18, R127. [Google Scholar] [CrossRef]
- Yang, M.L.; Chen, Y.H.; Wang, S.W.; Huang, Y.J.; Leu, C.H.; Yeh, N.C.; Chu, C.Y.; Lin, C.C.; Shieh, G.S.; Chen, Y.L.; et al. Galectin-1 binds to influenza virus and ameliorates influenza virus pathogenesis. J. Virol. 2011, 85, 10010–10020. [Google Scholar] [CrossRef]
- Chen, Y.; Zhou, J.; Cheng, Z.; Yang, S.; Chu, H.; Fan, Y.; Li, C.; Wong, B.H.; Zheng, S.; Zhu, Y.; et al. Functional variants regulating LGALS1 (Galectin 1) expression affect human susceptibility to influenza A(H7N9). Sci. Rep. 2015, 5, 8517. [Google Scholar] [CrossRef]
- Hatesuer, B.; Bertram, S.; Mehnert, N.; Bahgat, M.M.; Nelson, P.S.; Pohlmann, S.; Schughart, K. Tmprss2 is essential for influenza H1N1 virus pathogenesis in mice. PLoS Pathog. 2013, 9, e1003774. [Google Scholar] [CrossRef]
- Cheng, Z.; Zhou, J.; To, K.K.; Chu, H.; Li, C.; Wang, D.; Yang, D.; Zheng, S.; Hao, K.; Bosse, Y.; et al. Identification of TMPRSS2 as a Susceptibility Gene for Severe 2009 Pandemic A(H1N1) Influenza and A(H7N9) Influenza. J. Infect. Dis. 2015, 212, 1214–1221. [Google Scholar] [CrossRef]
- Bartoszko, J.J.; McNamara, I.F.; Aras, O.A.Z.; Hylton, D.A.; Zhang, Y.B.; Malhotra, D.; Hyett, S.L.; Morassut, R.E.; Rudziak, P.; Loeb, M. Does consecutive influenza vaccination reduce protection against influenza: A systematic review and meta-analysis. Vaccine 2018, 36, 3434–3444. [Google Scholar] [CrossRef]
- Belongia, E.A.; Skowronski, D.M.; McLean, H.Q.; Chambers, C.; Sundaram, M.E.; De Serres, G. Repeated annual influenza vaccination and vaccine effectiveness: Review of evidence. Expert Rev. Vaccines 2017, 16, 723–736. [Google Scholar] [CrossRef]
- Leidner, A.J.; Murthy, N.; Chesson, H.W.; Biggerstaff, M.; Stoecker, C.; Harris, A.M.; Acosta, A.; Dooling, K.; Bridges, C.B. Cost-effectiveness of adult vaccinations: A systematic review. Vaccine 2019, 37, 226–234. [Google Scholar] [CrossRef]
- Egli, A.; Santer, D.M.; O’Shea, D.; Barakat, K.; Syedbasha, M.; Vollmer, M.; Baluch, A.; Bhat, R.; Groenendyk, J.; Joyce, M.A.; et al. IL-28B is a key regulator of B- and T-cell vaccine responses against influenza. PLoS Pathog. 2014, 10, e1004556. [Google Scholar] [CrossRef]
- Gelder, C.M.; Lambkin, R.; Hart, K.W.; Fleming, D.; Williams, O.M.; Bunce, M.; Welsh, K.I.; Marshall, S.E.; Oxford, J. Associations between human leukocyte antigens and nonresponsiveness to influenza vaccine. J. Infect. Dis. 2002, 185, 114–117. [Google Scholar] [CrossRef]
- Linnik, J.E.; Egli, A. Impact of host genetic polymorphisms on vaccine induced antibody response. Hum. Vaccin Immunother. 2016, 12, 907–915. [Google Scholar] [CrossRef]
- Poland, G.A.; Ovsyannikova, I.G.; Jacobson, R.M. Immunogenetics of seasonal influenza vaccine response. Vaccine 2008, 26, D35–D40. [Google Scholar] [CrossRef]
- Burleson, G.R. Immunological Variation Due to Genetics of Inflammatory SNPs and Age and Impact on Disease Manifestation. Toxicol. Pathol. 2017, 45, 146–149. [Google Scholar] [CrossRef]
- Castiblanco, J.; Anaya, J.M. Genetics and vaccines in the era of personalized medicine. Curr. Genom. 2015, 16, 47–59. [Google Scholar] [CrossRef]
- Ellwanger, J.H.; Chies, J.A.B. Host genetic factors can impact vaccine immunogenicity and effectiveness. Lancet Infect. Dis. 2019, 19, 359–360. [Google Scholar] [CrossRef]
- Zeng, M.; Nourishirazi, E.; Guinet, E.; Nouri-Shirazi, M. The genetic background influences the cellular and humoral immune responses to vaccines. Clin. Exp. Immunol. 2016, 186, 190–204. [Google Scholar] [CrossRef]
- Allcock, R.J. The major histocompatibility complex: A paradigm for studies of the human genome. Methods Mol. Biol. 2012, 882, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Apanius, V.; Penn, D.; Slev, P.R.; Ruff, L.R.; Potts, W.K. The Nature of Selection on the Major Histocompatibility Complex. Crit. Rev. Immunol. 2017, 37, 75–120. [Google Scholar] [CrossRef] [PubMed]
- Garred, P.; Genster, N.; Pilely, K.; Bayarri-Olmos, R.; Rosbjerg, A.; Ma, Y.J.; Skjoedt, M.O. A journey through the lectin pathway of complement-MBL and beyond. Immunol. Rev. 2016, 274, 74–97. [Google Scholar] [CrossRef] [PubMed]
- Turner, M.W. The role of mannose-binding lectin in health and disease. Mol. Immunol. 2003, 40, 423–429. [Google Scholar] [CrossRef]
- Tang, Y.W.; Li, H.; Wu, H.; Shyr, Y.; Edwards, K.M. Host single-nucleotide polymorphisms and altered responses to inactivated influenza vaccine. J. Infect. Dis. 2007, 196, 1021–1025. [Google Scholar] [CrossRef] [PubMed][Green Version]



{kind=link}
{kind=link}
| Gene | Function | SNPs (Type) | References |
|---|---|---|---|
| TLR-3 | Recognizes dsRNA, triggering IFN production. | rs not annotated; F303S (NonSyn). rs5743313 (NCR). | [98] [99,100] |
| RIG-I | Detects dsRNA and 5′- triphosphates of the negative ssRNA IAV genome, leading to innate immune responses activation. | rs72710678; R71H (NonSyn). rs138425677; P885S (NonSyn). | [109] |
| IRF-7 | Transcription factor that increases IFN production in response to viruses. | rs786205223; F410V (NonSyn) rs375323253; Q421X (NonSyn) | [113] |
| IRF-9 | Transcription factor essential for IFN signaling and the transcriptional induction of ISGs. | c.991G>A occurred in the final nucleotide of exon 7 and disrupted the essential splice site at the boundary of exon 7 and intron 7 (NonSyn). c.577+1G>T, was localized in the donor splice site of introns 5 and 6 and led to transcripts lacking exon 5 (NonSyn). | [115] [116] |
| IFITM3 | ISG which abrogates the release of IAV content from late endosomes into the cytoplasm. IFITM3 increases the survival of mouse lung-resident CD8+ T cells after IAV infection, which can help clear the infection. | rs12252, leading to an alteration of the first splice acceptor site, leading to an IFITM3 protein lacking the first 21 amino acids (NonSyn). rs34481144, is located in the 5′-UTR and affects IFITM3 expression with the risk allele showing lower mRNA expression (NCR). | [100,118,120,122] [123] |
| IL-1B | Inflammatory cytokine involved in the development of adaptive immune responses. Furthermore, accumulating data has suggested that IL-1A and IL-1B have critical roles in innate immunity against viral infections. | rs1143627, located 31 base pairs upstream from the transcription start site, on the IL-1B promoter. This nucleotide change is located in a TATA-box motif of IL-1B, affecting the transcription activity of IL-1B (NCR). | [128,129] |
| IL-1A | Inflammatory cytokine that plays important roles in the development of adaptive immune responses. Moreover, multiple pieces of evidence have suggested that IL-1A and IL-1B play relevant roles in innate immunity against viral infections. | rs17561; A114S (NonSyn). | [126] |
| TNF-α | Pro-inflammatory cytokine which orchestrates the host´s defense. | rs361525, a minor allele (A) at position 238 (NCR). | [92,130,131] |
| CCR5 | Cytokine receptor which has a role in mediating leukocyte migration in response to its ligands, including MIP-1a, MIP-1b, and RANTES. Furthermore, it can help direct many immune cell subsets, including regulatory T cells and Th17 cells to sites of infection, supporting the antiviral immune response. | CCR5-Δ32 allele (NonSyn). | [135] |
| CD55 | Blocks C3 and C5 activation by inhibiting the formation of new C3 and C5 convertases, which are two proteases involved in complement activation and inflammation. CD55 functions to protect cells from complement attack and decreases the amplification of the complement cascade | rs2564978, resides in the minimal promoter region, affecting gene expression (NCR). | [47,79,100,137,138] |
| C1QBP | Binds to the globular heads of C1q molecules activating the classical pathway of complement. | rs3786054, localized in an intron (NCR). | [138,140] |
| SFTPA2 | Soluble pattern-recognition molecule that may neutralize IAV infection. | -rs1965708; Q223K (NonSyn). -rs1059046; T9N (NonSyn). | [145] |
| Galectin-1 | Recognizes galactose-containing oligosaccharides present in the cellular plasma membranes and in viruses, such as IAV. | -rs4820294 (NCR). -rs13057866 (NCR). | [147] |
| TMPRSS2 | Type II transmembrane serine protease family member which activates HA proteins of diverse human IAV in tissue culture cells. Deletion of Tmprss2 in mice impairs the spread of H1N1 influenza viruses, including the pH1N1. Moreover, body weight loss and survival were less severe in Tmprss2 mutant mice compared to wild type mice after infection with H3N2 IAV. | -rs2070788, localized in an intron (NCR). -rs383510, localized in an intron (NCR). | [149] |
| Gene | Function | SNPs (Type) | Vaccine | Reference |
|---|---|---|---|---|
| IL-6 | Cytokine expressed as a response to infections or tissue injuries. It plays an important role in host defense through the stimulation of acute-phase responses. | -rs1800796 (NCR). - rs2069861 (NCR). | IIV | [156] |
| IL-12B | Cytokine that serves as a crucial inducer of Th1 cell development. | rs3212227, located in 3´UTR (NCR). | IIV | [156] |
| IFN-B1 | Cytokine released as part of the innate immune response against infection by viruses or other pathogens. | rs1364613 (NCR). | IIV | [156] |
| TNFRSF1A | Cytokine receptor, its interaction with TNF-α control cell survival, apoptosis, and inflammation. | rs4149621 (NCR). | IIV | [156] |
| IL-1R1 | Cytokine receptor involved in inflammatory and immune responses. | rs3732131, located in 3´UTR (NCR). | IIV | [156] |
| IL-10RB | Cytokine receptor that mediates the activation of the JAK/STAT signaling pathway leading to the expression of ISG. | rs3171425, located in 3´UTR (NCR). | IIV | [156] |
| IL-2RA | This cytokine receptor is important for the signaling pathway leading to immune cell differentiation and function. | -rs2228150 (Syn). -rs12722605 (NCR). | IIV | [156] |
| IL-10RA | Cytokine receptor that is involved in the inhibition of the synthesis of several proinflammatory cytokines. | -rs4252249 (Syn) -rs4252243 (NCR). | IIV | [156] |
| IL-12RB2 | Cytokine receptor that plays a role in Th1 cell differentiation. | rs2307153; D465G (NonSyn). | IIV | [156] |
| IL-1RN | Cytokine receptor which modulates a variety of immune and inflammatory responses related with IL-1. | -rs315952 (Syn). -rs315951 located in 3´UTR (NCR). | IIV | [156] |
| TNFRSF1B | Cytokine receptor involved in the recruitment of anti-apoptotic proteins. | rs5746026; K232E (NonSyn) | IIV | [156] |
| MBL-2 | This calcium-dependent protein that plays an important role in innate immunity, and activates the lectin complement pathway. | rs1800450; G54D (NonSyn) | IIV | [165] |
| IL-28B (IFNL3) | Type III IFN molecule, with brad functions in antiviral responses | rs8099917 (NCR). | IIV | [153] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nogales, A.; L. DeDiego, M. Host Single Nucleotide Polymorphisms Modulating Influenza A Virus Disease in Humans. Pathogens 2019, 8, 168. https://doi.org/10.3390/pathogens8040168
Nogales A, L. DeDiego M. Host Single Nucleotide Polymorphisms Modulating Influenza A Virus Disease in Humans. Pathogens. 2019; 8(4):168. https://doi.org/10.3390/pathogens8040168
Chicago/Turabian StyleNogales, Aitor, and Marta L. DeDiego. 2019. "Host Single Nucleotide Polymorphisms Modulating Influenza A Virus Disease in Humans" Pathogens 8, no. 4: 168. https://doi.org/10.3390/pathogens8040168
APA StyleNogales, A., & L. DeDiego, M. (2019). Host Single Nucleotide Polymorphisms Modulating Influenza A Virus Disease in Humans. Pathogens, 8(4), 168. https://doi.org/10.3390/pathogens8040168