Manipulation of Innate and Adaptive Immunity by Staphylococcal Superantigens


Abstract
1. Introduction
2. The Staphylococcal Superantigen Family
3. Staphylococcal Superantigens and Disease
4. Superantigens in S. aureus Pathogenesis
4.1. T cells in S. aureus Immunity
4.2. Conventional T cell Responses to SAgs
4.3. SAgs and Phagocytic Cells
4.4. Unconventional T Cell Responses to SAgs
4.5. SAgs in Colonisation
4.6. SAgs’ Cooperation with Other S. aureus Virulence Determinants
5. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Tong, S.Y.C.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef] [PubMed]
- Foster, T.J. Immune evasion by staphylococci. Nat. Rev. Microbiol. 2005. [Google Scholar] [CrossRef] [PubMed]
- Spaulding, A.R.; Salgado-Pabón, W.; Kohler, P.L.; Horswill, A.R.; Leung, D.Y.M.; Schlievert, P.M. Staphylococcal and streptococcal superantigen exotoxins. Clin. Microbiol. Rev. 2013, 26, 422–447. [Google Scholar] [CrossRef] [PubMed]
- Ross Fitzgerald, J.; Nutbeam-Tuffs, S.; Richardson, E.; Wilson, G.J.; O’Gara, J.P.; Corander, J.; McAdam, P.R.; Richards, A.C.; Lee, C.Y.; Spoor, L.E.; et al. Recombination-mediated remodelling of host–pathogen interactions during Staphylococcus aureus niche adaptation. Microb. Genom. 2015, 1. [Google Scholar] [CrossRef]
- Ono, H.K.; Sato’o, Y.; Narita, K.; Naito, I.; Hirose, S.; Hisatsune, J.; Asano, K.; Hu, D.L.; Omoe, K.; Sugai, M.; et al. Identification and characterization of a novel staphylococcal emetic toxin. Appl. Environ. Microbiol. 2015, 81, 7034–7040. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.W.; Lee, G.J.; Lee, S.Y.; Park, C.; Yoo, J.H.; Park, H.M. Prevalence of genes for enterotoxins, toxic shock syndrome toxin 1 and exfoliative toxin among clinical isolates of Staphylococcus pseudintermedius from canine origin. Vet. Dermatol. 2010, 21, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Madhusoodanan, J.; Seo, K.S.; Remortel, B.; Park, J.Y.; Hwang, S.Y.; Fox, L.K.; Park, Y.H.; Deobald, C.F.; Wang, D.; Liu, S.; et al. An enterotoxin-bearing Pathogenicity Island in Staphylococcus epidermidis. J. Bacteriol. 2011, 193, 1854–1862. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Fox, L.K.; Seo, K.S.; McGuire, M.A.; Park, Y.H.; Rurangirwa, F.R.; Sischo, W.M.; Bohach, G.A. Detection of classical and newly described staphylococcal superantigen genes in coagulase-negative staphylococci isolated from bovine intramammary infections. Vet. Microbiol. 2011, 147, 149–154. [Google Scholar] [CrossRef] [PubMed]
- Proft, T.; Fraser, J.D. Bacterial superantigens. Clin. Exp. Immunol. 2003, 133, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Fraser, J.D.; Proft, T. The bacterial superantigen and superantigen-like proteins. Immunol. Rev. 2008, 225, 226–243. [Google Scholar] [CrossRef] [PubMed]
- Argudín, M.Á.; Mendoza, M.C.; Rodicio, M.R. Food poisoning and Staphylococcus aureus enterotoxins. Toxins (Basel) 2010, 2, 1751–1773. [Google Scholar] [CrossRef] [PubMed]
- Omoe, K.; Hu, D.L.; Ono, H.K.; Shimizu, S.; Takahashi-Omoe, H.; Nakane, A.; Uchiyama, T.; Shinagawa, K.; Imanishi, K. Emetic potentials of newly identified staphylococcal enterotoxin-like toxins. Infect. Immun. 2013, 81, 3627–3631. [Google Scholar] [CrossRef] [PubMed]
- Seo, K.S. Superantigens. Methods Mol. Biol. 2016, 1396. [Google Scholar] [CrossRef]
- Thomas, D.Y.; Jarraud, S.; Lemercier, B.; Cozon, G.; Echasserieau, K.; Etienne, J.; Gougeon, M.-L.; Lina, G.; Vandenesch, F. Staphylococcal enterotoxin-like toxins U2 and V, two new staphylococcal superantigens arising from recombination within the enterotoxin gene cluster. Infect. Immun. 2006, 74, 4724–4734. [Google Scholar] [CrossRef] [PubMed]
- Seo, K.S.; Park, J.Y.; Terman, D.S.; Bohach, G.A. A quantitative real time PCR method to analyze T cell receptor Vβ subgroup expansion by staphylococcal superantigens. J. Transl. Med. 2010, 8, 2. [Google Scholar] [CrossRef] [PubMed]
- Omoe, K.; Hu, D.-L.; Takahashi-Omoe, H.; Nakane, A.; Shinagawa, K. Comprehensive analysis of classical and newly described staphylococcal superantigenic toxin genes in Staphylococcus aureus isolates. FEMS Microbiol. Lett. 2005, 246, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Alibayov, B.; Baba-Moussa, L.; Sina, H.; Zdeňková, K.; Demnerová, K. Staphylococcus aureus mobile genetic elements. Mol. Biol. Rep. 2014, 41, 5005–5018. [Google Scholar] [CrossRef] [PubMed]
- Arad, G.; Levy, R.; Nasie, I.; Hillman, D.; Rotfogel, Z.; Barash, U.; Supper, E.; Shpilka, T.; Minis, A.; Kaempfer, R. Binding of superantigen toxins into the CD28 homodimer interface Is essential for induction of cytokine genes that mediate lethal shock. PLoS Biol. 2011, 9. [Google Scholar] [CrossRef] [PubMed]
- Levy, R.; Rotfogel, Z.; Hillman, D.; Popugailo, A.; Arad, G.; Supper, E.; Osman, F.; Kaempfer, R. Superantigens hyperinduce inflammatory cytokines by enhancing the B7-2/CD28 costimulatory receptor interaction. Proc. Natl. Acad. Sci. USA 2016, 113, E6437–E6446. [Google Scholar] [CrossRef] [PubMed]
- Bueno, C.; Lemke, C.D.; Criado, G.; Baroja, M.L.; Ferguson, S.S.G.; Rahman, A.K.M.N.-U.; Tsoukas, C.D.; McCormick, J.K.; Madrenas, J. Bacterial superantigens bypass Lck-dependent T cell receptor signaling by activating a Galpha11-dependent, PLC-beta-mediated pathway. Immunity 2006, 25, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zeppa, J.J.; Hancock, M.A.; McCormick, J.K.; Doherty, T.M.; Hendy, G.N.; Madrenas, J. Staphylococcal superantigens use LAMA2 as a coreceptor to activate T cells. J. Immunol. 2018, 200, 1471–1479. [Google Scholar] [CrossRef] [PubMed]
- Petersson, K.; Pettersson, H.; Skartved, N.J.; Walse, B.; Forsberg, G. Staphylococcal enterotoxin H induces Vα-specific expansion of T cells. J. Immunol. 2003, 170, 4148–4154. [Google Scholar] [CrossRef] [PubMed]
- Fields, B.A.; Malchiodi, E.L.; Li, H.; Ysern, X.; Stauffacher, C.V.; Schlievert, P.M.; Karjalainen, K.; Mariuzza, R.A. Crystal structure of a T-cell receptor β-chain complexed with a superantigen. Nature 1996, 384, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Llera, A.; Malchiodi, E.L.; Mariuzza, R.A. The structural basis of T cell activation by superantigens. Annu. Rev. Immunol. 1999, 17, 435–466. [Google Scholar] [CrossRef] [PubMed]
- Nur-ur Rahman, A.K.M.; Bonsor, D.A.; Herfst, C.A.; Pollard, F.; Peirce, M.; Wyatt, A.W.; Kasper, K.J.; Madrenas, J.; Sundberg, E.J.; McCormick, J.K. The T cell receptor β-chain second complementarity determining region loop (CDR2β) governs T cell activation and Vβ specificity by bacterial superantigens. J. Biol. Chem. 2011, 286, 4871–4881. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.D.; Acharya, K.R. Superantigens: Structure-function relationships. Int. J. Med. Microbiol. 2004, 293, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Papageorgiou, A.C.; Acharya, K.R. Microbial superantigens: From structure to function. Trends Microbiol. 2000, 8, 369–375. [Google Scholar] [CrossRef]
- Papageorgiou, A.C.; Acharya, K.R. Superantigens as immunomodulators: Recent structural insights. Structure 1997, 5, 991–996. [Google Scholar] [CrossRef]
- Lina, G.; Bohach, G.A.; Nair, S.P.; Hiramatsu, K.; Jouvin-Marche, E.; Mariuzza, R. Standard nomenclature for the superantigens expressed by Staphylococcus. J. Infect. Dis. 2004, 189, 2334–2336. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.L.; Nakane, A. Mechanisms of staphylococcal enterotoxin-induced emesis. Eur. J. Pharmacol. 2014, 722, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Sergelidis, D.; Angelidis, A.S. Methicillin-resistant Staphylococcus aureus: A controversial food-borne pathogen. Lett. Appl. Microbiol. 2017, 64, 409–418. [Google Scholar] [CrossRef] [PubMed]
- McCormick, J.K.; Yarwood, J.M.; Schlievert, P.M. Toxic shock syndrome and bacterial superantigens: An update. Annu. Rev. Microbiol. 2001, 55, 77–104. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-J.; Hu, D.-L.; Maina, E.K.; Shinagawa, K.; Omoe, K.; Nakane, A. Superantigenic activity of toxic shock syndrome toxin-1 is resistant to heating and digestive enzymes. J. Appl. Microbiol. 2011, 110, 729–736. [Google Scholar] [CrossRef] [PubMed]
- Langley, R.; Wines, B.; Willoughby, N.; Basu, I.; Proft, T.; Fraser, J.D. The staphylococcal superantigen-like protein 7 binds IgA and complement C5 and inhibits IgA-Fc alpha RI binding and serum killing of bacteria. J. Immunol. 2005, 174, 2926–2933. [Google Scholar] [CrossRef] [PubMed]
- Bestebroer, J.; Poppelier, M.J.J.G.; Ulfman, L.H.; Lenting, P.J.; Denis, C.V.; van Kessel, K.P.M.; van Strijp, J.A.G.; de Haas, C.J.C. Staphylococcal superantigen-like 5 binds PSGL-1 and inhibits P-selectin-mediated neutrophil rolling. Blood 2007, 109, 2936–2943. [Google Scholar] [CrossRef] [PubMed]
- Koymans, K.J.; Bisschop, A.; Vughs, M.M.; van Kessel, K.P.M.; de Haas, C.J.C.; van Strijp, J.A.G. Staphylococcal Superantigen-Like Protein 1 and 5 (SSL1 & SSL5) Limit Neutrophil Chemotaxis and Migration through MMP-Inhibition. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef]
- Xu, S.X.; McCormick, J.K. Staphylococcal superantigens in colonization and disease. Front. Cell. Infect. Microbiol. 2012, 2, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wilson, G.J.; Seo, K.S.; Cartwright, R.A.; Connelley, T.; Chuang-Smith, O.N.; Merriman, J.A.; Guinane, C.M.; Park, J.Y.; Bohach, G.A.; Schlievert, P.M.; et al. A novel core genome-encoded superantigen contributes to lethality of community-associated MRSA necrotizing pneumonia. PLoS Pathog. 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Okumura, K.; Shimomura, Y.; Murayama, S.Y.; Yagi, J.; Ubukata, K.; Kirikae, T.; Miyoshi-Akiyama, T. Evolutionary paths of streptococcal and staphylococcal superantigens. BMC Genom. 2012, 13, 404. [Google Scholar] [CrossRef] [PubMed]
- Roetzer, A.; Haller, G.; Beyerly, J.; Geier, C.B.; Wolf, H.M.; Gruener, C.S.; Model, N.; Eibl, M.M. Genotypic and phenotypic analysis of clinical isolates of Staphylococcus aureus revealed production patterns and hemolytic potentials unlinked to gene profiles and source. BMC Microbiol. 2016, 16. [Google Scholar] [CrossRef] [PubMed]
- Van Wamel, W.J.B.; Rooijakkers, S.H.M.; Ruyken, M.; Van Kessel, K.P.M.; Van Strijp, J.A.G. The innate immune modulators staphylococcal complement inhibitor and chemotaxis inhibitory protein of Staphylococcus aureus are located on β-hemolysin-converting bacteriophages. J. Bacteriol. 2006, 188, 1310–1315. [Google Scholar] [CrossRef] [PubMed]
- Van Belkum, A.; Melles, D.C.; Snijders, S.V.; Van Leeuwen, W.B.; Wertheim, H.F.L.; Nouwen, J.L.; Verbrugh, H.A.; Etienne, J. Clonal distribution and differential occurrence of the enterotoxin gene cluster, egc, in carriage-versus bacteremia-associated isolates of Staphylococcus aureus. J. Clin. Microbiol. 2006, 44, 1555–1557. [Google Scholar] [CrossRef] [PubMed]
- Letertre, C.; Perelle, S.; Dilasser, F.; Fach, P. Identification of a new putative enterotoxin SEU encoded by the egc cluster of Staphylococcus aureus. J. Appl. Microbiol. 2003, 95, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Recsei, P.; Kreiswirth, B.; O’Reilly, M.; Schlievert, P.; Gruss, A.; Novick, R.P. Regulation of exoprotein gene expression in Staphylococcus aureus by agr. Mol. Gen. Genet. 1986, 202, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Baroja, M.L.; Herfst, C.A.; Kasper, K.J.; Xu, S.X.; Gillett, D.A.; Li, J.; Reid, G.; McCormick, J.K. The SaeRS two-component system is a direct and dominant transcriptional activator of toxic shock syndrome toxin 1 in Staphylococcus aureus. J. Bacteriol. 2016, 198, 2732–2742. [Google Scholar] [CrossRef] [PubMed]
- Andrey, D.O.; Renzoni, A.; Monod, A.; Lew, D.P.; Cheung, A.L.; Kelley, W.L. Control of the Staphylococcus aureus toxic shock tst promoter by the global regulator SarA. J. Bacteriol. 2010, 192, 6077–6085. [Google Scholar] [CrossRef] [PubMed]
- Andrey, D.O.; Jousselin, A.; Villanueva, M.; Renzoni, A.; Monod, A.; Barras, C.; Rodriguez, N.; Kelley, W.L. Impact of the regulators SigB, Rot, SarA and SarS on the toxic shock tst promoter and TSST-1 expression in Staphylococcus aureus. PLoS ONE 2015, 10, e0135579. [Google Scholar] [CrossRef] [PubMed]
- Seidl, K.; Bischoff, M.; Berger-Bächi, B. CcpA mediates the catabolite repression of tst in Staphylococcus aureus. Infect. Immun. 2008, 76, 5093–5099. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.A.; Donegan, N.P.; Kwan, W.A., Jr.; Cheung, A. Influences of σB and agr on expression of staphylococcal enterotoxin B (seb) in Staphylococcus aureus. Can. J. Microbiol. 2004, 50, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Tseng, C.W.; Stewart, G.C. Rot repression of enterotoxin B expression in Staphylococcus aureus. J. Bacteriol. 2005, 187, 5301–5309. [Google Scholar] [CrossRef] [PubMed]
- Sato ’o, Y.; Hisatsune, J.; Nagasako, Y.; Ono, H.K.; Omoe, K.; Sugai, M. Positive Regulation of Staphylococcal Enterotoxin H by Rot (Repressor of Toxin) Protein and Its Importance in Clonal Complex 81 Subtype 1 Lineage-Related Food Poisoning. Appl. Environ. Microbiol. 2015, 81, 7782–7790. [Google Scholar] [CrossRef] [PubMed]
- Kusch, K.; Hanke, K.; Holtfreter, S.; Schmudde, M.; Kohler, C.; Erck, C.; Wehland, J.; Hecker, M.; Ohlsen, K.; Bröker, B.; et al. The influence of SaeRS and σB on the expression of superantigens in different Staphylococcus aureus isolates. Int. J. Med. Microbiol. 2011, 301, 488–499. [Google Scholar] [CrossRef] [PubMed]
- Nygaard, T.K.; Pallister, K.B.; Ruzevich, P.; Griffith, S.; Vuong, C.; Voyich, J.M. SaeR Binds a Consensus Sequence within Virulence Gene Promoters to Advance USA300 Pathogenesis. J. Infect. Dis. 2010, 201, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Langley, R.J.; Tian, Y.; Clow, F.; Young, P.G.; Radcliff, F.J.; Choi, M.; Sequeira, R.P.; Holtfreter, S.; Baker, H.; Fraser, J.D. Staphylococcal enterotoxin-like X (SElX) is a unique superantigen with functional features of two major families of staphylococcal virulence factors. PLoS Pathog. 2017, 13, e1006549. [Google Scholar] [CrossRef] [PubMed]
- Stach, C.S.; Vu, B.G.; Merriman, J.A.; Herrera, A.; Cahill, M.P.; Schlievert, P.M.; Salgado-Pabón, W. Novel tissue level effects of the Staphylococcus aureus enterotoxin gene cluster are essential for infective endocarditis. PLoS ONE 2016, 11, e0154762. [Google Scholar] [CrossRef] [PubMed]
- Collery, M.M.; Smyth, C.J. Rapid differentiation of Staphylococcus aureus isolates harbouring egc loci with pseudogenes ψent1 and ψent2 and the selu or seluv gene using PCR-RFLP. J. Med. Microbiol. 2007, 56, 208–216. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Dauwalder, O.; Brun, V.; Badiou, C.; Ferry, T.; Etienne, J.; Vandenesch, F.; Lina, G. Staphylococcus aureus superantigens elicit redundant and extensive human Vβ patterns. Infect. Immun. 2009, 77, 2043–2050. [Google Scholar] [CrossRef] [PubMed]
- Grumann, D.; Scharf, S.S.; Holtfreter, S.; Kohler, C.; Steil, L.; Engelmann, S.; Hecker, M.; Volker, U.; Broker, B.M. Immune cell activation by enterotoxin gene cluster (egc)-encoded and non-egc superantigens from Staphylococcus aureus. J. Immunol. 2008, 181, 5054–5061. [Google Scholar] [CrossRef] [PubMed]
- Scholl, P.R.; Diez, A.; Geha, R.S. Staphylococcal enterotoxin B and toxic shock syndrome toxin-1 bind to distinct sites on HLA-DR and HLA-DQ molecules. J. Immunol. 1989, 143, 2583–2588. [Google Scholar] [PubMed]
- Scholl, P.R.; Diez, A.; Karr, R.; Sekaly, R.P.; Trowsdale, J.; Geha, R.S. Effect of isotypes and allelic polymorphism on the binding of staphylococcal exotoxins to MHC class II molecules. J. Immunol. 1990, 144, 226–230. [Google Scholar] [PubMed]
- Krogman, A.; Tilahun, A.; David, C.S.; Chowdhary, V.R.; Alexander, M.P.; Rajagopalan, G. HLA-DR polymorphisms influence in vivo responses to staphylococcal toxic shock syndrome toxin-1 in a transgenic mouse model. Hla 2017, 89, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, G. Gram-positive and gram-negative bacterial toxins in sepsis: A brief review. Virulence 2014, 5, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Lappin, E.; Ferguson, A.J. Gram-positive toxic shock syndromes. Lancet Infect. Dis. 2009, 9, 281–290. [Google Scholar] [CrossRef]
- Davis, C.C.; Baccam, M.; Mantz, M.J.; Osborn, T.W.; Hill, D.R.; Squier, C.A. Use of porcine vaginal tissue ex-vivo to model environmental effects on vaginal mucosa to toxic shock syndrome toxin-1. Toxicol. Appl. Pharmacol. 2014, 274, 240–248. [Google Scholar] [CrossRef] [PubMed]
- MacPhee, R.A.; Miller, W.L.; Gloor, G.B.; McCormick, J.K.; Hammond, J.A.; Burton, J.P.; Reid, G. Influence of the vaginal microbiota on toxic shock syndrome toxin 1 production by Staphylococcus aureus. Appl. Environ. Microbiol. 2013, 79, 1835–1842. [Google Scholar] [CrossRef] [PubMed]
- Elkins, C.A.; Munoz, M.E.; Mullis, L.B.; Stingley, R.L.; Hart, M.E. Lactobacillus-mediated inhibition of clinical toxic shock syndrome Staphylococcus aureus strains and its relation to acid and peroxide production. Anaerobe 2008, 14, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, W.; Xu, S.X.; Magarvey, N.A.; McCormick, J.K. Lactobacillus reuteri-produced cyclic dipeptides quench agr-mediated expression of toxic shock syndrome toxin-1 in staphylococci. Proc. Natl. Acad. Sci. USA 2011, 108, 3360–3365. [Google Scholar] [CrossRef] [PubMed]
- Gajer, P.; Brotman, R.M.; Bai, G.; Sakamoto, J.; Schütte, U.M.; Zhong, X.; Koenig, S.S.; Fu, L.; Ma, Z.; Zhou, X.; et al. Temporal dynamics of the human vaginal microbiota. Sci. Transl. Med. 2012, 4, 132–152. [Google Scholar] [CrossRef] [PubMed]
- Peterson, M.L.; Ault, K.; Kremer, M.J.; Klingelhutz, A.J.; Davis, C.C.; Squier, C.A.; Schlievert, P.M. The innate immune system is activated by stimulation of vaginal epithelial cells with Staphylococcus aureus and toxic shock syndrome toxin 1. Infect. Immun. 2005, 73, 2164–2174. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.C.; Kremer, M.J.; Schlievert, P.M.; Squier, C.A. Penetration of toxic shock syndrome toxin-1 across porcine vaginal mucosa ex vivo: Permeability characteristics, toxin distribution, and tissue damage. Am. J. Obstet. Gynecol. 2003, 189, 1785–1791. [Google Scholar] [CrossRef]
- Hajjeh, R.A.; Reingold, A.; Weil, A.; Shutt, K.; Schuchat, A.; Perkins, B.A. Toxic shock syndrome in the United States: Surveillance update, 1979-1996. Emerg. Infect. Dis. 1999, 5, 807–810. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.A.; Bisch, S.; Arntfield, S.; Hosseini-Moghaddam, S.M. A confirmed case of toxic shock syndrome associated with the use of a menstrual cup. Can. J. Infect. Dis. Med. Microbiol. 2015, 26, 218–220. [Google Scholar] [CrossRef] [PubMed]
- Chatzopoulou, M.; Koufakis, T.; Ntava, E.; Gabranis, I.; Tsiakalou, M. Intense, flu-like symptoms in women using menstrual devices: Always think of staphylococcal Toxic Shock Syndrome. Oxford Med. Case Rep. 2017, 5, 70–72. [Google Scholar] [CrossRef]
- Strandberg, K.L.; Rotschafer, J.H.; Vetter, S.M.; Buonpane, R.A.; Kranz, D.M.; Schlievert, P.M. Staphylococcal superantigens cause lethal pulmonary disease in rabbits. J. Infect. Dis. 2010, 202, 1690–1697. [Google Scholar] [CrossRef] [PubMed]
- Tuffs, S.W.; James, D.B.A.; Bestebroer, J.; Richards, A.C.; Goncheva, M.I.; O’Shea, M.; Wee, B.A.; Seo, K.S.; Schlievert, P.M.; Lengeling, A.; van Strijp, J.A.; Torres, V.J.; Fitzgerald, J.R. The Staphylococcus aureus superantigen SElX is a bifunctional toxin that inhibits neutrophil function. PLoS Pathog. 2017, 13, e1006461. [Google Scholar] [CrossRef] [PubMed]
- Salgado-Pabón, W.; Breshears, L.; Spaulding, A.R.; Merriman, J.A.; Stach, C.S.; Horswill, A.R.; Peterson, M.L.; Schlievert, P.M. Superantigens are critical for Staphylococcus aureus infective endocarditis, sepsis, and acute kidney injury. mBio 2013, 4. [Google Scholar] [CrossRef] [PubMed]
- Krakauer, T.; Stiles, B.G. The staphylococcal enterotoxin (SE) family. Virulence 2013, 4, 759–773. [Google Scholar] [CrossRef] [PubMed]
- Chowdhary, V.R.; Tilahun, A.Y.; Clark, C.R.; Grande, J.P.; Rajagopalan, G. Chronic exposure to staphylococcal superantigen elicits a systemic inflammatory disease mimicking lupus. J. Immunol. 2012, 189, 2054–2062. [Google Scholar] [CrossRef] [PubMed]
- Dar, S.A.; Janahi, E.M.A.; Haque, S.; Akhter, N.; Jawed, A.; Wahid, M.; Ramachandran, V.G.; Bhattacharya, S.N.; Banerjee, B.D.; Das, S. Superantigen influence in conjunction with cytokine polymorphism potentiates autoimmunity in systemic lupus erythematosus patients. Immunol. Res. 2016, 64, 1001–1012. [Google Scholar] [CrossRef] [PubMed]
- MacIas, E.S.; Pereira, F.A.; Rietkerk, W.; Safai, B. Superantigens in dermatology. J. Am. Acad. Dermatol. 2011, 64, 455–472. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, K.; Fukaya, T. The role of superantigens of group A Streptococcus and Staphylococcus aureus in Kawasaki disease. Curr. Opin. Infect. Dis. 2007, 20, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Vu, B.G.; Stach, C.S.; Kulhankova, K.; Salgado-Pabón, W.; Klingelhutz, A.J.; Schlievert, P.M. Chronic superantigen exposure induces systemic inflammation, elevated bloodstream endotoxin, and abnormal glucose tolerance in rabbits: Possible role in diabetes. mBio 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Banke, E.; Rödström, K.; Ekelund, M.; Dalla-Riva, J.; Lagerstedt, J.O.; Nilsson, S.; Degerman, E.; Lindkvist-Petersson, K.; Nilson, B. Superantigen activates the gp130 receptor on adipocytes resulting in altered adipocyte metabolism. Metabolism 2014, 63, 831–840. [Google Scholar] [CrossRef] [PubMed]
- Peton, V.; Le Loir, Y. Staphylococcus aureus in veterinary medicine. Infect. Genet. Evol. 2014, 21, 602–615. [Google Scholar] [CrossRef] [PubMed]
- Smyth, D.S.; Hartigan, P.J.; Meaney, W.J.; Fitzgerald, J.R.; Deobald, C.F.; Bohach, G.A.; Smyth, C.J. Superantigen genes encoded by the egc cluster and SaPlbov are predominant among Staphylococcus aureus isolates from cows, goats, sheep, rabbits and poultry. J. Med. Microbiol. 2005, 54, 401–411. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, J.R.; Hartigan, P.J.; Meaney, W.J.; Smyth, C.J. Molecular population and virulence factor analysis of Staphylococcus aureus from bovine intramammary infection. J. Appl. Microbiol. 2000, 88, 1028–1037. [Google Scholar] [CrossRef] [PubMed]
- Haveri, M.; Roslöf, A.; Rantala, L.; Pyörälä, S. Virulence genes of bovine Staphylococcus aureus from persistent and nonpersistent intramammary infections with different clinical characteristics. J. Appl. Microbiol. 2007, 103, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, J.R.; Monday, S.R.; Foster, T.J.; Bohach, G.A.; Hartigan, P.J.; Meaney, W.J.; Smyth, C.J. Characterization of a putative pathogenicity island from bovine Staphylococcus aureus encoding multiple superantigens. J. Bacteriol. 2001, 183, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Kuroishi, T.; Komine, K.; Kai, K.; Itagaki, M.; Kobayashi, J.; Ohta, M.; Kamata, S.; Kumagai, K. Concentrations and specific antibodies to staphylococcal enterotoxin-C and toxic shock syndrome toxin-1 in bovine mammary gland secretions, and inflammatory response to the intramammary inoculation of these toxins. J. Vet. Med. Sci. 2003, 65, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Allard, M.; Ster, C.; Jacob, C.L.; Scholl, D.; Diarra, M.S.; Lacasse, P.; Malouin, F. The expression of a putative exotoxin and an ABC transporter during bovine intramammary infection contributes to the virulence of Staphylococcus aureus. Vet. Microbiol. 2013, 162, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Bröker, B.M.; Holtfreter, S.; Bekeredjian-Ding, I. Immune control of Staphylococcus aureus—Regulation and counter-regulation of the adaptive immune response. Int. J. Med. Microbiol. 2014, 304, 204–214. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, G.; Tilahun, A.Y.; Asmann, Y.W.; David, C.S. Early gene expression changes induced by the bacterial superantigen staphylococcal enterotoxin B and its modulation by a proteasome inhibitor. Physiol. Genom. 2009, 37, 279–293. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Brown, A.F.; Murphy, A.G.; Lalor, S.J.; Leech, J.M.; O’Keeffe, K.M.; Mac Aogáin, M.; O’Halloran, D.P.; Lacey, K.A.; Tavakol, M.; Hearnden, C.H.; et al. Memory Th1 cells are protective in invasive Staphylococcus aureus infection. PLoS Pathog. 2015, 11, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Ibrahim, A.S.; Xu, X.; Farber, J.M.; Avanesian, V.; Baquir, B.; Fu, Y.; French, S.W.; Edwards, J.E.; Spellberg, B. Th1-Th17 cells mediate protective adaptive immunity against Staphylococcus aureus and Candida albicans infection in mice. PLoS Pathog. 2009, 5. [Google Scholar] [CrossRef] [PubMed]
- Salgado-Pabón, W.; Schlievert, P.M. Models matter: The search for an effective Staphylococcus aureus vaccine. Nat. Rev. Microbiol. 2014, 12, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Yeung, R.S.; Penninger, J.M.; Kündig, T.; Khoo, W.; Ohashi, P.S.; Kroemer, G.; Mak, T.W. Human CD4 and human major histocompatibility complex class II (DQ6) transgenic mice: Supersensitivity to superantigen-induced septic shock. Eur. J. Immunol. 1996, 26, 1074–1082. [Google Scholar] [CrossRef] [PubMed]
- Mollick, J.A.; Chintagumpala, M.; Cook, R.G.; Rich, R.R. Staphylococcal exotoxin activation of T cells. Role of exotoxin-MHC class II binding affinity and class II isotype. J. Immunol. 1991, 146, 463–468. [Google Scholar] [PubMed]
- Szabo, P.A.; Rudak, P.T.; Choi, J.; Xu, S.X.; Schaub, R.; Singh, B.; McCormick, J.K.; Haeryfar, S.M.M. Invariant NKT cells are pathogenic in the HLA-DR4-transgenic humanized mouse model of toxic shock syndrome and can be targeted to reduce morbidity. J. Infect. Dis. 2016, 215, 824–829. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xu, S.X.; Gilmore, K.J.; Szabo, P.A.; Zeppa, J.J.; Baroja, M.L.; Haeryfar, S.M.M.; McCormick, J.K. Superantigens subvert the neutrophil response to promote abscess formation and enhance Staphylococcus aureus survival in vivo. Infect. Immun. 2014, 82, 3588–3598. [Google Scholar] [CrossRef] [PubMed]
- Norrby-Teglund, A.; Nepom, G.T.; Kotb, M. Differential presentation of group A streptococcal superantigens by HLA class II DQ and DR alleles. Eur. J. Immunol. 2002, 32, 2570–2577. [Google Scholar] [CrossRef]
- Krakauer, T.; Pradhan, K.; Stiles, B.G. Staphylococcal superantigens spark host-mediated danger signals. Front. Immunol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Islander, U.; Andersson, A.; Lindberg, E.; Adlerberth, I.; Wold, A.E.; Rudin, A. Superantigenic Staphylococcus aureus stimulates production of interleukin-17 from memory but not naive T cells. Infect. Immun. 2010, 78, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Szabo, P.A.; Goswami, A.; Mazzuca, D.M.; Kim, K.; O’Gorman, D.B.; Hess, D.A.; Welch, I.D.; Young, H.A.; Singh, B.; McCormick, J.K.; et al. Rapid and rigorous IL-17A production by a distinct subpopulation of effector memory T lymphocytes constitutes a novel mechanism of toxic shock syndrome immunopathology. J. Immunol. 2017, 198, 2805–2818. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, L.; Cooper, A.; Fantino, C.; Altmann, D.M.; Sriskandan, S. The mechanism of superantigen-mediated toxic shock: Not a simple Th1 cytokine storm. J. Immunol. 2005, 175, 6870–6877. [Google Scholar] [CrossRef] [PubMed]
- Plaza, R.; Rodriguez-Sanchez, J.L.; Juarez, C. Staphylococcal enterotoxin B in vivo modulates both gamma interferon receptor expression and ligand-induced activation of signal transducer and activator of transcription 1 in T cells. Infect. Immun. 2007, 75, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Björkander, S.; Hell, L.; Johansson, M.A.; Forsberg, M.M.; Lasaviciute, G.; Roos, S.; Holmlund, U.; Sverremark-Ekström, E. Staphylococcus aureus-derived factors induce IL-10, IFN-γ and IL-17A-expressing FOXP3+CD161+T-helper cells in a partly monocyte-dependent manner. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.L.; Cross, E.L.A.; Llewelyn, M.J. Induction of contact-dependent CD8 + regulatory T cells through stimulation with staphylococcal and streptococcal superantigens. Immunology 2012, 135, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Taylor, A.L.; Llewelyn, M.J. Superantigen-induced proliferation of human CD4+CD25- T cells is followed by a switch to a functional regulatory phenotype. J. Immunol. 2010, 185, 6591–6598. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Park, N.; Park, J.Y.; Kaplan, B.L.F.; Pruett, S.B.; Park, J.W.; Park, Y.H.; Seo, K.S. Induction of immunosuppressive CD8 + CD25 + FOXP3 + regulatory T cells by suboptimal stimulation with staphylococcal enterotoxin C1. J. Immunol. 2018, 200, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Leech, J.M.; Lacey, K.A.; Mulcahy, M.E.; Medina, E.; McLoughlin, R.M. IL-10 plays opposing roles during Staphylococcus aureus systemic and localized infections. J. Immunol. 2017, 198, 2352–2365. [Google Scholar] [CrossRef] [PubMed]
- Chau, T.A.; McCully, M.L.; Brintnell, W.; An, G.; Kasper, K.J.; Vinés, E.D.; Kubes, P.; Haeryfar, S.M.M.; McCormick, J.K.; Cairns, E.; et al. Toll-like receptor 2 ligands on the staphylococcal cell wall downregulate superantigen-induced T cell activation and prevent toxic shock syndrome. Nat. Med. 2009, 15, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Tilahun, A.Y.; Chowdhary, V.R.; David, C.S.; Rajagopalan, G. Systemic inflammatory response elicited by superantigen destabilizes T regulatory cells, rendering them ineffective during toxic shock syndrome. J. Immunol. 2014, 193, 2919–2930. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.R.O.; Janik, D.K.; Lee, W.T. Superantigen-induced CD4 memory T cell anergy. I. Staphylococcal enterotoxin B induces Fyn-mediated negative signaling. Cell. Immunol. 2012, 276, 16–25. [Google Scholar] [CrossRef] [PubMed]
- William T Lee, D.K.J. Staphylococcal enterotoxin B (SEB) induces memory CD4 T cell anergy in vivo and impairs eecall immunity to unrelated antigens. J. Clin. Cell. Immunol. 2015, 06, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kolata, J.B.; Kühbandner, I.; Link, C.; Normann, N.; Vu, C.H.; Steil, L.; Weidenmaier, C.; Bröker, B.M. The fall of a Dogma? Unexpected high T-cell memory response to Staphylococcus aureus in humans. J. Infect. Dis. 2015, 212, 830–838. [Google Scholar] [CrossRef] [PubMed]
- Holtfreter, S.; Bauer, K.; Thomas, D.; Feig, C.; Lorenz, V.; Roschack, K.; Friebe, E.; Selleng, K.; Lövenich, S.; Greve, T.; et al. egc-encoded superantigens from Staphylococcus aureus are neutralized by human sera much less efficiently than are classical staphylococcal enterotoxins or toxic shock syndrome toxin. Infect. Immun. 2004, 72, 4061–4071. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-Y.; Kim, J.-S.; Woo, H. Prevalence of antibody to toxic shock syndrome toxin-1 in burn patients. Ann. Lab. Med. 2015, 35, 89–93. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Parsonnet, J.; Goering, R.V.; Hansmann, M.A.; Jones, M.B.; Ohtagaki, K.; Davis, C.C.; Totsuka, K. Prevalence of toxic shock syndrome toxin 1 (TSST-1)-producing strains of Staphylococcus aureus and antibody to TSST-1 among healthy Japanese women. J. Clin. Microbiol. 2008, 46, 2731–2738. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.E.; Borgogna, T.R.; Patel, D.M.; Sward, E.W.; Voyich, J.M. Epic immune battles of history: Neutrophils vs. Staphylococcus aureus. Front. Cell. Infect. Microbiol. 2017, 7, 1–19. [Google Scholar] [CrossRef]
- McGuinness, W.; Kobayashi, S.; DeLeo, F. Evasion of neutrophil killing by Staphylococcus aureus. Pathogens 2016, 5, 32. [Google Scholar] [CrossRef] [PubMed]
- Tzianabos, A.O.; Mcloughlin, R.M.; Lee, J.C.; Kasper, D.L. IFN-γ regulated chemokine production determines the outcome of Staphylococcus aureus infection. J. Immunol. 2008, 181, 1323–1332. [Google Scholar] [CrossRef]
- Szabo, P.A.; Goswami, A.; Memarnejadian, A.; Mallett, C.L.; Foster, P.J.; McCormick, J.K.; Haeryfar, S.M.M. Swift intrahepatic accumulation of granulocytic myeloid-derived suppressor cells in a humanized mouse model of toxic shock syndrome. J. Infect. Dis. 2016, 213, 1990–1995. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A. The growing diversity and spectrum of action of myeloid-derived suppressor cells. Eur. J. Immunol. 2010, 40, 3317–3320. [Google Scholar] [CrossRef] [PubMed]
- Brosnahan, A.J.; Schlievert, P.M. Gram-positive bacterial superantigen outside-in signaling causes toxic shock syndrome. FEBS J. 2011, 278, 4649–4667. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Yoon, Y.H.; Kwak, S.Y.; Rha, K.-S.; Kim, Y.M. Staphylococcal enterotoxin B induced expression of IL-17A in nasal epithelial cells and its association with pathogenesis of nasal polyposis. Eur. Arch. Oto-Rhino-Laryngol. 2013, 271, 525–534. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.-L.; Dong, Z. Proinflammatory impact of Staphylococcus aureus enterotoxin B on human nasal epithelial cells and inhibition by dexamethasone. Am. J. Rhinol. Allergy 2009, 23, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Derycke, L.; Zhang, N.; Holtappels, G.; Dutré, T.; Bachert, C. IL-17A as a regulator of neutrophil survival in nasal polyp disease of patients with and without cystic fibrosis. J. Cyst. Fibros. 2012, 11, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Kissner, T.L.; Ruthel, G.; Alam, S.; Ulrich, R.G.; Fernandez, S.; Saikh, K.U. Activation of MyD88 signaling upon staphylococcal enterotoxin binding to MHC class II molecules. PLoS ONE 2011, 6. [Google Scholar] [CrossRef] [PubMed]
- Bright, J.J.; Xin, Z.; Sriram, S. Superantigens augment antigen-specific Th1 responses by inducing IL-12 production in macrophages. J. Leukoc. Biol. 1999, 65, 665–670. [Google Scholar] [CrossRef] [PubMed]
- Flammier, S.; Rasigade, J.P.; Badiou, C.; Henry, T.; Vandenesch, F.; Laurent, F.; Trouillet-Assant, S. Human monocyte-derived osteoclasts are targeted by staphylococcal pore-forming toxins and superantigens. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- King, J.M.; Kulhankova, K.; Stach, C.S.; Vu, B.G.; Salgado-Pabón, W. Phenotypes and virulence among Staphylococcus aureus USA100, USA200, USA300, USA400, and USA600 clonal lineages. mSphere 2016, 1, e00071-16. [Google Scholar] [CrossRef] [PubMed]
- Shaler, C.R.; Choi, J.; Rudak, P.T.; Memarnejadian, A.; Szabo, P.A.; Tun-Abraham, M.E.; Rossjohn, J.; Corbett, A.J.; McCluskey, J.; McCormick, J.K.; et al. MAIT cells launch a rapid, robust and distinct hyperinflammatory response to bacterial superantigens and quickly acquire an anergic phenotype that impedes their cognate antimicrobial function: Defining a novel mechanism of superantigen-induced immunopatho. PLoS Biol. 2017, 15, e2001930. [Google Scholar] [CrossRef] [PubMed]
- Hayworth, J.L.; Mazzuca, D.M.; Vareki, S.M.; Welch, I.; McCormick, J.K.; Haeryfar, S.M. CD1d-independent activation of mouse and human iNKT cells by bacterial superantigens. Immunol. Cell Biol. 2012, 90, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Brennan, P.J.; Brigl, M.; Brenner, M.B. Invariant natural killer T cells: An innate activation scheme linked to diverse effector functions. Nat. Rev. Immunol. 2013, 13, 101–117. [Google Scholar] [CrossRef] [PubMed]
- Salou, M.; Franciszkiewicz, K.; Lantz, O. MAIT cells in infectious diseases. Curr. Opin. Immunol. 2017, 48, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Kjer-Nielsen, L.; Patel, O.; Corbett, A.J.; Le Nours, J.; Meehan, B.; Liu, L.; Bhati, M.; Chen, Z.; Kostenko, L.; Reantragoon, R.; et al. MR1 presents microbial vitamin B metabolites to MAIT cells. Nature 2012, 491, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, J.K.; Norrby-Teglund, A.; Leeansyah, E. Bacterial deception of MAIT cells in a cloud of superantigen and cytokines. PLoS Biol. 2017, 15. [Google Scholar] [CrossRef] [PubMed]
- Pasman, L.; Kasper, D.L. Building conventions for unconventional lymphocytes. Immunol. Rev. 2017, 279, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Lawand, M.; Déchanet-Merville, J.; Dieu-Nosjean, M.C. Key features of γδ T-cell subsets in human diseases and their immunotherapeutic implications. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Kalyan, S.; Chow, A.W. Human peripheral γδ T cells potentiate the early proinflammatory cytokine response to staphylococcal toxic shock syndrome toxin-1. J. Infect. Dis. 2004, 189, 1892–1896. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stinissen, P.; Vandevyver, C.; Raus, J.; Zhang, J. Superantigen reactivity of γδ T cell clones isolated from patients with multiple sclerosis and controls. Cell. Immunol. 1995, 166, 227–235. [Google Scholar] [CrossRef] [PubMed]
- Morita, C.T.; Li, H.; Lamphear, J.G.; Rich, R.R.; Fraser, J.D.; Mariuzza, R.A.; Lee, H.K. Superantigen recognition by γδ T cells: SEA recognition site for human Vγ2 T cell receptors. Immunity 2001, 14, 331–344. [Google Scholar] [CrossRef]
- Pérez-Bosque, A.; Miró, L.; Polo, J.; Russell, L.; Campbell, J.; Weaver, E.; Crenshaw, J.; Moretó, M. Dietary plasma proteins modulate the immune response of diffuse gut-associated lymphoid tissue in rats challenged with Staphylococcus aureus enterotoxin B. J. Nutr. 2008, 138, 533–537. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Colpitts, S.L.; Ménoret, A.; Budelsky, A.L.; Lefrancois, L.; Vella, A.T. Rapid αβ T cell responses orchestrate innate immunity in response to Staphylococcal enterotoxin A. Mucosal Immunol. 2013, 6, 1006–1015. [Google Scholar] [CrossRef] [PubMed]
- Burian, M.; Grumann, D.; Holtfreter, S.; Wolz, C.; Goerke, C.; Bröker, B.M. Expression of staphylococcal superantigens during nasal colonization is not sufficient to induce a systemic neutralizing antibody response in humans. Eur. J. Clin. Microbiol. Infect. Dis. 2012, 31, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Verkaik, N.J.; de Vogel, C.P.; Boelens, H.A.; Grumann, D.; Hoogenboezem, T.; Vink, C.; Hooijkaas, H.; Foster, T.J.; Verbrugh, H.A.; van Belkum, A.; et al. Anti-staphylococcal humoral immune response in persistent nasal carriers and noncarriers of Staphylococcus aureus. J. Infect. Dis. 2009, 199, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.X.; Kasper, K.J.; Zeppa, J.J.; McCormick, J.K. Superantigens modulate bacterial density during Staphylococcus aureus nasal colonization. Toxins (Basel) 2015, 7, 1821–1836. [Google Scholar] [CrossRef] [PubMed]
- Krismer, B.; Peschel, A. Does Staphylococcus aureus nasal colonization involve biofilm formation? Future Microbiol. 2011, 6, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Archer, N.K.; Harro, J.M.; Shirtliff, M.E. Clearance of Staphylococcus aureus nasal carriage is T cell dependent and mediated through interleukin-17A expression and neutrophil influx. Infect. Immun. 2013, 81, 2070–2075. [Google Scholar] [CrossRef] [PubMed]
- Nurjadi, D.; Kain, M.; Marcinek, P.; Gaile, M.; Heeg, K.; Zanger, P. Ratio of T-helper type 1 (Th1) to Th17 cytokines in whole blood is associated with human β-defensin 3 expression in skin and persistent Staphylococcus aureus nasal carriage. J. Infect. Dis. 2016, 214, 1744–1751. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Smith, E.J.; Visai, L.; Kerrigan, S.W.; Speziale, P.; Foster, T.J. The Sbi protein is a multifunctional immune evasion factor of Staphylococcus aureus. Infect. Immun. 2011, 79, 3801–3809. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Thammavongsa, V. Recurrent infecitons and immune evasion strategies of Staphylococcus aureus. Curr. Opin. Microbiol. 2012, 15, 92–99. [Google Scholar] [CrossRef] [PubMed]
- O’Halloran, D.P.; Wynne, K.; Geoghegan, J.A. Protein A is released into the Staphylococcus aureus culture supernatant with an unprocessed sorting signal. Infect. Immun. 2015, 83, 1598–1609. [Google Scholar] [CrossRef] [PubMed]
- Ramsland, P.A.; Willoughby, N.; Trist, H.M.; Farrugia, W.; Hogarth, P.M.; Fraser, J.D.; Wines, B.D. Structural basis for evasion of IgA immunity by Staphylococcus aureus revealed in the complex of SSL7 with Fc of human IgA1. Proc. Natl. Acad. Sci. USA 2007, 104, 15051–15056. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Wines, B.D.; Langley, R.J.; Fraser, J.D. Specificity of staphylococcal superantigen-like protein 10 toward the human IgG1 Fc domain. J. Immunol. 2010, 184, 6283–6292. [Google Scholar] [CrossRef] [PubMed]
- Gillman, A.N.; Breshears, L.M.; Kistler, C.K.; Finnegan, P.M.; Torres, V.J.; Schlievert, P.M.; Peterson, M.L. Epidermal growth factor receptor signaling enhances the proinflammatory effects of Staphylococcus aureus gamma-toxin on the mucosa. Toxins (Basel) 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Brosnahan, A.J.; Mantz, M.J.; Squier, C.A.; Peterson, M.L.; Schlievert, P.M. Cytolysins augment superantigen penetration of stratified mucosa. J. Immunol. 2009, 182, 2364–2373. [Google Scholar] [CrossRef] [PubMed]
- Holzinger, D.; Gieldon, L.; Mysore, V.; Nippe, N.; Taxman, D.J.; Duncan, J.A.; Broglie, P.M.; Marketon, K.; Austermann, J.; Vogl, T.; Foell, D.; Niemann, S.; Peters, G.; Roth, J.; Loffler, B. Staphylococcus aureus Panton-Valentine leukocidin induces an inflammatory response in human phagocytes via the NLRP3 inflammasome. J. Leukoc. Biol. 2012, 92, 1069–1081. [Google Scholar] [CrossRef] [PubMed]
- Melehani, J.H.; James, D.B.A.; DuMont, A.L.; Torres, V.J.; Duncan, J.A. Staphylococcus aureus leukocidin A/B (LukAB) kills human monocytes via host NLRP3 and ASC when extracellular, but not intracellular. PLoS Pathog. 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Kebaier, C.; Chamberland, R.R.; Allen, I.C.; Gao, X.; Broglie, P.M.; Hall, J.D.; Jania, C.; Doerschuk, C.M.; Tilley, S.L.; Duncan, J.A. Staphylococcus aureus α-hemolysin mediates virulence in a murine model of severe pneumonia through activation of the NLRP3 inflammasome. J. Infect. Dis. 2012, 205, 807–817. [Google Scholar] [CrossRef] [PubMed]



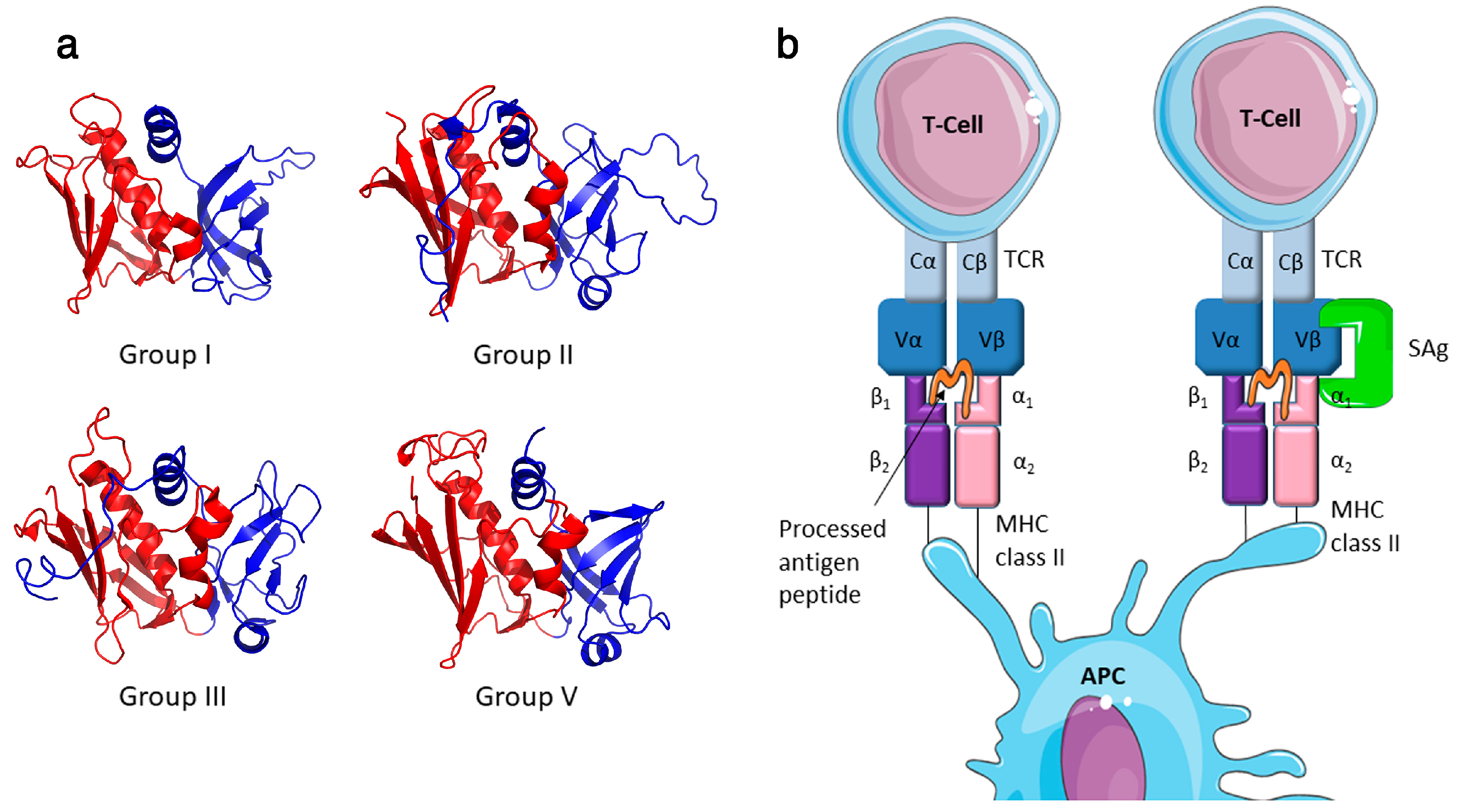{kind=link}
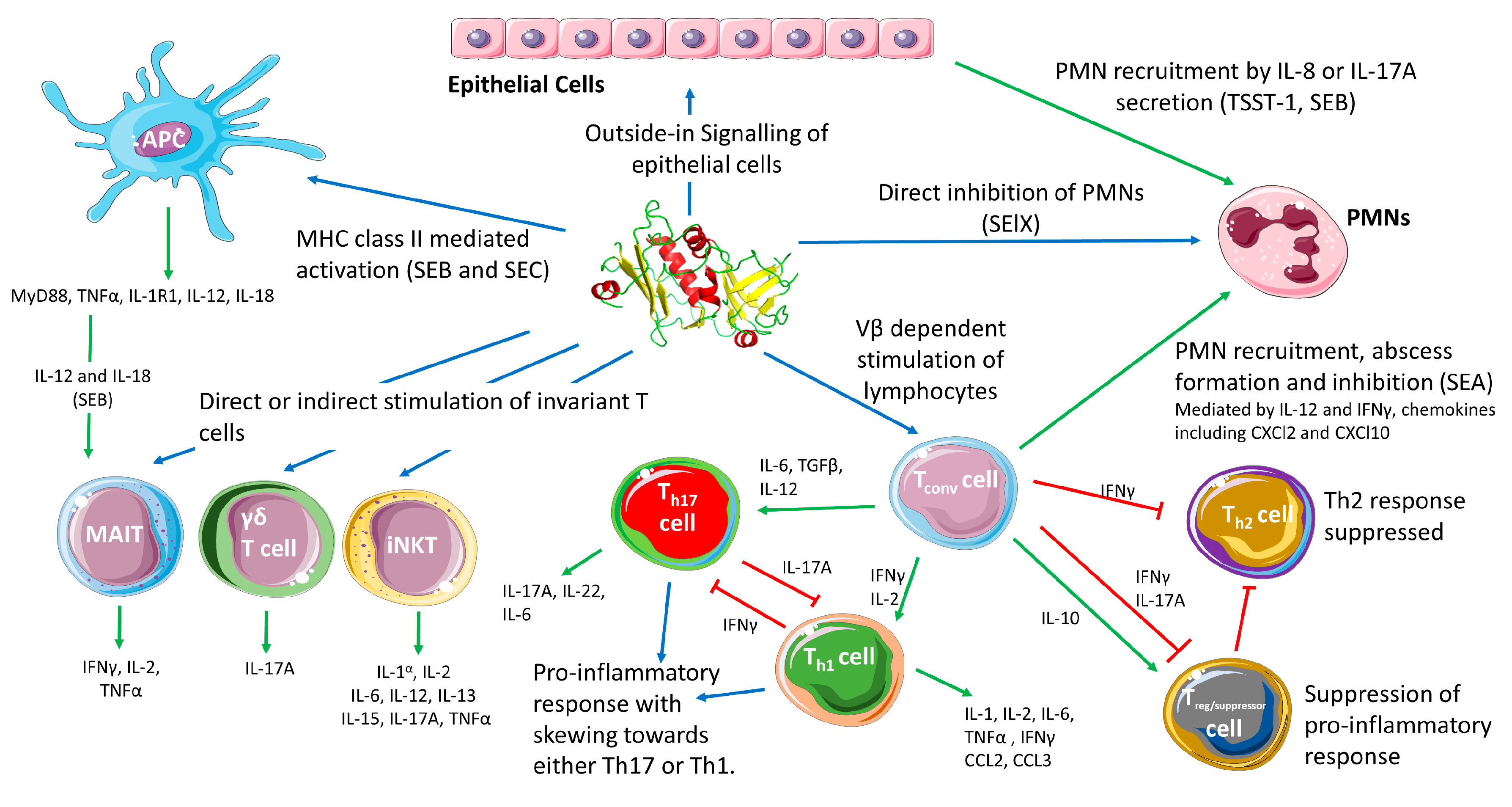{kind=link}
| SAg | Phylogenetic Group | Molecular Mass (kDa) | Emetic | Human Vβ Specificity | MHC Class II Binding a | Associated Mobile Genetic Elements |
|---|---|---|---|---|---|---|
| SEA | III | 27.1 | + | 1, 5, 6, 7, 9, 15, 16, 18, 21, 22, 24 | α + β | ɸSa3n |
| SEB | II | 28.3 | + | 1, 3, 6, 12, 13.2, 14, 15, 17, 20 | α | SaPI |
| SEC | II | 27.5 | + | 3, 12, 13.2, 14, 15, 17, 20 | α | SaPI |
| SED | III | 26.4 | + | 1, 3, 5, 8, 9, 12, 14 | α + β | Plasmid (pIB485-like) |
| SEE | III | 26.4 | + | 5, 6, 8, 9, 13.1, 16, 18, 21 | α + β | Integrated Plasmid |
| SEG | II | 27 | + | 3, 12, 13, 14, 15 | α | vSAβ (egc) |
| SEH | III | 25.2 | + | Vα8, Vα10 | β | ɸSa3mu |
| SEI | V | 24.9 | + b | 1, 5, 6, 23 | β | vSAβ (egc) |
| SElJ | III | 28.5 | NK | 8, 21 | α + β | SaPI/ɸSa3n/Plasmid (pF5/pIB485-like) |
| SElK | V | 26 | - | 1, 5, 6 | β | SaPI |
| SElL | V | 26 | - | 1, 5, 7, 16, 22, 23 | α + β | SaPI |
| SElM | V | 24.8 | + b | 8, 9, 18, 21 | α + β | vSAβ (egc) |
| SElN | III | 26.1 | + b | 7, 8, 9, 17 | α + β | vSAβ (egc) |
| SElO | III | 26.7 | + b | 5, 7 | α + β | vSAβ (egc) |
| SElP | III | 27 | + b | 5, 8, 16, 18, 21 | α + β | ɸSa3n |
| SElQ | V | 28 | - | 6, 21 | α + β | SaPI/ɸSa3n |
| SER | II | 27 | + | 3, 12, 14 | α | Plasmid (pF5/pIB485-like) |
| SES | III | 26.2 | + | 9, 16 | α + β | Plasmid (pF5) |
| SET | I | 22.6 | + b | NK | α | Plasmid (pF5) |
| SElU/U2 | II | 27.1 | NK | 13.2, 14 | α | vSAβ (egc) |
| SElV | V | 25 | NK | 6, 18, 21 | α + β | vSAβ (egc) |
| SElW | III | 27.3 | NK | NK | NK | Chromosomal (Core genome) |
| SElX | I | 19.3 | NK | 1, 6, 18, 21 | NK | Chromosomal (Core genome) |
| SElY | I | 22.5 | +c | NK | NK | Chromosomal |
| SElZ | II | 27.0 | NK | NK | NK | Chromosomal |
| TSST-1 | I | 22 | - | 2 | α | SaPI |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tuffs, S.W.; Haeryfar, S.M.M.; McCormick, J.K. Manipulation of Innate and Adaptive Immunity by Staphylococcal Superantigens. Pathogens 2018, 7, 53. https://doi.org/10.3390/pathogens7020053
Tuffs SW, Haeryfar SMM, McCormick JK. Manipulation of Innate and Adaptive Immunity by Staphylococcal Superantigens. Pathogens. 2018; 7(2):53. https://doi.org/10.3390/pathogens7020053
Chicago/Turabian StyleTuffs, Stephen W., S. M. Mansour Haeryfar, and John K. McCormick. 2018. "Manipulation of Innate and Adaptive Immunity by Staphylococcal Superantigens" Pathogens 7, no. 2: 53. https://doi.org/10.3390/pathogens7020053
APA StyleTuffs, S. W., Haeryfar, S. M. M., & McCormick, J. K. (2018). Manipulation of Innate and Adaptive Immunity by Staphylococcal Superantigens. Pathogens, 7(2), 53. https://doi.org/10.3390/pathogens7020053