Pharmacological Agents Targeting the Cellular Prion Protein

 , ,
, ,
Abstract
1. Introduction
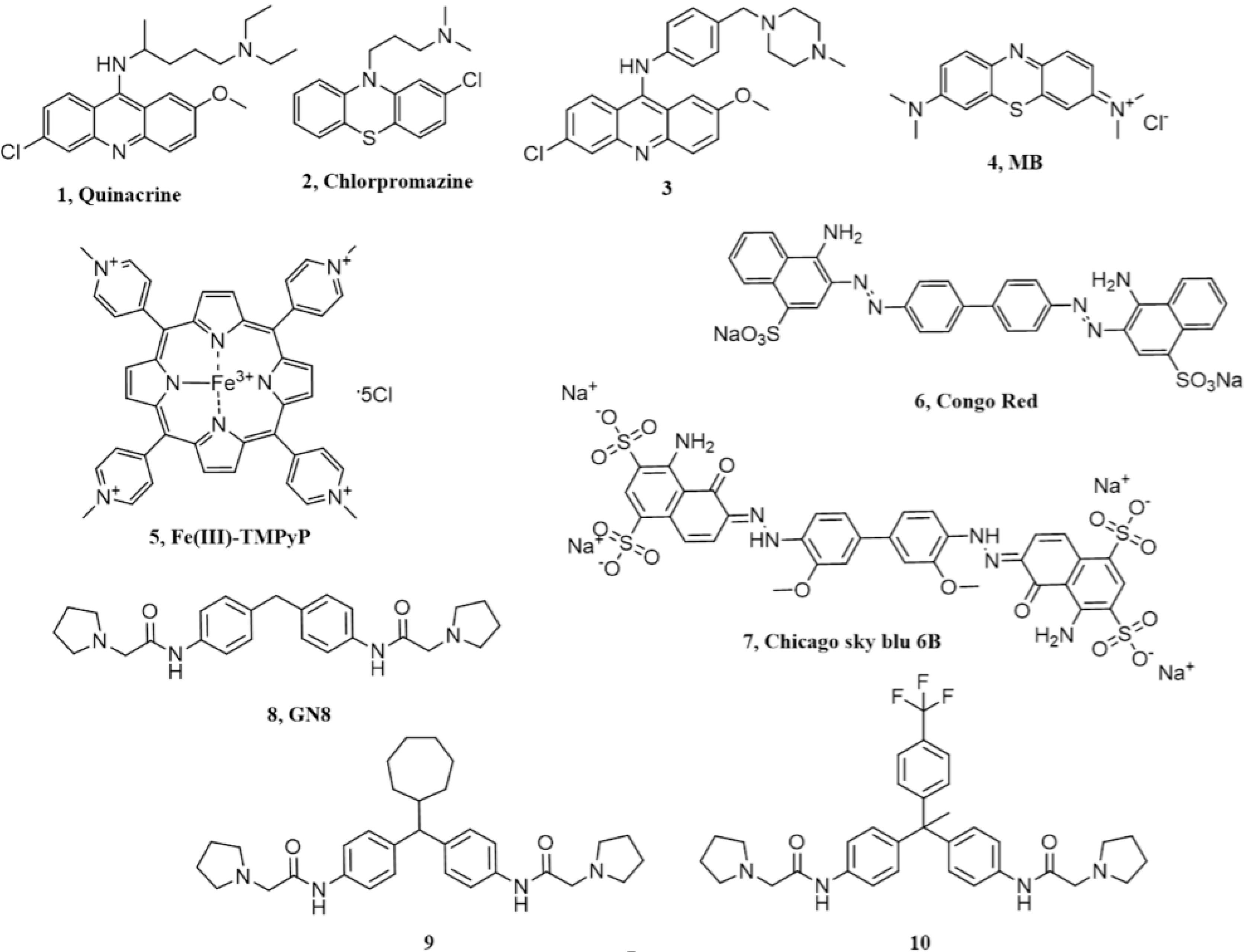
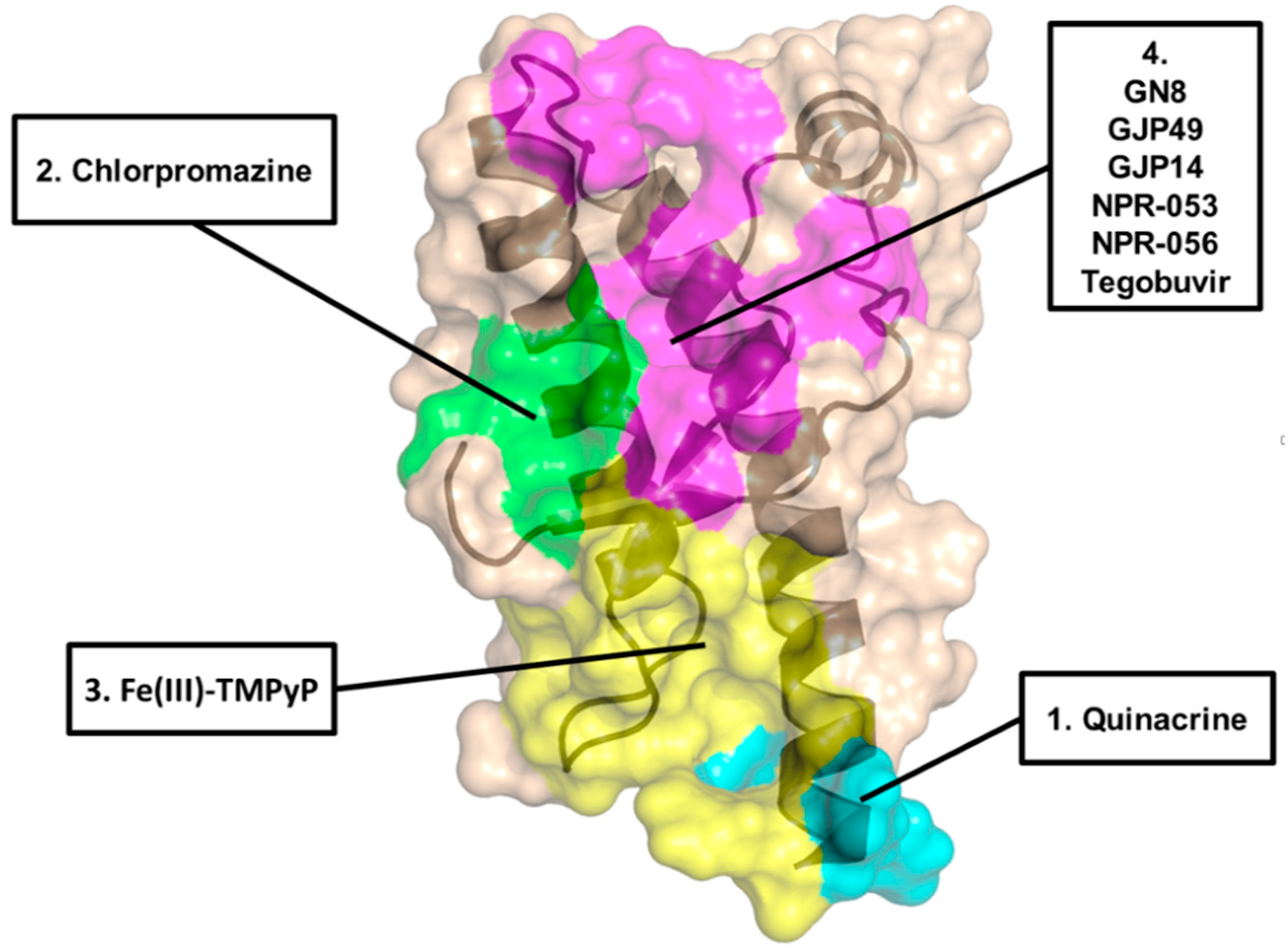
2. Acridine and Phenothiazine Derivatives
3. Cyclic Tetrapyrroles
4. Diazo Dyes
5. Chicago Sky Blue 6B
6. Diphenylmethane Derivatives
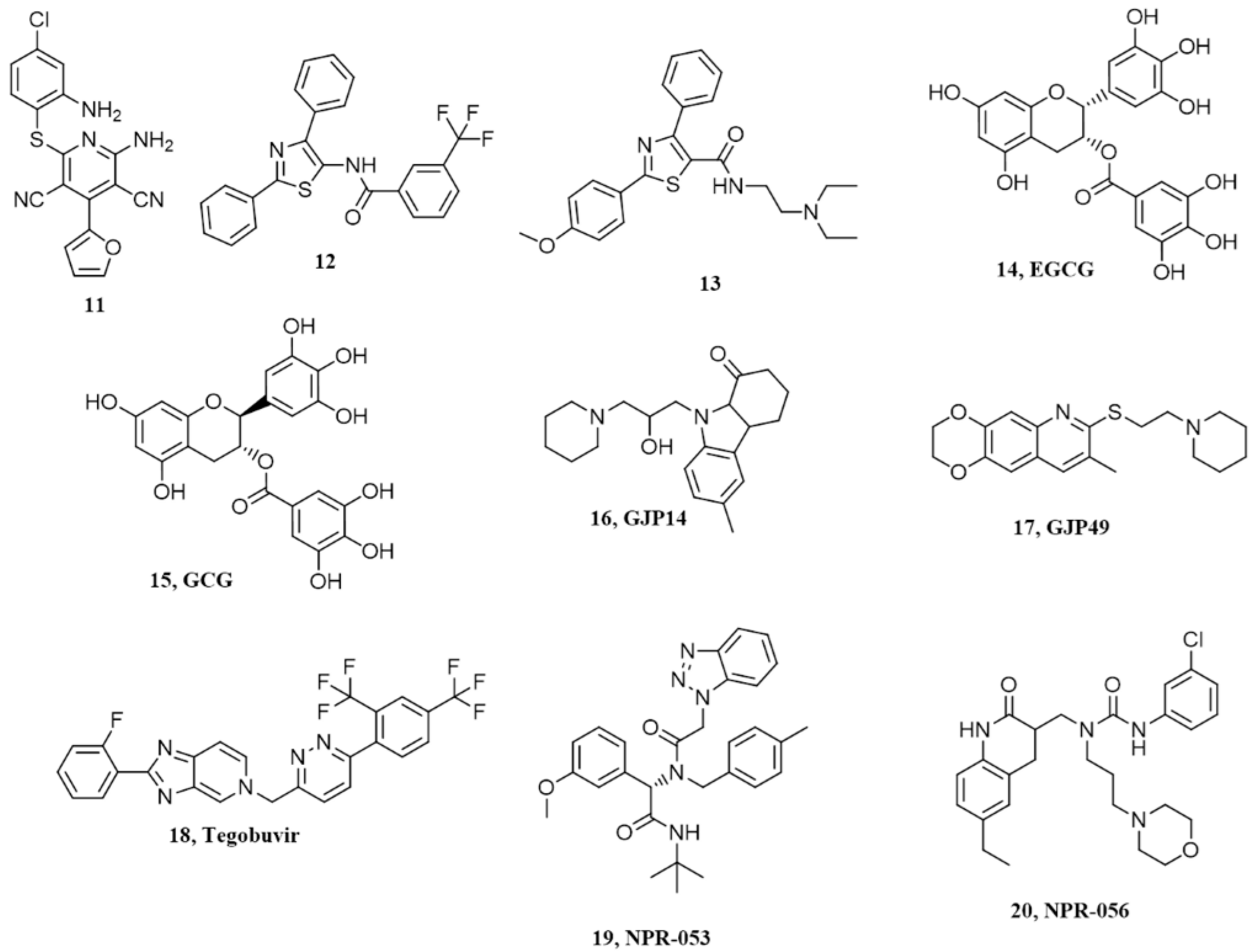
7. Pyridine Dicarbonitriles
8. Diarylthiazoles
9. Natural Polyphenols
10. Miscellanea
11. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chiti, F.; Dobson, C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006, 75, 333–366. [Google Scholar] [CrossRef] [PubMed]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Biology and genetics of prions causing neurodegeneration. Annu. Rev. Genet. 2013, 47, 601–623. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Giles, K.; Olson, S.H.; Prusiner, S.B. Developing therapeutics for prp prion diseases. Cold Spring Harbor Perspect. Med. 2017, 7, a023747. [Google Scholar] [CrossRef] [PubMed]
- Baskakov, I.V. The many shades of prion strain adaptation. Prion 2014, 8, 27836. [Google Scholar] [CrossRef]
- Espinosa, J.C.; Nonno, R.; Di Bari, M.; Aguilar-Calvo, P.; Pirisinu, L.; Fernandez-Borges, N.; Vanni, I.; Vaccari, G.; Marin-Moreno, A.; Frassanito, P.; et al. PrPc governs susceptibility to prion strains in bank vole, while other host factors modulate strain features. J. Virol. 2016, 90, 10660–10669. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J. Medicine. Prion strain mutation and selection. Science 2010, 328, 1111–1112. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Browning, S.; Mahal, S.P.; Oelschlegel, A.M.; Weissmann, C. Darwinian evolution of prions in cell culture. Science 2010, 327, 869–872. [Google Scholar] [CrossRef] [PubMed]
- Sim, V.L. Prion disease: Chemotherapeutic strategies. Infect. Disord. Drug Targets 2012, 12, 144–160. [Google Scholar] [CrossRef] [PubMed]
- Ghaemmaghami, S.; Ahn, M.; Lessard, P.; Giles, K.; Legname, G.; DeArmond, S.J.; Prusiner, S.B. Continuous quinacrine treatment results in the formation of drug-resistant prions. PLoS Pathog. 2009, 5, e1000673. [Google Scholar] [CrossRef] [PubMed]
- Giles, K.; Berry, D.B.; Condello, C.; Hawley, R.C.; Gallardo-Godoy, A.; Bryant, C.; Oehler, A.; Elepano, M.; Bhardwaj, S.; Patel, S.; et al. Different 2-aminothiazole therapeutics produce distinct patterns of scrapie prion neuropathology in mouse brains. J. Pharmacol. Exp. Ther. 2015, 355, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Riek, R.; Hornemann, S.; Wider, G.; Billeter, M.; Glockshuber, R.; Wüthrich, K. NMR structure of the mouse prion protein domain PrP(121–231). Nature 1996, 382, 180–182. [Google Scholar] [CrossRef] [PubMed]
- Zahn, R.; Liu, A.; Luhrs, T.; Riek, R.; von Schroetter, C.; Lopez Garcia, F.; Billeter, M.; Calzolai, L.; Wider, G.; Wüthrich, K. NMR solution structure of the human prion protein. Proc. Natl. Acad. Sci. USA 2000, 97, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Antonyuk, S.V.; Trevitt, C.R.; Strange, R.W.; Jackson, G.S.; Sangar, D.; Batchelor, M.; Cooper, S.; Fraser, C.; Jones, S.; Georgiou, T.; et al. Crystal structure of human prion protein bound to a therapeutic antibody. Proc. Natl. Acad. Sci. USA 2009, 106, 2554–2558. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, A.J.; Collinge, J. Preventing prion pathogenicity by targeting the cellular prion protein. Infect. Disord. Drug Targets 2009, 9, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Mallucci, G.R.; White, M.D.; Farmer, M.; Dickinson, A.; Khatun, H.; Powell, A.D.; Brandner, S.; Jefferys, J.G.; Collinge, J. Targeting cellular prion protein reverses early cognitive deficits and neurophysiological dysfunction in prion-infected mice. Neuron 2007, 53, 325–335. [Google Scholar] [CrossRef] [PubMed]
- Brandner, S.; Isenmann, S.; Raeber, A.; Fischer, M.; Sailer, A.; Kobayashi, Y.; Marino, S.; Weissmann, C.; Aguzzi, A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature 1996, 379, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Biasini, E.; Medrano, A.Z.; Thellung, S.; Chiesa, R.; Harris, D.A. Multiple biochemical similarities between infectious and non-infectious aggregates of a prion protein carrying an octapeptide insertion. J. Neurochem. 2008, 104, 1293–1308. [Google Scholar] [CrossRef] [PubMed]
- Biasini, E.; Seegulam, M.E.; Patti, B.N.; Solforosi, L.; Medrano, A.Z.; Christensen, H.M.; Senatore, A.; Chiesa, R.; Williamson, R.A.; Harris, D.A. Non-infectious aggregates of the prion protein react with several PrPsc-directed antibodies. J. Neurochem. 2008, 105, 2190–2204. [Google Scholar] [CrossRef] [PubMed]
- Chiesa, R.; Harris, D.A. Prion diseases: What is the neurotoxic molecule? Neurobiol. Dis. 2001, 8, 743–763. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Aguzzi, A.; Falsig, J. Prion propagation, toxicity and degradation. Nat. Neurosci. 2012, 15, 936–939. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, M.K.; Al-Doujaily, H.; Sharps, B.; Clarke, A.R.; Collinge, J. Prion propagation and toxicity in vivo occur in two distinct mechanistic phases. Nature 2011, 470, 540–542. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Ryazanov, S.; Leonov, A.; Levin, J.; Shi, S.; Schmidt, F.; Prix, C.; Pan-Montojo, F.; Bertsch, U.; Mitteregger-Kretzschmar, G.; et al. ANLE138B: A novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson’s disease. Acta Neuropathol. 2013, 125, 795–813. [Google Scholar] [CrossRef] [PubMed]
- Elezgarai, S.R.; Biasini, E. Common therapeutic strategies for prion and alzheimer’s diseases. Biol. Chem. 2016, 397, 1115–1124. [Google Scholar] [CrossRef] [PubMed]
- Lauren, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature 2009, 457, 1128–1132. [Google Scholar] [CrossRef] [PubMed]
- Biasini, E.; Harris, D.A. Targeting the cellular prion protein to treat neurodegeneration. Future Med. Chem. 2012, 4, 1655–1658. [Google Scholar] [CrossRef] [PubMed]
- Biasini, E.; Turnbaugh, J.A.; Unterberger, U.; Harris, D.A. Prion protein at the crossroads of physiology and disease. Trends Neurosci. 2012, 35, 92–103. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.G.; Temido-Ferreira, M.; Miranda, H.V.; Batalha, V.L.; Coelho, J.E.; Szego, E.M.; Marques-Morgado, I.; Vaz, S.H.; Rhee, J.S.; Schmitz, M.; et al. Alpha-synuclein interacts with PrP(c) to induce cognitive impairment through mGluR5 and NMDAR2B. Nat. Neurosci. 2017, 20, 1569–1579. [Google Scholar] [CrossRef] [PubMed]
- Minikel, E.V.; Vallabh, S.M.; Lek, M.; Estrada, K.; Samocha, K.E.; Sathirapongsasuti, J.F.; McLean, C.Y.; Tung, J.Y.; Yu, L.P.; Gambetti, P.; et al. Quantifying prion disease penetrance using large population control cohorts. Sci. Transl. Med. 2016, 20, 322–323. [Google Scholar] [CrossRef] [PubMed]
- Doh-Ura, K.; Iwaki, T.; Caughey, B. Lysosomotropic agents and cysteine protease inhibitors inhibit scrapie-associated prion protein accumulation. J. Virol. 2000, 74, 4894–4897. [Google Scholar] [CrossRef] [PubMed]
- Korth, C.; May, B.C.; Cohen, F.E.; Prusiner, S.B. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc. Natl. Acad. Sci. USA 2001, 98, 9836–9841. [Google Scholar] [CrossRef] [PubMed]
- Ryou, C.; Legname, G.; Peretz, D.; Craig, J.C.; Baldwin, M.A.; Prusiner, S.B. Differential inhibition of prion propagation by enantiomers of quinacrine. Lab. Investig. 2003, 83, 837–843. [Google Scholar] [CrossRef] [PubMed]
- Collins, S.J.; Lewis, V.; Brazier, M.; Hill, A.F.; Fletcher, A.; Masters, C.L. Quinacrine does not prolong survival in a murine Creutzfeldt-JaKob disease model. Ann. Neurol. 2002, 52, 503–506. [Google Scholar] [CrossRef] [PubMed]
- Barret, A.; Tagliavini, F.; Forloni, G.; Bate, C.; Salmona, M.; Colombo, L.; De Luigi, A.; Limido, L.; Suardi, S.; Rossi, G.; et al. Evaluation of quinacrine treatment for prion diseases. J. Virol. 2003, 77, 8462–8469. [Google Scholar] [CrossRef] [PubMed]
- Collinge, J.; Gorham, M.; Hudson, F.; Kennedy, A.; Keogh, G.; Pal, S.; Rossor, M.; Rudge, P.; Siddique, D.; Spyer, M.; et al. Safety and efficacy of quinacrine in human prion disease (prion-1 study): A patient-preference trial. Lancet Neurol. 2009, 8, 334–344. [Google Scholar] [CrossRef]
- Geschwind, M.D.; Kuo, A.L.; Wong, K.S.; Haman, A.; Devereux, G.; Raudabaugh, B.J.; Johnson, D.Y.; Torres-Chae, C.C.; Finley, R.; Garcia, P.; et al. Quinacrine treatment trial for sporadic Creutzfeldt-JaKob disease. Neurology 2013, 81, 2015–2023. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.; Ghaemmaghami, S.; Huang, Y.; Phuan, P.W.; May, B.C.; Giles, K.; DeArmond, S.J.; Prusiner, S.B. Pharmacokinetics of quinacrine efflux from mouse brain via the P-glycoprotein efflux transporter. PLoS ONE 2012, 7, e39112. [Google Scholar] [CrossRef] [PubMed]
- Vogtherr, M.; Grimme, S.; Elshorst, B.; Jacobs, D.M.; Fiebig, K.; Griesinger, C.; Zahn, R. Antimalarial drug quinacrine binds to C-terminal helix of cellular prion protein. J. Med. Chem. 2003, 46, 3563–3564. [Google Scholar] [CrossRef] [PubMed]
- Kawatake, S.; Nishimura, Y.; Sakaguchi, S.; Iwaki, T.; Doh-ura, K. Surface plasmon resonance analysis for the screening of anti-prion compounds. Biol. Pharm. Bull. 2006, 29, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Touil, F.; Pratt, S.; Mutter, R.; Chen, B. Screening a library of potential prion therapeutics against cellular prion proteins and insights into their mode of biological activities by surface plasmon resonance. J. Pharm. Biomed. Anal. 2006, 40, 822–832. [Google Scholar] [CrossRef] [PubMed]
- Georgieva, D.; Schwark, D.; von Bergen, M.; Redecke, L.; Genov, N.; Betzel, C. Interactions of recombinant prions with compounds of therapeutical significance. Biochem. Biophys. Res. Commun. 2006, 344, 463–470. [Google Scholar] [CrossRef] [PubMed]
- Cope, H.; Mutter, R.; Heal, W.; Pascoe, C.; Brown, P.; Pratt, S.; Chen, B. Synthesis and SAR study of acridine, 2-methylquinoline and 2-phenylquinazoline analogues as anti-prion agents. Eur. J. Med. Chem. 2006, 41, 1124–1143. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Okochi, H.; May, B.C.; Legname, G.; Prusiner, S.B.; Benet, L.Z.; Guglielmo, B.J.; Lin, E.T. Quinacrine is mainly metabolized to mono-desethyl quinacrine by CYP3A4/5 and its brain accumulation is limited by P-glycoprotein. Drug Metab. Dispos. 2006, 34, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.; Sakasegawa, Y.; Doh-Ura, K.; Go, M.L. Anti-prion activities and drug-like potential of functionalized quinacrine analogs with basic phenyl residues at the 9-amino position. Eur. J. Med. Chem. 2011, 46, 2917–2929. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.H.; Lee, C.Y.; Teruya, K.; Ong, W.Y.; Doh-ura, K.; Go, M.L. Antiprion activity of functionalized 9-aminoacridines related to quinacrine. Bioorg. Med. Chem. 2008, 16, 6737–6746. [Google Scholar] [CrossRef] [PubMed]
- Kamatari, Y.O.; Hayano, Y.; Yamaguchi, K.; Hosokawa-Muto, J.; Kuwata, K. Characterizing antiprion compounds based on their binding properties to prion proteins: Implications as medical chaperones. Protein Sci. 2013, 22, 22–34. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, A.J.; Trevitt, C.R.; Tattum, M.H.; Risse, E.; Quarterman, E.; Ibarra, A.A.; Wright, C.; Jackson, G.S.; Sessions, R.B.; Farrow, M.; et al. Pharmacological chaperone for the structured domain of human prion protein. Proc. Natl. Acad. Sci. USA 2010, 107, 17610–17615. [Google Scholar] [CrossRef] [PubMed]
- Baral, P.K.; Swayampakula, M.; Rout, M.K.; Kav, N.N.; Spyracopoulos, L.; Aguzzi, A.; James, M.N. Structural basis of prion inhibition by phenothiazine compounds. Structure 2014, 22, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Stincardini, C.; Massignan, T.; Biggi, S.; Elezgarai, S.R.; Sangiovanni, V.; Vanni, I.; Pancher, M.; Adami, V.; Moreno, J.; Stravalaci, M.; et al. An antipsychotic drug exerts anti-prion effects by altering the localization of the cellular prion protein. PLoS ONE 2017, 12, e0182589. [Google Scholar] [CrossRef] [PubMed]
- Daniel, J.A.; Chau, N.; Abdel-Hamid, M.K.; Hu, L.; von Kleist, L.; Whiting, A.; Krishnan, S.; Maamary, P.; Joseph, S.R.; Simpson, F.; et al. Phenothiazine-derived antipsychotic drugs inhibit dynamin and clathrin-mediated endocytosis. Traffic 2015, 16, 635–654. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, T.; Suzuki, A.; Hasebe, R.; Horiuchi, M. Comparison of the anti-prion mechanism of four different anti-prion compounds, anti-PrP monoclonal antibody 44B1, pentosan polysulfate, chlorpromazine, and u18666a, in prion-infected mouse neuroblastoma cells. PLoS ONE 2014, 9, e106516. [Google Scholar] [CrossRef] [PubMed]
- Cavaliere, P.; Torrent, J.; Prigent, S.; Granata, V.; Pauwels, K.; Pastore, A.; Rezaei, H.; Zagari, A. Binding of methylene blue to a surface cleft inhibits the oligomerization and fibrillization of prion protein. Biochim. Biophys. Acta 2013, 1832, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Koyama, N.; Segawa, T.; Maeda, M.; Maruyama, N.; Kinoshita, N.; Hou, H.; Tan, J.; Town, T. Methylene blue modulates beta-secretase, reverses cerebral amyloidosis, and improves cognition in transgenic mice. J. Biol. Chem. 2014, 289, 30303–30317. [Google Scholar] [CrossRef] [PubMed]
- Sontag, E.M.; Lotz, G.P.; Agrawal, N.; Tran, A.; Aron, R.; Yang, G.; Necula, M.; Lau, A.; Finkbeiner, S.; Glabe, C.; et al. Methylene blue modulates huntingtin aggregation intermediates and is protective in huntington’s disease models. J. Neurosci. 2012, 32, 11109–11119. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.M.; Edwards, P.C.; Lai, R.Y.; Roth, M.; Harrington, C.R. Selective inhibition of alzheimer disease-like tau aggregation by phenothiazines. Proc. Natl. Acad. Sci. USA 1996, 93, 11213–11218. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M.; Nonaka, T.; Arai, T.; Kametani, F.; Buchman, V.L.; Ninkina, N.; Bachurin, S.O.; Akiyama, H.; Goedert, M.; Hasegawa, M. Methylene blue and dimebon inhibit aggregation of TDP-43 in cellular models. FEBS Lett. 2009, 583, 2419–2424. [Google Scholar] [CrossRef] [PubMed]
- Caughey, W.S.; Raymond, L.D.; Horiuchi, M.; Caughey, B. Inhibition of protease-resistant prion protein formation by porphyrins and phthalocyanines. Proc. Natl. Acad. Sci. USA 1998, 95, 12117–12122. [Google Scholar] [CrossRef] [PubMed]
- Priola, S.A.; Raines, A.; Caughey, W.S. Porphyrin and phthalocyanine antiscrapie compounds. Science 2000, 287, 1503–1506. [Google Scholar] [CrossRef] [PubMed]
- Massignan, T.; Cimini, S.; Stincardini, C.; Cerovic, M.; Vanni, I.; Elezgarai, S.R.; Moreno, J.; Stravalaci, M.; Negro, A.; Sangiovanni, V.; et al. A cationic tetrapyrrole inhibits toxic activities of the cellular prion protein. Sci. Rep. 2016, 6, 23180. [Google Scholar] [CrossRef] [PubMed]
- Kocisko, D.A.; Caughey, W.S.; Race, R.E.; Roper, G.; Caughey, B.; Morrey, J.D. A porphyrin increases survival time of mice after intracerebral prion infection. Antimicrob. Agents Chemother. 2006, 50, 759–761. [Google Scholar] [CrossRef] [PubMed]
- Rajora, M.A.; Lou, J.W.H.; Zheng, G. Advancing porphyrin’s biomedical utility via supramolecular chemistry. Chem. Soc. Rev. 2017, 46, 6433–6469. [Google Scholar] [CrossRef] [PubMed]
- Caspi, S.; Halimi, M.; Yanai, A.; Sasson, S.B.; Taraboulos, A.; Gabizon, R. The anti-prion activity of congo red. Putative mechanism. J. Biol. Chem. 1998, 273, 3484–3489. [Google Scholar] [CrossRef] [PubMed]
- Milhavet, O.; Mange, A.; Casanova, D.; Lehmann, S. Effect of congo red on wild-type and mutated prion proteins in cultured cells. J. Neurochem. 2000, 74, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.; Brown, K.; Raymond, G.J.; Katzenstein, G.E.; Thresher, W. Binding of the protease-sensitive form of prp (prion protein) to sulfated glycosaminoglycan and congo red [corrected]. J. Virol. 1994, 68, 2135–2141. [Google Scholar] [PubMed]
- Caughey, B.; Ernst, D.; Race, R.E. Congo red inhibition of scrapie agent replication. J. Virol. 1993, 67, 6270–6272. [Google Scholar] [PubMed]
- Ingrosso, L.; Ladogana, A.; Pocchiari, M. Congo red prolongs the incubation period in scrapie-infected hamsters. J. Virol. 1995, 69, 506–508. [Google Scholar] [PubMed]
- Rudyk, H.; Knaggs, M.H.; Vasiljevic, S.; Hope, J.; Birkett, C.; Gilbert, I.H. Synthesis and evaluation of analogues of congo red as potential compounds against transmissible spongiform encephalopathies. Eur. J. Med. Chem. 2003, 38, 567–579. [Google Scholar] [CrossRef]
- Rudyk, H.; Vasiljevic, S.; Hennion, R.M.; Birkett, C.R.; Hope, J.; Gilbert, I.H. Screening congo red and its analogues for their ability to prevent the formation of PrP-res in scrapie-infected cells. J. Gen. Virol. 2000, 81, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Sellarajah, S.; Lekishvili, T.; Bowring, C.; Thompsett, A.R.; Rudyk, H.; Birkett, C.R.; Brown, D.R.; Gilbert, I.H. Synthesis of analogues of congo red and evaluation of their anti-prion activity. J. Med. Chem. 2004, 47, 5515–5534. [Google Scholar] [CrossRef] [PubMed]
- Risse, E.; Nicoll, A.J.; Taylor, W.A.; Wright, D.; Badoni, M.; Yang, X.; Farrow, M.A.; Collinge, J. Identification of a compound that disrupts binding of amyloid-beta to the prion protein using a novel fluorescence-based assay. J. Biol. Chem. 2015, 290, 17020–17028. [Google Scholar] [CrossRef] [PubMed]
- Kuwata, K.; Nishida, N.; Matsumoto, T.; Kamatari, Y.O.; Hosokawa-Muto, J.; Kodama, K.; Nakamura, H.K.; Kimura, K.; Kawasaki, M.; Takakura, Y.; et al. Hot spots in prion protein for pathogenic conversion. Proc. Natl. Acad. Sci. USA 2007, 104, 11921–11926. [Google Scholar] [CrossRef] [PubMed]
- Kimura, T.; Hosokawa-Muto, J.; Kamatari, Y.O.; Kuwata, K. Synthesis of GN8 derivatives and evaluation of their antiprion activity in TSE-infected cells. Bioorg. Med. Chem. Lett. 2011, 21, 1502–1507. [Google Scholar] [CrossRef] [PubMed]
- Perrier, V.; Wallace, A.C.; Kaneko, K.; Safar, J.; Prusiner, S.B.; Cohen, F.E. Mimicking dominant negative inhibition of prion replication through structure-based drug design. Proc. Natl. Acad. Sci. USA 2000, 97, 6073–6078. [Google Scholar] [CrossRef] [PubMed]
- Guo, K.; Mutter, R.; Heal, W.; Reddy, T.R.; Cope, H.; Pratt, S.; Thompson, M.J.; Chen, B. Synthesis and evaluation of a focused library of pyridine dicarbonitriles against prion disease. Eur. J. Med. Chem. 2008, 43, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Reddy, T.R.; Mutter, R.; Heal, W.; Guo, K.; Gillet, V.J.; Pratt, S.; Chen, B. Library design, synthesis, and screening: Pyridine dicarbonitriles as potential prion disease therapeutics. J. Med. Chem. 2006, 49, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Heal, W.; Thompson, M.J.; Mutter, R.; Cope, H.; Louth, J.C.; Chen, B. Library synthesis and screening: 2,4-diphenylthiazoles and 2,4-diphenyloxazoles as potential novel prion disease therapeutics. J. Med. Chem. 2007, 50, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Thompson, M.J.; Louth, J.C.; Greenwood, G.K.; Sorrell, F.J.; Knight, S.G.; Adams, N.B.; Chen, B. Improved 2,4-diarylthiazole-based antiprion agents: Switching the sense of the amide group at C5 leads to an increase in potency. ChemMedChem 2010, 5, 1476–1488. [Google Scholar] [CrossRef] [PubMed]
- Porat, Y.; Abramowitz, A.; Gazit, E. Inhibition of amyloid fibril formation by polyphenols: Structural similarity and aromatic interactions as a common inhibition mechanism. Chem. Biol. Drug Des. 2006, 67, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Rambold, A.S.; Miesbauer, M.; Olschewski, D.; Seidel, R.; Riemer, C.; Smale, L.; Brumm, L.; Levy, M.; Gazit, E.; Oesterhelt, D.; et al. Green tea extracts interfere with the stress-protective activity of PrP and the formation of PrP. J. Neurochem. 2008, 107, 218–229. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa-Muto, J.; Kamatari, Y.O.; Nakamura, H.K.; Kuwata, K. Variety of antiprion compounds discovered through an in silico screen based on cellular-form prion protein structure: Correlation between antiprion activity and binding affinity. Antimicrob. Agents Chemother. 2009, 53, 765–771. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kimura, T.; Hosokawa-Muto, J.; Asami, K.; Murai, T.; Kuwata, K. Synthesis of 9-substituted 2,3,4,9-tetrahydro-1H-carbazole derivatives and evaluation of their anti-prion activity in tse-infected cells. Eur. J. Med. Chem. 2011, 46, 5675–5679. [Google Scholar] [CrossRef] [PubMed]
- Hyeon, J.W.; Choi, J.; Kim, S.Y.; Govindaraj, R.G.; Jam Hwang, K.; Lee, Y.S.; An, S.S.; Lee, M.K.; Joung, J.Y.; No, K.T.; et al. Discovery of novel anti-prion compounds using in silico and in vitro approaches. Sci. Rep. 2015, 5, 14944. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Yamaguchi, K.; Fukuoka, M.; Kuwata, K. Logical design of anti-prion agents using nagara. Biochem. Biophys. Res. Commun. 2016, 469, 930–935. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, D.; Nakagaki, T.; Ishikawa, T.; Atarashi, R.; Watanabe, K.; Cruz, F.A.; Hamada, T.; Nishida, N. Structure-based drug discovery for prion disease using a novel binding simulation. EBioMedicine 2016, 9, 238–249. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Chemical Scaffold | Compound (Figure 1) | KD * | EC50 ** | Effect In Vivo *** | Conclusions |
|---|---|---|---|---|---|
| Acridine derivatives | 1 | ~1mM | ~0.3 µM | Not significant | Primary effects are PrP-independent |
| Phenothiazine derivatives | 2 | >400 µM | ~3 µM | Not significant | Likely acting by inducing PrPC re-localization from the cell surface |
| Tetrapyrroles | 5 | 4.52 µM | 1.6 µM | Prolongation of survival time in prion-infected mice | Low specificity and possible poor pharmacokinetics |
| Diazo dyes | 6 | 1.6 µM | 0.015 µM | Not available | Low specificity |
| Chicago sky blue 6B | 7 | 0.55 µM | Low µM | Not available | Need confirmation |
| Diphenylmethane derivatives | 8 | 5 µM | 1.35 µM | Prolongation of survival time in prion-infected mice | PrPC binding not reproduced in some study |
| Pyridine Dicarbonitriles | 11 | ~20µM | 18.6 µM | Not available | No correlation between anti-prion activity and binding to PrPC |
| Diarylthiazoles | 13 | 3.8 µM | 4 µM | Not available | No correlation between anti-prion activity and binding to PrPC |
| Natural polyphenols | 14 | 0.13 µM | - | Not available | Possible non-specific interaction with PrPC |
| Miscellanea | 17 | 50.8 µM | Not available | Not available | Need confirmation |
| 20 | 19 µM | 3.72 µM | Not available | Need confirmation |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barreca, M.L.; Iraci, N.; Biggi, S.; Cecchetti, V.; Biasini, E. Pharmacological Agents Targeting the Cellular Prion Protein. Pathogens 2018, 7, 27. https://doi.org/10.3390/pathogens7010027
Barreca ML, Iraci N, Biggi S, Cecchetti V, Biasini E. Pharmacological Agents Targeting the Cellular Prion Protein. Pathogens. 2018; 7(1):27. https://doi.org/10.3390/pathogens7010027
Chicago/Turabian StyleBarreca, Maria Letizia, Nunzio Iraci, Silvia Biggi, Violetta Cecchetti, and Emiliano Biasini. 2018. "Pharmacological Agents Targeting the Cellular Prion Protein" Pathogens 7, no. 1: 27. https://doi.org/10.3390/pathogens7010027
APA StyleBarreca, M. L., Iraci, N., Biggi, S., Cecchetti, V., & Biasini, E. (2018). Pharmacological Agents Targeting the Cellular Prion Protein. Pathogens, 7(1), 27. https://doi.org/10.3390/pathogens7010027