Listeria monocytogenes: The Impact of Cell Death on Infection and Immunity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Necrosis and Necroptosis
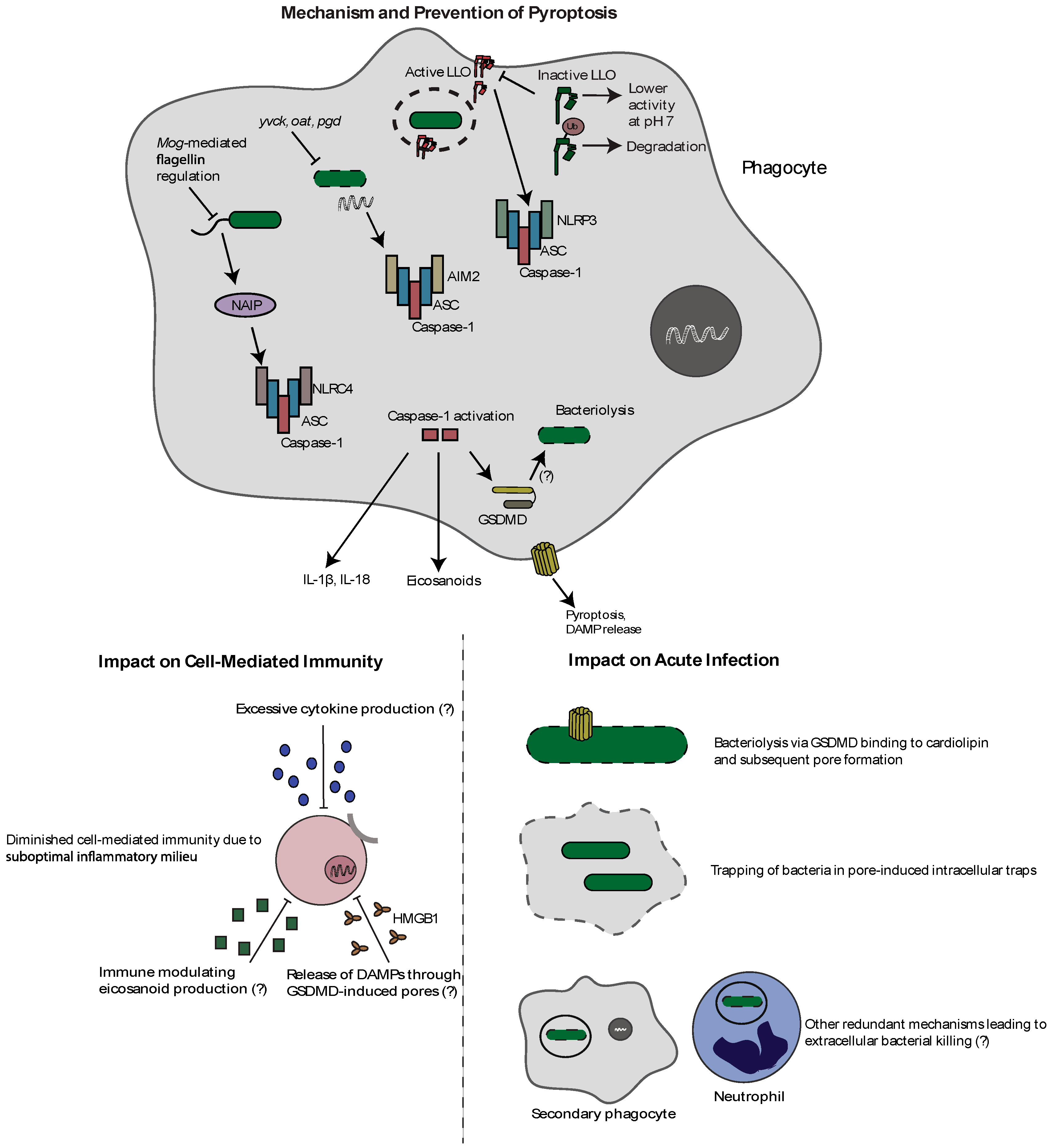
3. Pyroptosis
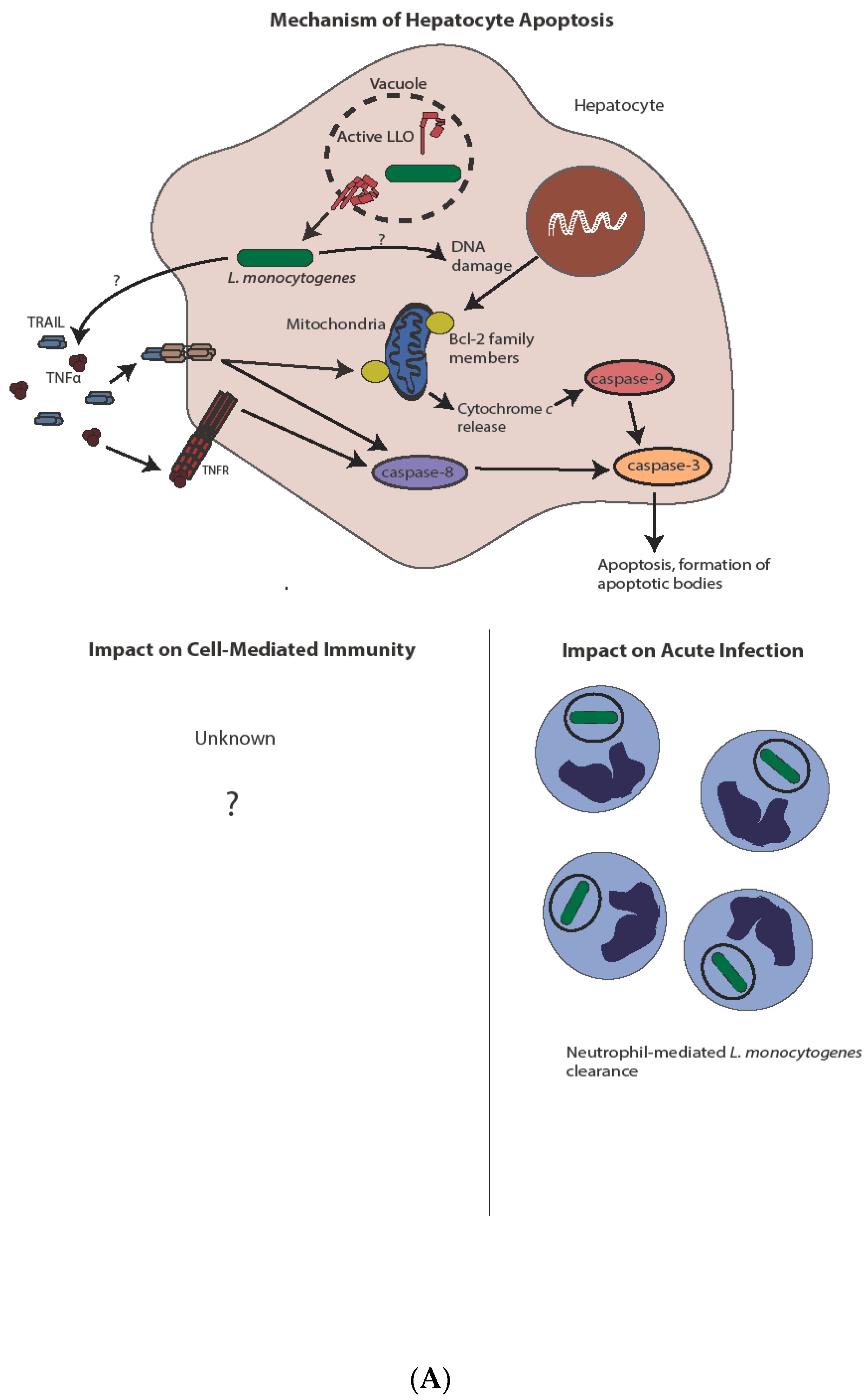
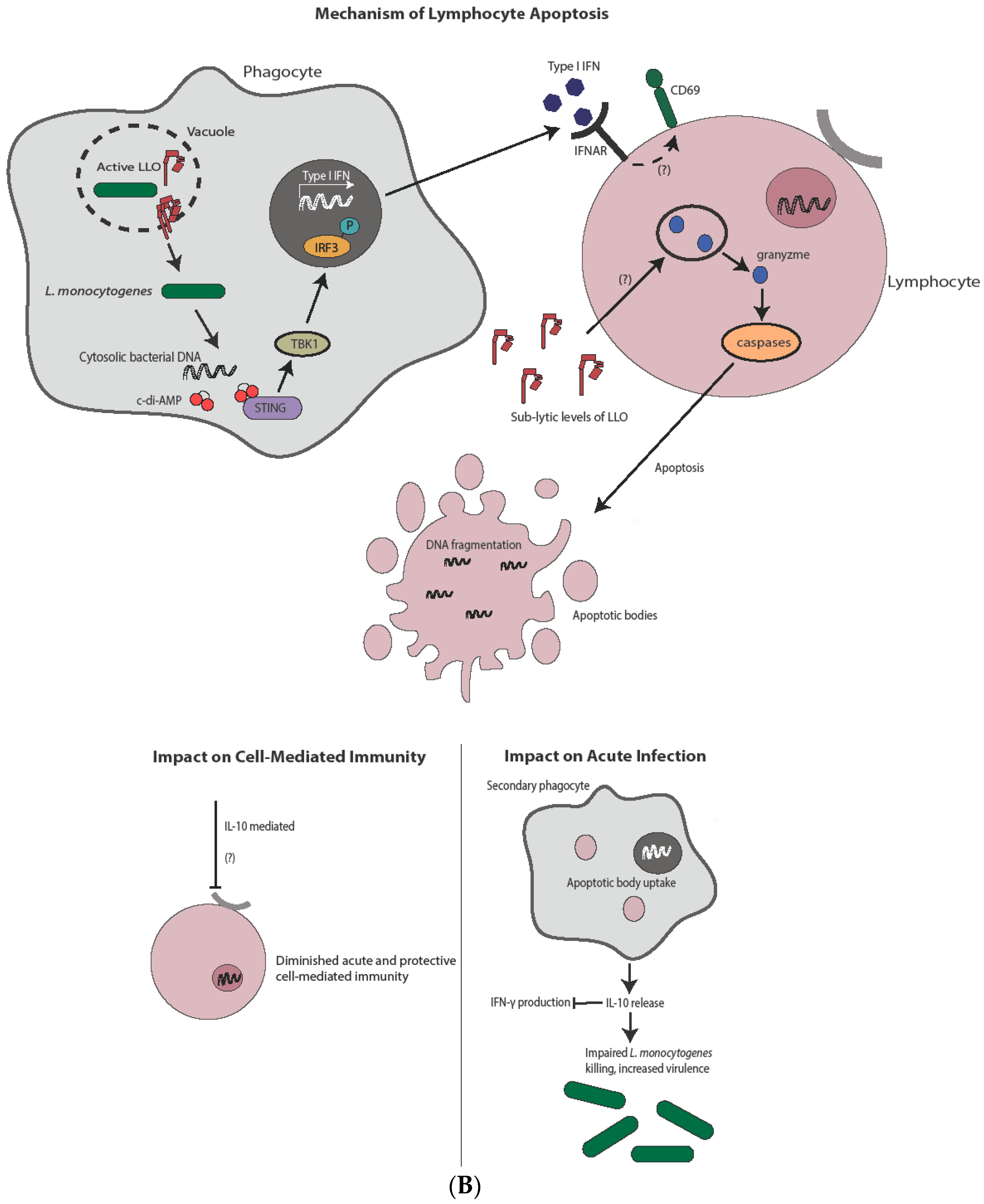
4. Apoptosis
5. Conclusions and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ferreira, V.; Wiedmann, M.; Teixeira, P.; Stasiewicz, M.J. Listeria monocytogenes Persistence in Food-Associated Environments: Epidemiology, Strain Characteristics, and Implications for Public Health. J. Food Prot. 2014, 77, 150–170. [Google Scholar] [CrossRef] [PubMed]
- Swaminathan, B.; Gerner-Smidt, P. The epidemiology of human listeriosis. Microbes Infect. 2007, 9, 1236–1243. [Google Scholar] [CrossRef] [PubMed]
- Mengaud, J.; Ohayon, H.; Gounon, P.; Mege, R.-M.; Cossart, P. E-cadherin is the receptor for internalin, a surface protein required for entry of L. monocytogenes into epithelial cells. Cell 1996, 84, 923–932. [Google Scholar] [CrossRef]
- Shen, Y.; Naujokas, M.; Park, M.; Ireton, K. InIB-dependent internalization of Listeria is mediated by the Met receptor tyrosine kinase. Cell 2000, 103, 501–510. [Google Scholar] [CrossRef]
- Portnoy, D.A.; Jacks, P.S.; Hinrichs, D.J. Role of hemolysin for the intracellular growth of Listeria monocytogenes. J. Exp. Med. 1988, 167, 1459–1471. [Google Scholar] [CrossRef] [PubMed]
- Hamon, M.A.; Ribet, D.; Stavru, F.; Cossart, P. Listeriolysin O: The Swiss army knife of Listeria. Trends Microbiol. 2012, 20, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Kocks, C.; Gouin, E.; Tabouret, M.; Berche, P.; Ohayon, H.; Cossart, P. L. monocytogenes-induced actin assembly requires the actA gene product, a surface protein. Cell 1992, 68, 521–531. [Google Scholar] [CrossRef]
- Carr, K.D.; Sieve, A.N.; Indramohan, M.; Break, T.J.; Lee, S.; Berg, R.E. Specific depletion reveals a novel role for neutrophil-mediated protection in the liver during Listeria monocytogenes infection. Eur. J. Immunol. 2011, 41, 2666–2676. [Google Scholar] [CrossRef] [PubMed]
- Baker, L.A.; Campbell, P.A.; Hollister, J.R. Chemotaxigenesis and complement fixation by Listeria monocytogenes cell wall fractions. J. Immunol. 1977, 119, 1723–1726. [Google Scholar] [PubMed]
- Shaughnessy, L.M.; Swanson, J.A. The role of the activated macrophage in clearing Listeria monocytogenes infection. Front. Biosci. 2007, 12, 2683–2692. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Dubenksy, T.W.; Brockstedt, D.G. Clinical development of Listeria monocytogenes-based immunotherapies. Semin. Oncol. 2012, 39, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Bahjat, K.S.; Liu, W.; Lemmens, E.E.; Schoenberger, S.P.; Portnoy, D.A.; Dubensky, T.W., Jr.; Brockstedt, D.G. Cytosolic entry controls CD8+-T-cell potency during bacterial infection. Infect. Immun. 2006, 74, 6387–6397. [Google Scholar] [CrossRef] [PubMed]
- Aduro BioTech. LADD Engineering Listeria Mononcytogenes Bacteria. 2017. Available online: http://www.aduro.com/technology/ladd/ (accessed on 17 May 2017).
- Lm Technology—Advaxis. 2017. Available online: https://www.advaxis.com/lm-technology/ (accessed on 15 November 2017).
- Blériot, C.; Lecuit, M. The interplay between regulated necrosis and bacterial infection. Cell. Mol. Life Sci. 2016, 73, 2369–2378. [Google Scholar] [CrossRef] [PubMed]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, T.; Kaiser, W.J.; Bertrand, M.J.; Vandenabeele, P. Molecular crosstalk between apoptosis, necroptosis, and survival signaling. Mol. Cell. Oncol. 2015, 2, e975093. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Kokkola, R.; Andersson, A.; Mullins, G.; Ostberg, T.; Treutiger, C.-J.; Arnold, B.; Nawroth, P.; Andersson, U.; Harris, R.A.; Harris, H.E. RAGE is the Major Receptor for the Proinflammatory Activity of HMGB1 in Rodent Macrophages. Scand. J. Immunol. 2005, 61, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Scaffidi, P.; Misteli, T.; Bianchi, M.E. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 2002, 418, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Seveau, S. Multifaceted activity of listeriolysin O, the cholesterol-dependent cytolysin of Listeria monocytogenes. Subcell. Biochem. 2014, 80, 161–195. [Google Scholar] [PubMed]
- Barsig, J.; Kaufmann, S.H. The mechanism of cell death in Listeria monocytogenes-infected murine macrophages is distinct from apoptosis. Infect. Immun. 1997, 65, 4075–4081. [Google Scholar] [PubMed]
- González-Juarbe, N.; Gilley, R.P.; Hinojosa, C.A.; Bradley, K.M.; Kamei, A.; Gao, G.; Dube, P.H.; Bergman, M.A.; Orihuela, C.J. Pore-Forming Toxins Induce Macrophage Necroptosis during Acute Bacterial Pneumonia. PLoS Pathog. 2015, 11, e1005337. [Google Scholar] [CrossRef] [PubMed]
- Geoffroy, C.; Gaillard, J.-L.; Alouf, J.E.; Berche, P. Purification, Characterization, and Toxicity of the Sulfhydryl-Activated Hemolysin Listeriolysin 0 from Listeria monocytogenes. Infect. Immun. 1987, 55, 1641–1646. [Google Scholar] [PubMed]
- Glomski, I.J.; Decatur, A.L.; Portnoy, D.A. Listeria monocytogenes mutants that fail to compartmentalize listerolysin O activity are cytotoxic, avirulent, and unable to evade host extracellular defenses. Infect. Immun. 2003, 71, 6754–6765. [Google Scholar] [CrossRef] [PubMed]
- Glomski, I.J.; Gedde, M.M.; Tsang, A.W.; Swanson, J.A.; Portnoy, D.A. The Listeria monocytogenes hemolysin has an acidic pH optimum to compartmentalize activity and prevent damage to infected host cells. J. Cell Biol. 2002, 156, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Schnupf, P.; Portnoy, D.A.; Decatur, A.L. Phosphorylation, ubiquitination and degradation of listeriolysin O in mammalian cells: Role of the PEST-like sequence. Cell Microbiol. 2006, 8, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Schuerch, D.W.; Wilson-Kubalek, E.M.; Tweten, R.K. Molecular basis of listeriolysin O pH dependence. Proc. Natl. Acad. Sci. USA 2005, 102, 12537–12542. [Google Scholar] [CrossRef] [PubMed]
- Schnupf, P.; Hofmann, J.; Norseen, J.; Glomski, I.J.; Schwartzstein, H.; Decatur, A.L. Regulated translation of listeriolysin O controls virulence of Listeria monocytogenes. Mol. Microbiol. 2006, 61, 999–1012. [Google Scholar] [CrossRef] [PubMed]
- Bavdek, A.; Gekara, N.O.; Priselac, D.; Gutiérrez Aguirre, I.; Darji, A.; Chakraborty, T.; Maček, P.; Lakey, J.H.; Weiss, S.; Anderluh, G. Sterol and pH Interdependence in the Binding, Oligomerization, and Pore Formation of Listeriolysin O. Biochemistry 2007, 46, 4425–4437. [Google Scholar] [CrossRef] [PubMed]
- Blériot, C.; Dupuis, T.; Jouvion, G.; Eberl, G.; Disson, O.; Lecuit, M. Liver-resident macrophage necroptosis orchestrates type 1 microbicidal inflammation and type-2-mediated tissue repair during bacterial infection. Immunity 2015, 42, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Di Paolo, N.C.; Doronin, K.; Baldwin, L.K.; Papayannopoulou, T.; Shayakhmetov, D.M. The transcription factor IRF3 triggers defensive suicide necrosis in response to viral and bacterial pathogens. Cell Rep. 2013, 3, 1840–1846. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, R.M.; Saha, S.K.; Vaidya, S.A.; Bruhn, K.W.; Miranda, G.A.; Zarnegar, B.; Perry, A.K.; Nguyen, B.O.; Lane, T.F.; Taniguchi, T.; et al. Type I Interferon Production Enhances Susceptibility to Listeria monocytogenes Infection. J. Exp. Med. 2004, 200, 437–445. [Google Scholar] [CrossRef] [PubMed]
- Auerbuch, V.; Brockstedt, D.G.; Meyer-Morse, N.; O’Riordan, M.; Portnoy, D.A. Mice lacking the type I interferon receptor are resistant to Listeria monocytogenes. J. Exp. Med. 2004, 200, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, A.; Nishimura, K.; Nakaima, Y.; Oh, T.; Noguchi, S.; Taniguchi, T.; Tamura, T. Cell type-dependent proapoptotic role of Bcl2L12 revealed by a mutation concomitant with the disruption of the juxtaposed Irf3 gene. Proc. Natl. Acad. Sci. USA 2009, 106, 12448–12452. [Google Scholar] [CrossRef] [PubMed]
- Barker, R.N.; Erwig, L.-P.; Pearce, W.P.; Devine, A.; Rees, A.J. Differential Effects of Necrotic or Apoptotic Cell Uptake on Antigen Presentation by Macrophages. Pathobiology 1999, 67, 302–305. [Google Scholar] [CrossRef] [PubMed]
- Festjens, N.; Vanden Berghe, T.; Vandenabeele, P. Necrosis, a well-orchestrated form of cell demise: Signalling cascades, important mediators and concomitant immune response. Biochim. Biophys. Acta Bioenerg. 2006, 1757, 1371–1387. [Google Scholar] [CrossRef] [PubMed]
- Sauter, B.; Albert, M.L.; Francisco, L.; Larsson, M.; Somersan, S.; Bhardwaj, N. Consequences of cell death: exposure to necrotic tumor cells, but not primary tissue cells or apoptotic cells, induces the maturation of immunostimulatory dendritic cells. J. Exp. Med. 2000, 191, 423–434. [Google Scholar] [CrossRef] [PubMed]
- Theisen, E.; Sauer, J.-D. Listeria monocytogenes-Induced Cell Death Inhibits the Generation of Cell-Mediated Immunity. Infect. Immun. 2017, 85, e00733-16. [Google Scholar] [CrossRef] [PubMed]
- Janda, J.; Schoneberger, P.; Skoberne, M.; Messerle, M.; Russmann, H.; Geginat, G. Cross-presentation of Listeria-derived CD8 T cell epitopes requires unstable bacterial translation products. J. Immunol. 2004, 173, 5644–5651. [Google Scholar] [CrossRef] [PubMed]
- Reinicke, A.T.; Omilusik, K.D.; Basha, G.; Jefferies, W.A. Dendritic cell cross-priming is essential for immune responses to Listeria monocytogenes. PLoS ONE 2009, 4, e7210. [Google Scholar] [CrossRef] [PubMed]
- Von Moltke, J.; Ayres, J.S.; Kofoed, E.M.; Chavarria-Smith, J.; Vance, R.E. Recognition of bacteria by inflammasomes. Annu. Rev. Immunol. 2013, 31, 73–106. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Mayor, A.; Tschopp, J. The inflammasomes: Guardians of the body. Annu. Rev. Immunol. 2009, 27, 229–265. [Google Scholar] [CrossRef] [PubMed]
- DeYoung, K.L.; Ray, M.E.; Su, Y.A.; Anzick, S.L.; Johnstone, R.W.; Trapani, J.A.; Meltzer, P.S.; Trent, J.M. Cloning a novel member of the human interferon-inducible gene family associated with control of tumorigenicity in a model of human melanoma. Oncogene 1997, 15, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Inohara; Chamaillard; McDonald, C.; Nunez, G.; Inohara, N.; Chamaillard, M.; McDonald, C.; Nunez, G. NOD-LRR proteins: role in host-microbial interactions and inflammatory disease. Annu. Rev. Biochem. 2004, 74, 355–383. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Hofmann, K.; Tschopp, J. The pyrin domain: A possible member of the death domain-fold family implicated in apoptosis and inflammation. Curr. Biol. 2001, 11, R118–R120. [Google Scholar] [CrossRef]
- Masumoto, J.; Taniguchi, S.; Ayukawa, K.; Sarvotham, H.; Kishino, T.; Niikawa, N.; Hidaka, E.; Katsuyama, T.; Higuchi, T.; Sagara, J. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J. Biol. Chem. 1999, 274, 33835–33838. [Google Scholar] [CrossRef] [PubMed]
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schröder, G.F.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified Polymerization Mechanism for the Assembly of ASC-Dependent Inflammasomes. Cell 2014, 156, 1193–1206. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Yang, J.; Shi, J.; Gong, Y.-N.; Lu, Q.; Xu, H.; Liu, L.; Shao, F. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 2011, 477, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ablasser, A.; Charrel-Dennis, M.; Bauernfeind, F.; Horvath, G.; Caffrey, D.R.; Latz, E.; Fitzgerald, K.A. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009, 458, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Alnemri, T.; Yu, J.W.; Datta, P.; Wu, J.; Alnemri, E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Von Moltke, J.; Trinidad, N.J.; Moayeri, M.; Kintzer, A.F.; Wang, S.B.; van Rooijen, N.; Brown, C.R.; Krantz, B.A.; Leppla, S.H.; Gronert, K.; et al. Rapid induction of inflammatory lipid mediators by the inflammasome in vivo. Nature 2012, 490, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Sarkar, A.; Vande Walle, L.; Vitari, A.C.; Amer, A.O.; Wewers, M.D.; Tracey, K.J.; Kanneganti, T.-D.; Dixit, V.M. Inflammasome-dependent release of the alarmin HMGB1 in endotoxemia. J. Immunol. 2010, 185, 4385–4392. [Google Scholar] [CrossRef] [PubMed]
- Fink, S.L.; Cookson, B.T. Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell. Microbiol. 2006, 8, 1812–1825. [Google Scholar] [CrossRef] [PubMed]
- Mascarenhas, D.P.A.; Cerqueira, D.M.; Pereira, M.S.F.; Castanheira, F.V.S.; Fernandes, T.D.; Manin, G.Z.; Cunha, L.D.; Zamboni, D.S. Inhibition of caspase-1 or gasdermin-D enable caspase-8 activation in the Naip5/NLRC4/ASC inflammasome. PLOS Pathog. 2017, 13, e1006502. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and Functions of Inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, I.; Zhang, Y.; Krantz, B.A.; Miao, E.A. Pyroptosis triggers pore-induced intracellular traps (PITs) that capture bacteria and lead to their clearance by efferocytosis. J. Exp. Med. 2016, 213, 2113–2128. [Google Scholar] [CrossRef] [PubMed]
- Miao, E.A.; Leaf, I.A.; Treuting, P.M.; Mao, D.P.; Dors, M.; Sarkar, A.; Warren, S.E.; Wewers, M.D.; Aderem, A. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 2010, 11, 1136–1142. [Google Scholar] [CrossRef] [PubMed]
- Sauer, J.; Pereyre, S.; Archer, K.A.; Burke, T.P.; Hanson, B.; Lauer, P.; Portnoy, D.A. Listeria monocytogenes engineered to activate the Nlrc4 inflammasome are severely attenuated and are poor inducers of protective immunity. Proc. Natl. Acad. Sci. USA 2011, 108, 12419–12424. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Shen, A.; Higgins, D.E. The MogR transcriptional repressor regulates nonhierarchal expression of flagellar motility genes and virulence in Listeria monocytogenes. PLoS Pathog. 2006, 2, e30. [Google Scholar] [CrossRef] [PubMed]
- Peel, M.; Donachie, W.; Shaw, A. Temperature-dependent Expression of Flagella of Listeria manocytogenes Studied by Electron Microscopy, SDS-PAGE and Western Blotting. Microbiology 1988, 134, 2171–2178. [Google Scholar] [CrossRef] [PubMed]
- Warren, S.E.; Mao, D.P.; Rodriguez, A.E.; Miao, E.A.; Aderem, A. Multiple Nod-like receptors activate caspase 1 during Listeria monocytogenes infection. J. Immunol. 2008, 180, 7558–7564. [Google Scholar] [CrossRef] [PubMed]
- Warren, S.E.; Duong, H.; Mao, D.P.; Armstrong, A.; Rajan, J.; Miao, E.A.; Aderem, A. Generation of a Listeria vaccine strain by enhanced caspase-1 activation. Eur. J. Immunol. 2011, 41, 1934–1940. [Google Scholar] [CrossRef] [PubMed]
- Sauer, J.D.; Witte, C.E.; Zemansky, J.; Hanson, B.; Lauer, P.; Portnoy, D.A. Listeria monocytogenes triggers AIM2-mediated pyroptosis upon infrequent bacteriolysis in the macrophage cytosol. Cell Host Microbe 2010, 7, 412–419. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.W.; Kayagaki, N.; Broz, P.; Henry, T.; Newton, K.; O’Rourke, K.; Chan, S.; Dong, J.; Qu, Y.; Roose-Girma, M.; et al. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc. Natl. Acad. Sci. USA 2010, 107, 9771–9776. [Google Scholar] [CrossRef] [PubMed]
- Pensinger, D.A.; Boldon, K.M.; Chen, G.Y.; Vincent, W.J.B.; Sherman, K.; Xiong, M.; Schaenzer, A.J.; Forster, E.R.; Coers, J.; Striker, R.; et al. The Listeria monocytogenes PASTA Kinase PrkA and Its Substrate YvcK Are Required for Cell Wall Homeostasis, Metabolism, and Virulence. PLOS Pathog. 2016, 12, e1006001. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.Y.; McDougal, C.E.; D’Antonio, M.A.; Portman, J.L.; Sauer, J.-D. A Genetic Screen Reveals that Synthesis of 1,4-Dihydroxy-2-Naphthoate (DHNA), but Not Full-Length Menaquinone, Is Required for Listeria monocytogenes Cytosolic Survival. MBio 2017, 8, e00119-17. [Google Scholar] [CrossRef] [PubMed]
- Rae, C.S.; Geissler, A.; Adamson, P.C.; Portnoy, D.A. Mutations of the Listeria monocytogenes Peptidoglycan N-Deacetylase and O-Acetylase Result in Enhanced Lysozyme Sensitivity, Bacteriolysis, and Hyperinduction of Innate Immune Pathways. Infect. Immun. 2011, 79, 3596–3606. [Google Scholar] [CrossRef] [PubMed]
- Meixenberger, K.; Pache, F.; Eitel, J.; Schmeck, B.; Hippenstiel, S.; Slevogt, H.; Guessan, P.N.; Witzenrath, M.; Netea, M.G.; Chakraborty, T.; et al. Listeria monocytogenes-Infected Human Peripheral Blood Mononuclear Cells Produce IL-1{beta}, Depending on Listeriolysin O and NLRP3. J. Immunol. 2009, 184, 922–930. [Google Scholar]
- Sakhon, O.S.; Victor, K.A.; Choy, A.; Tsuchiya, T.; Eulgem, T.; Pedra, J.H.F. NSD1 Mitigates Caspase-1 Activation by Listeriolysin O in Macrophages. PLoS ONE 2013, 8, e75911. [Google Scholar] [CrossRef] [PubMed]
- Hamon, M.A.; Cossart, P. K+ Efflux Is Required for Histone H3 Dephosphorylation by Listeria monocytogenes Listeriolysin O and Other Pore-Forming Toxins. Infect. Immun. 2011, 79, 2839–2846. [Google Scholar] [CrossRef] [PubMed]
- Williams, C.R.; Dustin, M.L.; Sauer, J.-D. Inflammasome-Mediated Inhibition of Listeria monocytogenes-Stimulated Immunity Is Independent of Myelomonocytic Function. PLoS ONE 2013, 8, e83191. [Google Scholar] [CrossRef] [PubMed]
- Mayer-Barber, K.D.; Andrade, B.B.; Oland, S.D.; Amaral, E.P.; Barber, D.L.; Gonzales, J.; Derrick, S.C.; Shi, R.; Kumar, N.P.; Wei, W.; et al. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature 2014, 511, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.H.; Perry, C.J.; Tsui, Y.-C.; Staron, M.M.; Parish, I.A.; Dominguez, C.X.; Rosenberg, D.W.; Kaech, S.M. Prostaglandin E2 and programmed cell death 1 signaling coordinately impair CTL function and survival during chronic viral infection. Nat. Med. 2015, 21, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Nataraj, C.; Thomas, D.W.; Tilley, S.L.; Nguyen, M.T.; Mannon, R.; Koller, B.H.; Coffman, T.M. Receptors for prostaglandin E2 that regulate cellular immune responses in the mouse. J. Clin. Investig. 2001, 108, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Maltez, V.I.; Tubbs, A.L.; Cook, K.D.; Aachoui, Y.; Falcone, E.L.; Holland, S.M.; Whitmire, J.K.; Miao, E.A. Inflammasomes Coordinate Pyroptosis and Natural Killer Cell Cytotoxicity to Clear Infection by a Ubiquitous Environmental Bacterium. Immunity 2015, 43, 987–997. [Google Scholar] [CrossRef] [PubMed]
- Kerr, J.F.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, A. Death receptor-induced cell killing. Cell Signal. 2004, 16, 139–144. [Google Scholar] [CrossRef] [PubMed]
- Riedl, S.J.; Salvesen, G.S. The apoptosome: Signalling platform of cell death. Nat. Rev. Mol. Cell Biol. 2007, 8, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Weinrauch, Y.; Zychlinsky, A. The Induction of Apoptosis by Bacterial Pathogens. Annu. Rev. Microbiol. 1999, 53, 155–187. [Google Scholar] [CrossRef] [PubMed]
- Rogers, H.W.; Callery, M.P.; Deck, B.; Unanue, E.R.; Rogers, H.W.; Gallery, M.P.; Deck, B.; Unanue2, E.R. Listeria monocytogenes induces apoptosis of infected hepatocytes. J. Immunol. 1996, 156, 679–684. [Google Scholar] [PubMed]
- Dos Santos, S.A.; de Andrade Júnior, D.R.; de Andrade, D.R. TNF-α production and apoptosis in hepatocytes after Listeria monocytogenes and Salmonella Typhimurium invasion. Rev. Inst. Med. Trop. Sao Paulo 2011, 53, 107–112. [Google Scholar] [CrossRef]
- Dos Santos, S.A.; de Andrade, D.R.; Andrade, J.D.R. Rat hepatocyte invasion by Listeria monocytogenes and analysis of TNF-alpha role in apoptosis. Rev. Inst. Med. Trop. Sao Paulo 2005, 47, 73–80. [Google Scholar] [CrossRef]
- Margaroli, C.; Oberle, S.; Lavanchy, C.; Scherer, S.; Rosa, M.; Strasser, A.; Pellegrini, M.; Zehn, D.; Acha-Orbea, H.; Ehirchiou, D. Role of proapoptotic BH3-only proteins in Listeria monocytogenes infection. Eur. J. Immunol. 2016, 46, 1427–1437. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.-J.; Jiang, J.; Shen, H.; Chen, Y.H. Reduced apoptosis and ameliorated listeriosis in TRAIL-null mice. J. Immunol. 2004, 173, 5652–5658. [Google Scholar] [CrossRef] [PubMed]
- Mandel, T.E.; Cheers, C. Resistance and Susceptibility of Mice to Bacterial Infection: Histopathology of Listeriosis in Resistant and Susceptible Strains. Infect. Immun. 1980, 30, 851–861. [Google Scholar] [PubMed]
- Merrick, J.C.; Edelson, B.T.; Bhardwaj, V.; Swanson, P.E.; Unanue, E.R. Lymphocyte apoptosis during early phase of Listeria infection in mice. Am. J. Pathol. 1997, 151, 785–792. [Google Scholar] [PubMed]
- Carrero, J.A.; Calderon, B.; Unanue, E.R. Lymphocytes are detrimental during the early innate immune response against Listeria monocytogenes. J. Exp. Med. 2006, 203, 933–940. [Google Scholar] [CrossRef] [PubMed]
- Carrero, J.A.; Vivanco-Cid, H.; Unanue, E.R. Granzymes drive a rapid listeriolysin O-induced T cell apoptosis. J. Immunol. 2008, 181, 1365–1374. [Google Scholar] [CrossRef] [PubMed]
- Browne, K.A.; Blink, E.; Sutton, V.R.; Froelich, C.J.; Jans, D.A.; Trapani, J.A. Cytosolic delivery of granzyme B by bacterial toxins: evidence that endosomal disruption, in addition to transmembrane pore formation, is an important function of perforin. Mol. Cell. Biol. 1999, 19, 8604–8615. [Google Scholar] [CrossRef] [PubMed]
- Carrero, J.A.; Calderon, B.; Vivanco-Cid, H.; Unanue, E.R. Recombinant Listeria monocytogenes Expressing a Cell Wall-Associated Listeriolysin O Is Weakly Virulent but Immunogenic. Infect. Immun. 2009, 77, 4371–4382. [Google Scholar] [CrossRef] [PubMed]
- Sauer, J.-D.; Sotelo-Troha, K.; von Moltke, J.; Monroe, K.M.; Rae, C.S.; Brubaker, S.W.; Hyodo, M.; Hayakawa, Y.; Woodward, J.J.; Portnoy, D.A.; et al. The N-Ethyl-N-Nitrosourea-Induced Goldenticket Mouse Mutant Reveals an Essential Function of Sting in the In Vivo Interferon Response to Listeria monocytogenes and Cyclic Dinucleotides. Infect. Immun. 2011, 79, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Woodward, J.J.; Iavarone, A.T.; Portnoy, D.A. c-di-AMP Secreted by Intracellular Listeria monocytogenes Activates a Host Type I Interferon Response. Science 2010, 328, 1703–1705. [Google Scholar] [CrossRef] [PubMed]
- Hansen, K.; Prabakaran, T.; Laustsen, A.; Jørgensen, S.E.; Rahbæk, S.H.; Jensen, S.B.; Nielsen, R.; Leber, J.H.; Decker, T.; Horan, K.A.; et al. Listeria monocytogenes induces IFNβ expression through an IFI16-, cGAS- and STING-dependent pathway. EMBO J. 2014, 33, 1654–1666. [Google Scholar] [CrossRef] [PubMed]
- Carrero, J.A.; Calderon, B.; Unanue, E.R. Type I interferon sensitizes lymphocytes to apoptosis and reduces resistance to Listeria infection. J. Exp. Med. 2004, 200, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Voll, R.E.; Herrmann, M.; Roth, E.A.; Stach, C.; Kalden, J.R.; Girkontaite, I. Immunosuppressive effects of apoptotic cells. Nature 1997, 390, 350–351. [Google Scholar] [CrossRef] [PubMed]
- Dai, W.J.; Köhler, G.; Brombacher, F. Both innate and acquired immunity to Listeria monocytogenes infection are increased in IL-10-deficient mice. J. Immunol. 1997, 158, 2259–2267. [Google Scholar] [PubMed]
- Bancroft, G.J.; Bosma, M.J.; Bosma, G.C.; Unanue, E.R. Regulation of macrophage Ia expression in mice with severe combined immunodeficiency: Induction of Ia expression by a T cell-independent mechanism. J. Immunol. 1986, 137, 4–9. [Google Scholar] [PubMed]
- Bhardwaj, V.; Kanagawa, O.; Swanson, P.E.; Unanue, E.R. Chronic Listeria infection in SCID mice: Requirements for the carrier state and the dual role of T cells in transferring protection or suppression. J. Immunol. 1998, 160, 376–384. [Google Scholar] [PubMed]
- Archer, K.A.; Durack, J.; Portnoy, D.A. STING-dependent type I IFN production inhibits cell-mediated immunity to Listeria monocytogenes. PLoS Pathog. 2014, 10, e1003861. [Google Scholar] [CrossRef] [PubMed]
- Pattabiraman, G.; Palasiewicz, K.; Visvabharathy, L.; Freitag, N.E.; Ucker, D.S. Apoptotic cells enhance pathogenesis of Listeria monocytogenes. Microb. Pathog. 2017, 105, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Gougeon, M.L.; Lecoeur, H.; Dulioust, A.; Enouf, M.G.; Crouvoiser, M.; Goujard, C.; Debord, T.; Montagnier, L. Programmed cell death in peripheral lymphocytes from HIV-infected persons: increased susceptibility to apoptosis of CD4 and CD8 T cells correlates with lymphocyte activation and with disease progression. J. Immunol. 1996, 156, 3509–3520. [Google Scholar] [PubMed]
- Early, J.; Fischer, K.; Bermudez, L.E. Mycobacterium avium uses apoptotic macrophages as tools for spreading. Microb. Pathog. 2011, 50, 132–139. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McDougal, C.E.; Sauer, J.-D. Listeria monocytogenes: The Impact of Cell Death on Infection and Immunity. Pathogens 2018, 7, 8. https://doi.org/10.3390/pathogens7010008
McDougal CE, Sauer J-D. Listeria monocytogenes: The Impact of Cell Death on Infection and Immunity. Pathogens. 2018; 7(1):8. https://doi.org/10.3390/pathogens7010008
Chicago/Turabian StyleMcDougal, Courtney E., and John-Demian Sauer. 2018. "Listeria monocytogenes: The Impact of Cell Death on Infection and Immunity" Pathogens 7, no. 1: 8. https://doi.org/10.3390/pathogens7010008
APA StyleMcDougal, C. E., & Sauer, J.-D. (2018). Listeria monocytogenes: The Impact of Cell Death on Infection and Immunity. Pathogens, 7(1), 8. https://doi.org/10.3390/pathogens7010008