On the Demographic and Selective Forces Shaping Patterns of Human Cytomegalovirus Variation within Hosts

Abstract
:1. Introduction
2. HCMV Diversity within Hosts
3. Demographic Process Underlying Congenital Infection
4. The Role of Variable Mutation, Recombination, and Selection in HCMV Evolution
5. Clinical Implications
6. Conclusions and Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kenneson, A.; Cannon, M.J. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev. Med. Virol. 2007, 17, 253–276. [Google Scholar] [CrossRef] [PubMed]
- Boeckh, M.; Geballe, A.P. Science in medicine Cytomegalovirus: Pathogen, paradigm, and puzzle. J. Clin. Investig. 2011, 121, 1673–1680. [Google Scholar] [CrossRef] [PubMed]
- Manicklal, S.; Emery, V.C.; Lazzarotto, T.; Boppana, S.B.; Gupta, R.K. The “Silent” global burden of congenital cytomegalovirus. Clin. Microbiol. Rev. 2013, 26, 86–102. [Google Scholar] [CrossRef] [PubMed]
- Hassan, J.; Connell, J. Translational Mini-Review Series on Infectious Disease: Congenital cytomegalovirus infection: 50 Years on. Clin. Exp. Immunol. 2007, 149, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Anoh, A.E.; Akoua-Koffi, C.; Couacy-Hymann, E.; Pauly, M.; Schubert, G.; Mossoun, A.; Weiss, S.; Leendertz, S.A.J.; Jarvis, M.A.; Leendertz, F.H.; et al. Genetic identification of cytomegaloviruses in a rural population of Côte d’Ivoire. Virol. J. 2015, 12, 1–5. [Google Scholar] [CrossRef] [PubMed]
- McGeoch, D.J.; Rixon, F.J.; Davison, A.J. Topics in herpesvirus genomics and evolution. Virus Res. 2006, 117, 90–104. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.J. Evolution of sexually transmitted and sexually transmissible human herpesviruses. Ann. N. Y. Acad. Sci. 2011, 1230, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Spector, S.A.; Hirata, K.K.; Neuman, T.R. Identification of Multiple Cytomegalovirus Strains in Homosexual Men with Acquired Immunodeficiency Syndrome. J. Infect. Dis. 1984, 150, 953–956. [Google Scholar] [CrossRef] [PubMed]
- Meyer-König, U.; Ebert, K.; Schrage, B.; Pollak, S.; Hufert, F. Simultaneous infection of healthy people with multiple human cytomegalovirus strain. Lancet 1998, 352, 1280. [Google Scholar] [CrossRef]
- Drew, W.L.; Sweet, E.S.; Miner, R.C.; Mocarski, E.S. Multiple infections by cytomegalovirus in patients with acquired immunodeficiency syndrome: Documentation by Southern blot hybridization. J. Infect. Dis. 1984, 150, 952–953. [Google Scholar] [CrossRef] [PubMed]
- Haberland, M.; Meyer-Konig, U.; Hufert, F.T. Variation within the glycoprotein B gene of human cytomegalovirus is due to homologous recombination. J. Gen. Virol. 1999, 80 Pt 6, 1495–1500. [Google Scholar] [CrossRef] [PubMed]
- Faure-Della Corte, M.; Samot, J.; Garrigue, I.; Magnin, N.; Reigadas, S.; Couzi, L.; Dromer, C.; Velly, J.F.; Déchanet-Merville, J.; Fleury, H.J.A.; et al. Variability and recombination of clinical human cytomegalovirus strains from transplantation recipients. J. Clin. Virol. 2010, 47, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, Y.; Maeno, K.; Yoshida, S. Characterization of human cytomegalovirus-induced DNA polymerase and the associated 3′-to-5′, exonuclease. Virology 1983, 124, 221–231. [Google Scholar] [CrossRef]
- Trincado, D.E.; Scott, G.M.; White, P.A.; Hunt, C.; Rasmussen, L.; Rawlinson, W.D. Human cytomegalovirus strains associated with congenital and perinatal infections. J. Med. Virol. 2000, 61, 481–487. [Google Scholar] [CrossRef]
- Coaquette, A.; Bourgeois, A.; Dirand, C.; Varin, A.; Chen, W.; Herbein, G. Mixed cytomegalovirus glycoprotein B genotypes in immunocompromised patients. Clin. Infect. Dis. 2004, 39, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Pignatelli, S.; Dal Monte, P.; Rossini, G.; Chou, S.; Gojobori, T.; Hanada, K.; Guo, J.J.; Rawlinson, W.; Britt, W.; Mach, M.; et al. Human cytomegalovirus glycoprotein N (gpUL73-gN) genomic variants: Identification of a novel subgroup, geographical distribution and evidence of positive selective pressure. J. Gen. Virol. 2003, 84, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.A.; Novak, Z.; Pati, S.; Patro, R.K.; Blumenthal, J.; Danthuluri, V.R.; Ahmed, A.; Michaels, M.G.; Sánchez, P.J.; Bernstein, D.I.; et al. Mixed infection and strain diversity in congenital cytomegalovirus infection. J. Infect. Dis. 2011, 204, 1003–1007. [Google Scholar] [CrossRef] [PubMed]
- Zweygberg, W.B.; Brytting, M.; Linde, A.; Wahren, B.; Grillner, L. Sequence variation within three important cytomegalovirus gene regions in isolates from four different patient populations. J. Clin. Microbiol. 1998, 36, 3662–3669. [Google Scholar]
- Rasmussen, L.; Geissler, A.; Winters, M. Inter- and intragenic variations complicate the molecular epidemiology of human cytomegalovirus. J. Infect. Dis. 2003, 187, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Lasry, S.; Dény, P.; Asselot, C.; Rauzy, M.; Boucher, J.; Guyot, C.; Leroux, M.C.; Livartowski, A.; Reinert, P.; Nicolas, J.C. Interstrain variations in the cytomegalovirus (CMV) glycoprotein B gene sequence among CMV-infected children attending six day care centers. J. Infect. Dis. 1996, 174, 606–609. [Google Scholar] [CrossRef] [PubMed]
- Renzette, N.; Bhattacharjee, B.; Jensen, J.D.; Gibson, L.; Kowalik, T.F. Extensive Genome-Wide Variability of Human Cytomegalovirus in Congenitally Infected Infants. PLoS Pathog. 2011, 7. [Google Scholar] [CrossRef] [PubMed]
- Renzette, N.; Gibson, L.; Bhattacharjee, B.; Fisher, D.; Schleiss, M.R.; Jensen, J.D.; Kowalik, T.F. Rapid Intrahost Evolution of Human Cytomegalovirus Is Shaped by Demography and Positive Selection. PLoS Genet. 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Renzette, N.; Pokalyuk, C.; Gibson, L.; Bhattacharjee, B.; Schleiss, M.R.; Hamprecht, K.; Yamamoto, A.Y.; Mussi-Pinhata, M.M.; Britt, W.J.; Jensen, J.D.; et al. Limits and patterns of cytomegalovirus genomic diversity in humans. Proc. Natl. Acad. Sci. USA 2015, 112, E4120–E4128. [Google Scholar] [CrossRef] [PubMed]
- Hage, E.; Wilkie, G.S.; Linnenweber-Held, S.; Dhingra, A.; Suárez, N.M.; Schmidt, J.J.; Kay-Fedorov, P.C.; Mischak-Weissinger, E.; Heim, A.; Schwarz, A.; et al. Characterization of Human Cytomegalovirus Genome Diversity in Immunocompromised Hosts by Whole-Genome Sequencing Directly From Clinical Specimens. J. Infect. Dis. 2017, 215, 1673–1683. [Google Scholar] [CrossRef] [PubMed]
- Renzette, N.; Gibson, L.; Jensen, J.D.; Kowalik, T.F. Human cytomegalovirus intrahost evolution—A new avenue for understanding and controlling herpesvirus infections. Curr. Opin. Virol. 2014, 8, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Renzette, N.; Kowalik, T.F.; Jensen, J.D. On the relative roles of background selection and genetic hitchhiking in shaping human cytomegalovirus genetic diversity. Mol. Ecol. 2016, 25, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Renzette, N.; Pfeifer, S.P.; Matuszewski, S.; Kowalik, T.F.; Jensen, J.D. On the Analysis of Intrahost and Interhost Viral Populations: Human Cytomegalovirus as a Case Study of Pitfalls and Expectations. J. Virol. 2017, 91, e01976-16. [Google Scholar] [CrossRef] [PubMed]
- Pokalyuk, C.; Renzette, N.; Irwin, K.K.; Pfeifer, S.P.; Gibson, L.; Britt, W.J.; Yamamoto, A.Y.; Mussi-Pinhata, M.M.; Kowalik, T.F.; Jensen, J.D. Characterizing human cytomegalovirus reinfection in congenitally infected infants: An evolutionary perspective. Mol. Ecol. 2017, 26, 1980–1990. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Gonzalez, J.F.; Salazar, M.G.; Keele, B.F.; Learn, G.H.; Giorgi, E.E.; Li, H.; Decker, J.M.; Wang, S.; Baalwa, J.; Kraus, M.H.; et al. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J. Exp. Med. 2009, 206, 1273–1289. [Google Scholar] [CrossRef] [PubMed]
- Sowmya, P.; Madhavan, H.N. Analysis of mixed infections by multiple genotypes of human cytomegalovirus in immunocompromised patients. J. Med. Virol. 2009, 81, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Görzer, I.; Guelly, C.; Trajanoski, S.; Puchhammer-Stöckl, E. Deep sequencing reveals highly complex dynamics of human cytomegalovirus genotypes in transplant patients over time. J. Virol. 2010, 84, 7195–7203. [Google Scholar] [CrossRef] [PubMed]
- Sinzger, C.; Grefte, A.; Plachter, B.; Gouw, A.S.; The, T.H.; Jahn, G. Fibroblasts, epithelial cells, endothelial cells and smooth muscle cells are major targets of human cytomegalovirus infection in lung and gastrointestinal tissues. J. Gen. Virol. 1995, 76 Pt 4, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Pereira, L.; Maidji, E.; McDonagh, S.; Genbacev, O.; Fisher, S. Human cytomegalovirus transmission from the uterus to the placenta correlates with the presence of pathogenic bacteria and maternal immunity. J. Virol. 2003, 77, 13301–13314. [Google Scholar] [CrossRef] [PubMed]
- Lazzarotto, T.; Gabrielli, L.; Foschini, M.P.; Lanari, M.; Guerra, B.; Eusebi, V.; Landini, M.P. Congenital cytomegalovirus infection in twin pregnancies: Viral load in the amniotic fluid and pregnancy outcome. Pediatrics 2003, 112, E133–E137. [Google Scholar] [CrossRef]
- Stagno, S. Primary Cytomegalovirus Infection in Pregnancy. JAMA 1986, 256, 1904. [Google Scholar] [CrossRef] [PubMed]
- Fischer, W.; Ganusov, V.V.; Giorgi, E.E.; Hraber, P.T.; Keele, B.F.; Leitner, T.; Han, C.S.; Gleasner, C.D.; Green, L.; Lo, C.C.; et al. Transmission of single HIV-1 genomes and dynamics of early immune escape revealed by ultra-deep sequencing. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Keele, B.F.; Giorgi, E.E.; Salazar-Gonzalez, J.F.; Decker, J.M.; Pham, K.T.; Salazar, M.G.; Sun, C.; Grayson, T.; Wang, S.; Li, H.; et al. Identification and characterization of transmitted and early founder virus envelopes in primary HIV-1 infection. Proc. Natl. Acad. Sci. USA 2008, 105, 7552–7557. [Google Scholar] [CrossRef] [PubMed]
- Mayer, B.T.; Krantz, E.M.; Swan, D.; Ferrenberg, J.; Simmons, K.; Selke, S.; Huang, M.; Casper, C.; Corey, L.; Wald, A.; et al. Transient Oral Human Cytomegalovirus Infections Indicate Inefficient Viral Spread from Very Few Initially Infected Cells. J. Virol. 2017, 91, e00380-17. [Google Scholar] [CrossRef] [PubMed]
- Enders, G.; Daiminger, A.; Bäder, U.; Exler, S.; Enders, M. Intrauterine transmission and clinical outcome of 248 pregnancies with primary cytomegalovirus infection in relation to gestational age. J. Clin. Virol. 2011, 52, 244–246. [Google Scholar] [CrossRef] [PubMed]
- Numazaki, K. Human cytomegalovirus infection of breast milk. FEMS Immunol. Med. Microbiol. 1997, 18, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Begun, D.J.; Aquadro, C.F. Levels of naturally occurring DNA polymorphism correlate with recombination rates in D. melanogaster. Nature 1992, 356, 519–520. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, B.; Morgan, M.T.; Charlesworth, D. The effect of deleterious mutations on neutral molecular variation. Genetics 1993, 134, 1289–1303. [Google Scholar] [PubMed]
- Maynard Smith, J.; Haigh, J. The hitch-hiking effect of a favourable gene. Genet. Res. 1974, 23, 23–35. [Google Scholar] [CrossRef]
- Sanjuan, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral Mutation Rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.W.; Hwang, C.B.C. On the mutation rate of herpes simplex virus type 1. Genetics 2005, 170, 969–970. [Google Scholar] [CrossRef] [PubMed]
- Kropff, B.; Burkhardt, C.; Schott, J.; Nentwich, J.; Fisch, T.; Britt, W.; Mach, M. Glycoprotein N of Human Cytomegalovirus Protects the Virus from Neutralizing Antibodies. PLoS Pathog. 2012, 8. [Google Scholar] [CrossRef] [PubMed]
- Gardner, T.J.; Tortorella, D. Virion Glycoprotein-Mediated Immune Evasion by Human Cytomegalovirus: A Sticky Virus Makes a Slick Getaway. Microbiol. Mol. Biol. Rev. 2016, 80, 663–677. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.B.; Davies, A.A.; McSharry, B.P.; Wilkinson, G.W.; Sinclair, J.H. Complex I Binding by a Virally Encoded RNA Regulates Mitochondria-Induced Cell Death. Science 2007, 316, 1345–1348. [Google Scholar] [CrossRef] [PubMed]
- Kulesza, C.A.; Shenk, T. Murine cytomegalovirus encodes a stable intron that facilitates persistent replication in the mouse. Proc. Natl. Acad. Sci. USA 2006, 103, 18302–18307. [Google Scholar] [CrossRef] [PubMed]
- Pang, X.; Humar, A.; Preiksaitis, J.K. Concurrent genotyping and quantitation of cytomegalovirus gB genotypes in solid-organ-transplant recipients by use of a real-time PCR assay. J. Clin. Microbiol. 2008, 46, 4004–4010. [Google Scholar] [CrossRef] [PubMed]
- Puchhammer-Stockl, E.; Gorzer, I. Human cytomegalovirus: An enormous variety of strains and their possible clinical significance in the human host. Future Virol. 2011, 6, 259–271. [Google Scholar] [CrossRef]
- Manuel, O.; Åsberg, A.; Pang, X.; Rollag, H.; Emery, V.C.; Preiksaitis, J.K.; Kumar, D.; Pescovitz, M.D.; Bignamini, A.A.; Hartmann, A.; et al. Impact of Genetic Polymorphisms in Cytomegalovirus Glycoprotein B on Outcomes in Solid-Organ Transplant Recipients with Cytomegalovirus Disease. Clin. Infect. Dis. 2009, 49, 1160–1166. [Google Scholar] [CrossRef] [PubMed]
- Leach, C.T.; Detels, R.; Hennessey, K.; Liu, Z.; Visscher, B.R.; Dudley, J.P.; Cherry, J.D. A longitudinal study of cytomegalovirus infection in human immunodeficiency virus type 1-seropositive homosexual men: Molecular epidemiology and association with disease progression. J. Infect. Dis. 1994, 170, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Arav-Boger, R.; Willoughby, R.E.; Pass, R.F.; Zong, J.-C.; Jang, W.-J.; Alcendor, D.; Hayward, G.S. Polymorphisms of the cytomegalovirus (CMV)-encoded tumor necrosis factor-alpha and beta-chemokine receptors in congenital CMV disease. J. Infect. Dis. 2002, 186, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Britt, W.J. Congenital Human Cytomegalovirus Infection and the Enigma of Maternal Immunity. J. Virol. 2017, 91, e02392-16. [Google Scholar] [CrossRef] [PubMed]
- Tseng, T.C.; Kao, J.H. Elimination of Hepatitis B: Is It a Mission Possible? BMC Med. 2017, 15, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Voronin, Y.; Jani, I.; Graham, B.S.; Cunningham, C.K.; Mofenson, L.M.; Musoke, P.M.; Permar, S.R.; Scarlatti, G. Recent progress in immune-based interventions to prevent HIV-1 transmission to children. J. Int. AIDS Soc. 2017, 20, e25038. [Google Scholar] [CrossRef] [PubMed]
- Garrigue, I.; Moulinas, R.; Recordon-Pinson, P.; Delacour, M.L.; Essig, M.; Kaminski, H.; Rerolle, J.P.; Merville, P.; Fleury, H.; Alain, S. Contribution of next generation sequencing to early detection of cytomegalovirus UL97 emerging mutants and viral subpopulations analysis in kidney transplant recipients. J. Clin. Virol. 2016, 80, 74–81. [Google Scholar] [CrossRef] [PubMed]
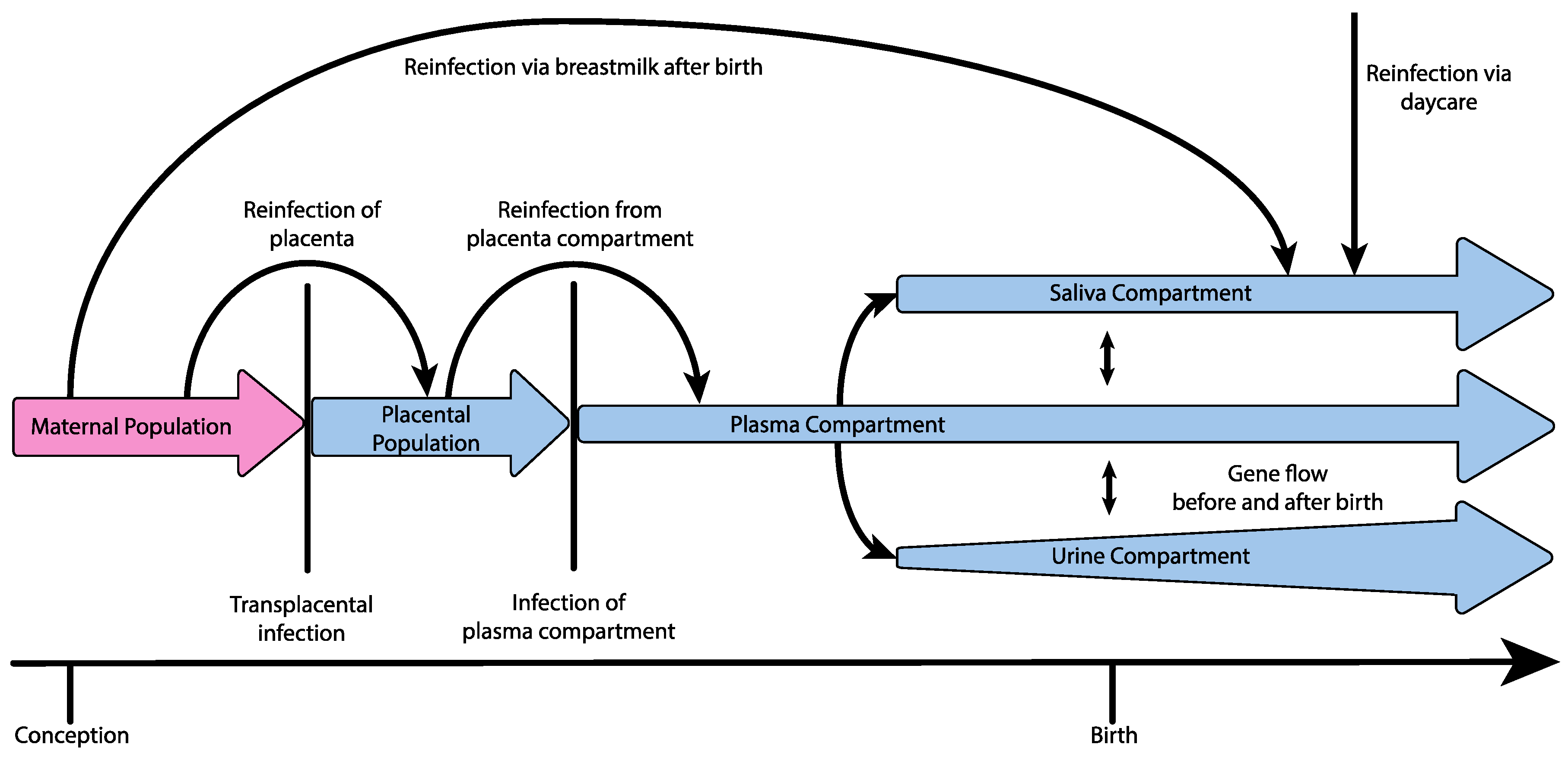

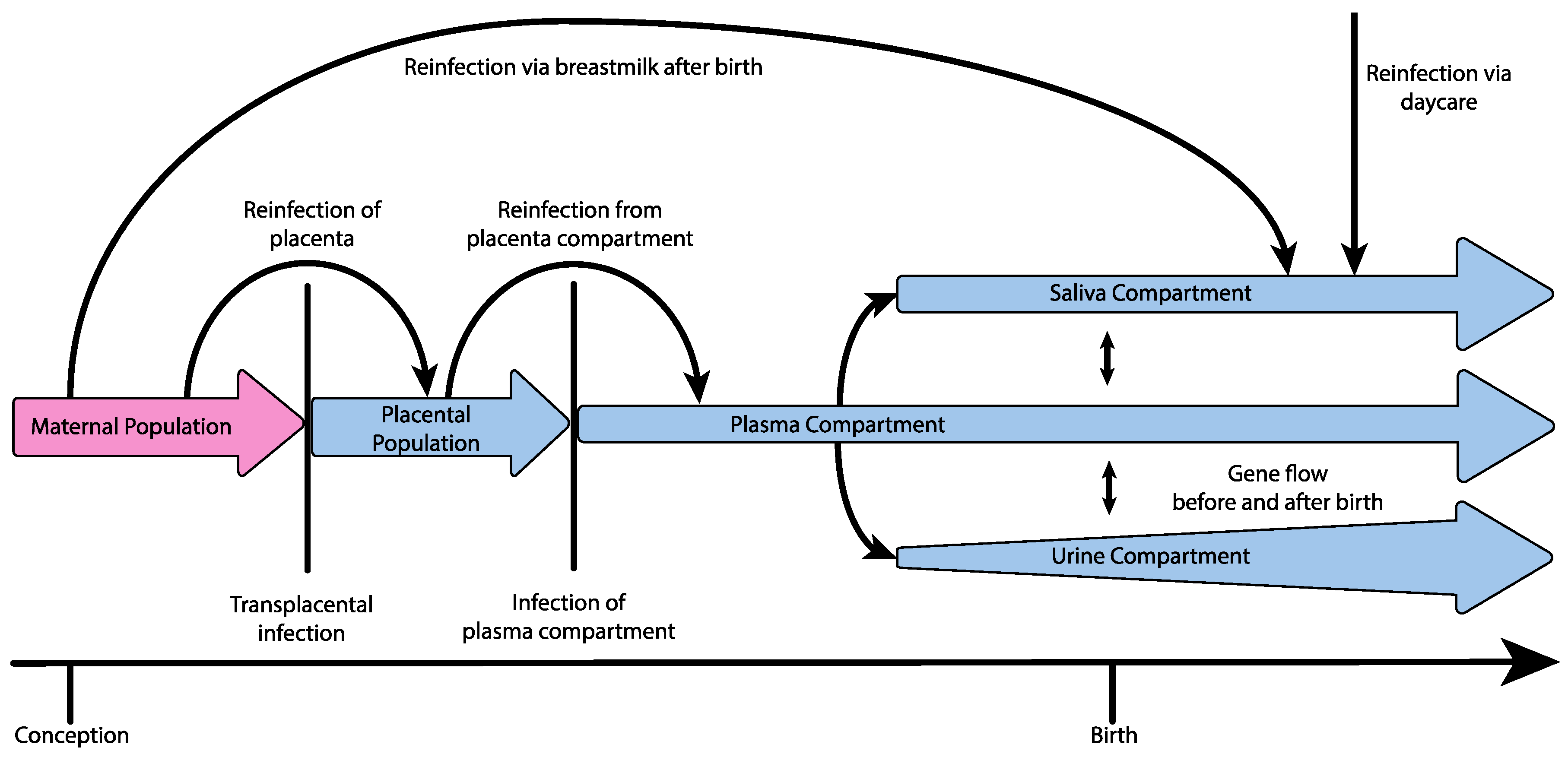{kind=link}
| Term | Definition |
|---|---|
| Population bottleneck | A sharp reduction in the size of a population, followed by recovery |
| Gene flow | The transfer of genetic variation between structured populations |
| Genetic hitchhiking | Changes in allele frequency at sites linked to positively selected loci |
| Background selection | Reduction of genetic diversity at sites linked to negatively selected loci |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sackman, A.M.; Pfeifer, S.P.; Kowalik, T.F.; Jensen, J.D. On the Demographic and Selective Forces Shaping Patterns of Human Cytomegalovirus Variation within Hosts. Pathogens 2018, 7, 16. https://doi.org/10.3390/pathogens7010016
Sackman AM, Pfeifer SP, Kowalik TF, Jensen JD. On the Demographic and Selective Forces Shaping Patterns of Human Cytomegalovirus Variation within Hosts. Pathogens. 2018; 7(1):16. https://doi.org/10.3390/pathogens7010016
Chicago/Turabian StyleSackman, Andrew M., Susanne P. Pfeifer, Timothy F. Kowalik, and Jeffrey D. Jensen. 2018. "On the Demographic and Selective Forces Shaping Patterns of Human Cytomegalovirus Variation within Hosts" Pathogens 7, no. 1: 16. https://doi.org/10.3390/pathogens7010016
APA StyleSackman, A. M., Pfeifer, S. P., Kowalik, T. F., & Jensen, J. D. (2018). On the Demographic and Selective Forces Shaping Patterns of Human Cytomegalovirus Variation within Hosts. Pathogens, 7(1), 16. https://doi.org/10.3390/pathogens7010016