
Genetic Fingerprint of Klebsiella pneumoniae Virulence: A Systematic Review
 , ,
, ,  , and
, and
Abstract
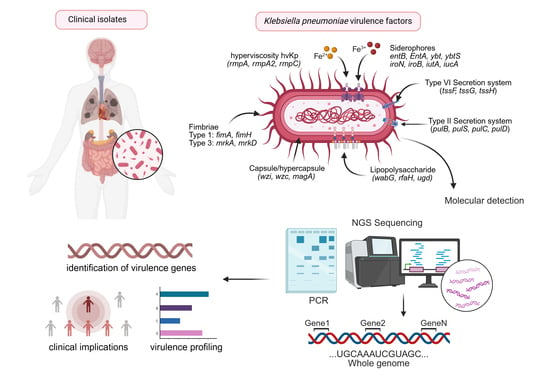
1. Introduction
2. Materials and Methods
2.1. Systematic Review
2.2. Search Strategy
2.3. Eligibility Criteria
2.4. Study Selection
2.5. Data Extraction
2.5.1. Study Variables and Outcomes
2.5.2. Effect Measures
2.5.3. Risk of Bias Assessment
2.5.4. Grouping Criteria for the Analyses
2.5.5. Handling Missing Data
2.5.6. Presentation of Results
2.6. Statistical Analysis
2.6.1. Narrative Summary of Results
2.6.2. Heterogeneity Analysis
2.6.3. Sensitivity Analysis
2.6.4. Bias Assessment
2.7. PROSPERO Registration
2.8. Ethical Approval
3. Results
3.1. Systematic Review and Characteristics of Included Studies
3.2. Genes Reported in the Period 2005–2010
3.3. Genes Reported in the Period 2011–2015
3.4. Genes Reported in the Period 2016–2020
3.5. Genes Reported in the Period 2021–2025
3.6. Genes Reported in cKp vs. hvKp
3.7. Comparative Analysis of Virulence-Associated Genes Between cKp and hvKp
3.8. Influence of Molecular Methods on Virulence Gene Detection
3.9. Regional Analysis of K. pneumoniae Virulence Genes
3.10. Results of Risk of Bias and Methodological Quality Assessment
3.11. Certainty of Evidence
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, T.-A.; Chuang, Y.-T.; Lin, C.-H. A Decade-Long Review of the Virulence, Resistance, and Epidemiological Risks of Klebsiella pneumoniae in ICUs. Microorganisms 2024, 12, 2548. [Google Scholar] [CrossRef]
- Rocha, J.; Henriques, I.; Gomila, M.; Manaia, C.M. Common and Distinctive Genomic Features of Klebsiella pneumoniae Thriving in the Natural Environment or in Clinical Settings. Sci. Rep. 2022, 12, 10441. [Google Scholar] [CrossRef]
- Ruekit, S.; Srijan, A.; Serichantalergs, O.; Margulieux, K.R.; Mc Gann, P.; Mills, E.G.; Stribling, W.C.; Pimsawat, T.; Kormanee, R.; Nakornchai, S.; et al. Molecular Characterization of Multidrug-Resistant ESKAPEE Pathogens from Clinical Samples in Chonburi, Thailand (2017–2018). BMC Infect. Dis. 2022, 22, 695. [Google Scholar] [CrossRef]
- Jesudason, T. WHO Publishes Updated List of Bacterial Priority Pathogens. Lancet Microbe 2024, 5, 100940. [Google Scholar] [CrossRef]
- Han, B.; Feng, C.; Jiang, Y.; Ye, C.; Wei, Y.; Liu, J.; Zeng, Z. Mobile Genetic Elements Encoding Antibiotic Resistance Genes and Virulence Genes in Klebsiella pneumoniae: Important Pathways for the Acquisition of Virulence and Resistance. Front. Microbiol. 2025, 16, 1529157. [Google Scholar] [CrossRef]
- Shapovalova, V.V.; Chulkova, P.S.; Ageevets, V.A.; Nurmukanova, V.; Verentsova, I.V.; Girina, A.A.; Protasova, I.N.; Bezbido, V.S.; Sergevnin, V.I.; Feldblum, I.V.; et al. High-Risk Lineages of Hybrid Plasmids Carrying Virulence and Carbapenemase Genes. Antibiotics 2024, 13, 1224. [Google Scholar] [CrossRef]
- Zaborskytė, G.; Hjort, K.; Lytsy, B.; Sandegren, L. Parallel Within-Host Evolution Alters Virulence Factors in an Opportunistic Klebsiella pneumoniae during a Hospital Outbreak. Nat. Commun. 2025, 16, 8727. [Google Scholar] [CrossRef] [PubMed]
- Fournier, P.-E.; Dubourg, G.; Raoult, D. Clinical Detection and Characterization of Bacterial Pathogens in the Genomics Era. Genome Med. 2014, 6, 114. [Google Scholar] [CrossRef] [PubMed]
- Mendes, G.; Santos, M.L.; Ramalho, J.F.; Duarte, A.; Caneiras, C. Virulence Factors in Carbapenem-Resistant Hypervirulent Klebsiella pneumoniae. Front. Microbiol. 2023, 14, 1325077. [Google Scholar] [CrossRef]
- Monteiro, A.D.S.S.; Cordeiro, S.M.; Reis, J.N. Virulence Factors in Klebsiella pneumoniae: A Literature Review. Indian J. Microbiol. 2024, 64, 389–401. [Google Scholar] [CrossRef]
- Singh, R.P.; Kapoor, A.; Sinha, A.; Ma, Y.; Shankar, M. Virulence Factors of Klebsiella pneumoniae: Insights into Canonical and Emerging Mechanisms Driving Pathogenicity and Drug Resistance. Microbe 2025, 7, 100289. [Google Scholar] [CrossRef]
- Zhu, J.; Wang, T.; Chen, L.; Du, H. Virulence Factors in Hypervirulent Klebsiella pneumoniae. Front. Microbiol. 2021, 12, 642484. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G. Preferred Reporting Items for Systematic Reviews and Meta-Analyses: The PRISMA Statement. BMJ 2009, 339, b2535. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. Declaración PRISMA 2020: Una Guía Actualizada Para La Publicación de Revisiones Sistemáticas. Rev. Esp. Cardiol. 2021, 74, 790–799. [Google Scholar] [CrossRef]
- Von Elm, E.; Altman, D.G.; Egger, M.; Pocock, S.J.; Gøtzsche, P.C.; Vandenbroucke, J.P. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) Statement: Guidelines for Reporting Observational Studies. J. Clin. Epidemiol. 2008, 61, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Bossuyt, P.M.; Reitsma, J.B.; Bruns, D.E.; Gatsonis, C.A.; Glasziou, P.P.; Irwig, L.; Lijmer, J.G.; Moher, D.; Rennie, D.; de Vet, H.C.W.; et al. STARD 2015: An Updated List of Essential Items for Reporting Diagnostic Accuracy Studies. BMJ 2015, 351, h5527. [Google Scholar] [CrossRef]
- QUADAS-2: A Revised Tool for the Quality Assessment of Diagnostic Accuracy Studies|Annals of Internal Medicine. Available online: https://www.acpjournals.org/doi/10.7326/0003-4819-155-8-201110180-00009 (accessed on 22 April 2026).
- Aromataris, E.; Lockwood, C.; Porritt, K.; Pilla, B.; Jordan, Z. (Eds.) JBI Manual for Evidence Synthesis; JBI: Adelaide, Australia, 2024; Available online: https://jbi-global-wiki.refined.site/space/jbi-global-wiki.refined.site (accessed on 22 April 2026).
- Wang, Q.; Ye, M.-Y.; Hong, C.; Li, Z.-P.; Lin, L. The Mechanisms of Resistance, Epidemiological Characteristics, and Molecular Evolution of Carbapenem-Resistant Hypervirulent Klebsiella pneumoniae. Lab. Med. 2025, 56, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Heng, H.; Sun, R.; Yang, X.; Ye, L.; Chen, K.; Li, J.; Chan, E.W.-C.; Li, R.; Zhang, R.; Chen, S. Profiling the Landscape of Carbapenem Resistance and Hypervirulence in Klebsiella pneumoniae: A Global Epidemiological Analysis of the Plasmidome. Drug Resist. Updates 2025, 81, 101254. [Google Scholar] [CrossRef] [PubMed]
- Fahy, S.; O’Connor, J.A.; Sleator, R.D.; Lucey, B. From Species to Genes: A New Diagnostic Paradigm. Antibiotics 2024, 13, 661. [Google Scholar] [CrossRef]
- Kot, B.; Piechota, M.; Szweda, P.; Mitrus, J.; Wicha, J.; Grużewska, A.; Witeska, M. Virulence Analysis and Antibiotic Resistance of Klebsiella pneumoniae Isolates from Hospitalised Patients in Poland. Sci. Rep. 2023, 13, 4448. [Google Scholar] [CrossRef]
- Marchetti, M.; De Bei, O.; Bettati, S.; Campanini, B.; Kovachka, S.; Gianquinto, E.; Spyrakis, F.; Ronda, L. Iron Metabolism at the Interface between Host and Pathogen: From Nutritional Immunity to Antibacterial Development. Int. J. Mol. Sci. 2020, 21, 2145. [Google Scholar] [CrossRef]
- Russo, T.A.; Olson, R.; Fang, C.-T.; Stoesser, N.; Miller, M.; MacDonald, U.; Hutson, A.; Barker, J.H.; La Hoz, R.M.; Johnson, J.R. Identification of Biomarkers for Differentiation of Hypervirulent Klebsiella pneumoniae from Classical K. pneumoniae. J. Clin. Microbiol. 2018, 56, e00776-18. [Google Scholar] [CrossRef]
- Russo, T.A.; Marr, C.M. Hypervirulent Klebsiella pneumoniae. Clin. Microbiol. Rev. 2019, 32, e00001-19. [Google Scholar] [CrossRef]
- Lam, M.M.C.; Wick, R.R.; Watts, S.C.; Cerdeira, L.T.; Wyres, K.L.; Holt, K.E. A Genomic Surveillance Framework and Genotyping Tool for Klebsiella pneumoniae and Its Related Species Complex. Nat. Commun. 2021, 12, 4188. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, M.; Cao, S.; Ishaq, H.M.; Zhao, H.; Yang, F.; Liu, L. The Biological and Regulatory Role of Type VI Secretion System of Klebsiella pneumoniae. Infect. Drug Resist. 2023, 16, 6911–6922. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Huang, H.-H.; Huang, Q.-S.; Fang-Ling, D.; Dan Wei, D.; La-Gen, W.; Xiang, T.-X.; Zhang, W.; Liu, Y. Distribution of Type VI Secretion System (T6SS) in Clinical Klebsiella pneumoniae Strains from a Chinese Hospital and Its Potential Relationship with Virulence and Drug Resistance. Microb. Pathog. 2022, 162, 105085. [Google Scholar] [CrossRef] [PubMed]
- Cianciotto, N.P.; White, R.C. Expanding Role of Type II Secretion in Bacterial Pathogenesis and Beyond. Infect. Immun. 2017, 85, e00014-17. [Google Scholar] [CrossRef]
- Li, Y.; Ni, M. Regulation of Biofilm Formation in Klebsiella pneumoniae. Front. Microbiol. 2023, 14, 1238482. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, Y.; Liu, P.; Jian, Z.; Yan, Q.; Tang, B.; Yang, A.; Liu, W. Genomic and Clinical Characterization of Klebsiella pneumoniae Carrying the Pks Island. Front. Microbiol. 2023, 14, 1189120. [Google Scholar] [CrossRef]
- Lu, M.-C.; Chen, Y.-T.; Chiang, M.-K.; Wang, Y.-C.; Hsiao, P.-Y.; Huang, Y.-J.; Lin, C.-T.; Cheng, C.-C.; Liang, C.-L.; Lai, Y.-C. Colibactin Contributes to the Hypervirulence of Pks+ K1 CC23 Klebsiella pneumoniae in Mouse Meningitis Infections. Front. Cell. Infect. Microbiol. 2017, 7, 103. [Google Scholar] [CrossRef]
- Siu, L.K.; Yeh, K.-M.; Lin, J.-C.; Fung, C.-P.; Chang, F.-Y. Klebsiella pneumoniae Liver Abscess: A New Invasive Syndrome. Lancet Infect. Dis. 2012, 12, 881–887. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2005–2010 | 2011–2015 | 2016–2020 | 2021–2025 | |
|---|---|---|---|---|
| Studies included | 8 | 34 | 160 | 474 |
| Total isolates | 416 | 2.803 | 13.561 | 52.387 |
| Distinct genes detected | 37 | 131 | 231 | 421 |
| Detection records | 42 | 169 | 2.197 | 6.672 |
| Distribution: strain type | cKp 54% hvKp 46% | cKp 65% hvKp 35% | cKp 53% hvKp 47% | cKp 54% hvKp 46% |
| Dominant technique | PCR (100%) | PCR (67.65%) WGS (17.65%) | WGS (47.50%) PCR (41.88%) | WGS (58.44%) PCR (32.49%) |
| Functional Group/Virulence Factors | N° of Genes | % | Genes |
|---|---|---|---|
| Structural genes of the K locus | 3 | 7.1% | wza, wzc, wzy |
| Capsular sugar synthesis | 2 | 7.1% | gnd, uge |
| Capsular serotype genes | 1 | 2.4% | wzi |
| Capsule regulation and hypermucoviscosity (hvKp) | 4 | 19.0% | rmpA, magA, rcsB, peg-344 |
| LPS/O-antigen/lipid A | 2 | 4.8% | wabG, wzx |
| Lipid A and charge modification | 0 | 0.0% | _ |
| Siderophores and iron uptake Aerobactin | 0 | 0.0% | _ |
| Siderophores and iron uptake Enterobactin | 1 | 2.4% | fepA |
| Siderophores and iron uptake Yersiniabactin | 1 | 2.4% | fyuA |
| Siderophores and iron uptake Salmochelin | 0 | 0.0% | _ |
| Other iron and heme uptake systems | 1 | 2.4% | kfu |
| Fimbriae and adhesins Type 1 fimbriae | 9 | 21.4% | fimA, fimB, fimC, fimD, fimE fimF, fimG, fimI, fimK |
| Fimbriae and adhesins Type 3 fimbriae | 5 | 11.9% | mrkA, mrkB, mrkC, mrkD, mrkF |
| Other fimbrial operons and pili | 0 | 0.0% | _ |
| Invasive/iron-regulated adhesins | 0 | 0.0% | _ |
| Matrix factors and biofilm regulation | 0 | 0.0% | _ |
| Type VI secretion system (T6SS) | 7 | 16.7% | impG, impH, impJ, icmF/tssM dotU/tssL, tli, impF |
| Type II secretion system (T2SS) | 0 | 0.0% | _ |
| Toxins and genotoxins colibactin | 0 | 0.0% | _ |
| Other toxins and cytotoxic factors | 0 | 0.0% | _ |
| Regulators, stress response and outer membrane | 1 | 2.4% | ompA |
| Toxin–antitoxin systems and persistence | 0 | 0.0% | _ |
| Nutrient acquisition and metabolism (excluding iron) | 0 | 0.0% | _ |
| Allantoin, purineand glycosylate metabolism | 0 | 0.0% | _ |
| Functional Group/Virulence Factors | No. of Genes | % | Genes |
|---|---|---|---|
| Structural genes of the K locus | 6 | 2.86% | wzc, wzb, wzy, wcaG, wcuF, wcoU |
| Capsular sugar synthesis | 3 | 1.63% | manB, uge, ugeE |
| Capsular serotype genes | 1 | 1.22% | wzi |
| Capsule regulation and hypermucoviscosity (hvKp) | 10 | 14.29% | rmpA, rmpA2, c-rmpA, magA, peg-344 orf2, glf, rcsB, ibrB, peg-589 |
| LPS/O-antigen/lipid A | 1 | 2.86% | wabG |
| Lipid A and charge modification | 0 | 0.00% | _ |
| Siderophores and iron uptake Aerobactin | 8 | 6.53% | iuc, iucA, iucB, iucC, iucD, iut, iutA, aero |
| Siderophores and iron uptake Enterobactin | 3 | 1.63% | ent, entB, fepA |
| Siderophores and iron uptake Yersiniabactin | 7 | 6.53% | ybt, ybtS, irp1, irp2, ybt9, fyu, fyuA |
| Siderophores and iron uptake Salmochelin | 5 | 4.08% | iro, iroB, iroC, iroD, iroN |
| Other iron and heme uptake systems | 20 | 11.84% | fec, fecA, fecI, fecR, fhuA, fhuB fhuC, fhuD, hmuR, hmuS, hmuT, hmuV hmuU, kfu, kfuB, kfuC, sitA, sitB, sitC, sitD |
| Fimbriae and adhesins Type 1 fimbriae | 12 | 9.39% | fim, fimA, fimB, fimC, fimD, fimE fimF, fimG, fimH, fimI, fimK, fimH-1 |
| Fimbriae and adhesins Type 3 fimbriae | 9 | 15.51% | mrk, mrkA, mrkB, mrkC, mrkD mrkF, mrkH, mrkI, mrkJ |
| Other fimbrial operons and pili | 13 | 5.31% | kpa, kpb, kpd, kpe, kpf, kpg, ecp ecpA, ecpB, ecpC, ecpD, ecpE, ecpR |
| Invasive/iron-regulated adhesins | 0 | 0.00% | _ |
| Matrix factors and biofilm regulation | 0 | 0.00% | _ |
| Type VI secretion system (T6SS) | 2 | 0.82% | vgrG/tssI, hcp/tssD |
| Type II secretion system (T2SS) | 0 | 0.00% | _ |
| Toxins and genotoxins colibactin | 3 | 1.63% | clb, clb2, clbR |
| Other toxins and cytotoxic factors | 2 | 0.82% | khe, hlyA |
| Regulators, stress response and outer membrane | 3 | 1.63% | ompA, pagO, traT |
| Toxin–antitoxin system and persistence | 0 | 0.00% | _ |
| Nutrient acquisition and metabolism (excluding iron) | 1 | 0.82% | ureA |
| Allantoin, purine and glycosylate metabolism | 22 | 10.61% | allA, allB, allC, allD, allR, allS, ybbW ybbY, ybbA, ylbE, ybbP, ybbQ, ybbS, ybbT ybbU, ybbX, glxK, glxR, fdrA, glc, hyi, ylbF |
| Functional Group/Virulence Factors | No. of Genes | % | Genes |
|---|---|---|---|
| Structural genes of the K locus | 12 | 1.88% | wza, wzc, wzm, wzb, wzc50, wzy cps, cpsA, cpsB, wcaG, wcaJ, wcbF |
| Capsular sugar synthesis | 7 | 1.17% | gmd, manB, falF, ugd, rmlB, uge, wbaP |
| Capsular serotype genes | 2 | 0.51% | wzi, wzi-705 |
| Capsule regulation and hypermucoviscosity (hvKp) | 12 | 11.19% | wmpA, wmpA2, p-rmpA, p-rmpA2, c-rmpA rmpC, magA, orf10, kvgS, rcsB, mviM, peg-344 |
| LPS/O-antigen/lipid A | 7 | 1.58% | waaE, wabG, wabN, wbbM, ofr, rfaH, wzx |
| Lipid A and charge modification | 0 | 0.00% | _ |
| Siderophores and iron uptake Aerobactin | 11 | 13.48% | iuc, iucA, iucB, iucC, iucD iuc1, iut, iutA, iutB, iutC, aero |
| Siderophores and iron uptake Enterobactin | 20 | 8.09% | ent, entA, entB, entC, entD, entE, entF entH, entS, fep, fepB, fepC, fepD, fepG fes, febI, febR, febD, febC, fepA |
| Siderophores and iron uptake Yersiniabactin | 30 | 16.33% | ybt, ybtA, ybtB, ybtE, ybtO, ybtQ, ybtS, ybtT ybtU, ybuD, ybtL, ybtX, irp, irp1, irp2 irp3, irp5, ybt0, ybt1, ybt4, ybt9, ybt10 ybt14, ybt16, ybt17, Airp1, Airp2, fyu, fyuA, ybtR, |
| Siderophores and iron uptake Salmochelin | 8 | 7.58% | iro, iro1, iro2, iroB, iroC, iroD, iroE, iroN |
| Other iron and heme uptake systems | 11 | 5.09% | fec, fecA, fecB, fecI, fecR ybdA, kfu, kfuA, kfuB, kfuC, kfuD |
| Fimbriae and adhesins Type 1 fimbriae | 12 | 6.31% | fim, fimA, fimB, fimC, fimD, fimE fimF, fimG, fimH, fimI, fimK, fimH-1 |
| Fimbriae and adhesins Type 3 fimbriae | 10 | 13.17% | mrk, mrkA, mrkB, mrkC, mrkD mrkF, mrkH, mrkI, mrkJ, mrkK |
| Other fimbrial operons and pili | 8 | 1.98% | kpn, ecpA, ecpB, ecpC, ecpD, ecpE, ecpR, pilW |
| Invasive/iron-regulated adhesins | 0 | 0.00% | _ |
| Matrix factors and biofilm regulation | 4 | 0.25% | pgaA, pgaB, pgaC, bcsA |
| Type VI secretion system (T6SS) | 8 | 0.46% | impG, impH, impJ, vasG icmF/tssM, dotU/tssL, impF, pld1 |
| Type II secretion system (T2SS) | 10 | 0.51% | gspE, gspG, pulB, pulC, pulD pulE, pulG, pulO, pulS, PulN |
| Toxins and genotoxins: colibactin | 22 | 5.80% | clb, clb2, clb3, clb5, clbA, clbB, clbC clbD, clbE, clbF, clbG, clbH, clbI, clbJ, clbK clbL, clbM, clbN, clbO, clbP, clbQ, clbR |
| Other toxins and cytotoxic factors | 5 | 0.36% | hly, CNF-1, khe, vatD, cnf |
| Regulators, stress response and outer membrane | 5 | 0.81% | ompA, pagO, traT, shiF, htrA |
| Toxin–antitoxin systems and persistence | 8 | 0.41% | hipA, hipB, vapB, vapC, hpd, doc, mazE, mazF |
| Nutrient acquisition and metabolism (excluding iron) | 3 | 0.66% | ureA, ureE, ureD |
| Allantoin, purine and glycosylate metabolism | 16 | 2.39% | allA, allB, allC, allD, allR, allS, ybbW ybbY, ylbE, glxK, glxR, fdrA, gcl, hyi, ylbF, arcC |
| Functional Group/Virulence Factors | No. of Genes | % | Genes |
|---|---|---|---|
| Structural genes of the K locus | 30 | 1.81% | wza, wzc, wzm, wzb, wzc50, wzxE, wzt, wzy cps, cpsA, cpsB, kps, KpsI, kpsM, KpsF, wcah wcai, wcaM, wcaG, wcaJ, wceN, wceM, wacJ, wckI wcqC, wcsF, wcsR, wcuT, wcoV, wcoU. |
| Capsular sugar synthesis | 23 | 1.48% | gmd, manC, manB, gnd, galF, galU, ugd, rmlB rmlC, rmlD, galA, galB, galC, galD, galE, galM galP, galR, galS, uge, ugeF, gmhA, wbaP. |
| Capsular serotype genes | 4 | 0.49% | wzi, wzi1, wzi23, wziK24. |
| Capsule regulation and hypermucoviscosity (hvKp) | 21 | 11.92% | rmpA, rmpA1, rmpA2, rmpA3, p-rmpA, p-rmpA2, c-rmpA rmpB, rmpC, rmpD, magA, kvrA, kvrB, kvgA, kvgS, rcs rcsB, capL, peg-344, peg-589, peg-1631. |
| LPS/O-antigen/lipid A | 17 | 1.08% | wabG, wabH, wabN, waaU, wbbO, wbbM, wbbN, wbbY rfaD, rfaE, rfaF, rfaQ, wecA, wzx, acpXL, pagP, wzzB. |
| Lipid A and charge modification | 9 | 0.15% | lpx, lpxA, lpxB, lpxC, lpxD, arnD, kds, kdsA, kdsB. |
| Siderophores and iron uptake Aerobactin | 18 | 14.15% | iuc, iucA, iucB, iucC, iucD, iucE, iucF, iuc19 iuc1, iuc2, iuc3, iuc4, iuc5, iut, iutA, iutB, Kvar_1549, aero. |
| Siderophores and iron uptake Enterobactin | 25 | 9.70% | ent, entA, entB, entB1, entB2, entC, entD, entE entF, entH, entS, fep, fepB, fepC, fepD fepG, fes, fesE, fesF, febA, febB, febF, febD, febC, fepA. |
| Siderophores and iron uptake Yersiniabactin | 30 | 15.53% | ybt, ybtA, ybtB, ybtE, ybtP, ybtQ, ybtS, ybtT ybtU, ybtD, ybtL, ybtX, irp, irp1, irp2 ybt1, ybt2, ybt9, ybt10, ybt12, ybt13, ybt14 ybt15, ybt16, ybt17, ybt19, Airp1, Airp2, fyu, fyuA. |
| Siderophores and iron uptake Salmochelin | 8 | 7.10% | iro, iro1, iro2, iroB, iroC, iroD, iroE, iroN. |
| Other iron and heme uptake systems | 31 | 2.28% | tonB, fecA, fecI, fecR, fur, feo, ybdA, ybdC ybdG, ybdK, ybdL, ybdM, ybdO, ybdZ, feoA, feoB feoC, fhuD, cirA, kfu, kfuA, kfuB, kfuC, kfuD sitA, sitB, sitC, sitD, ChuA, ChuS, ChuU. |
| Fimbriae and adhesins Type 1 fimbriae | 14 | 9.05% | fim, fimA, fimB, fimC, fimD, fimE, fimF fimG, fimH, fimM, fimI, fimJ, fimK, fimH-1. |
| Fimbriae and adhesins Type 3 fimbriae | 12 | 9.82% | mrk, mrkA, mrkB, mrkC, mrkD, mrkF mrkH, mrkI, mrkJ, mrkL, mrkK, mrkM. |
| Other fimbrial operons and pili | 25 | 2.53% | kpn, stbA, stbB, stbC, stbD, stbE, kpiA, kpiB kpiC, kpiD, kpiE, kpiF, kpiG, ecp, ecpA, ecpB, ecpC ecpD, ecpE, ecpR, pilR, pilT, pilD, pilW, pilU. |
| Invasive/iron-regulated adhesins | 15 | 0.24% | iha, air, ibeB, afa, papE, papF, papC papD, papX, focA, focC, focD, sfaD, sfaE, dfaF. |
| Matrix factors and biofilm regulation | 7 | 0.15% | pgaA, pgaB, pgaC, pgaD, agn43, bssS, clpK. |
| Type VI secretion system (T6SS) | 35 | 4.78% | tssA, tssB, tssC, tssD, tssE, tssF, tssG, tssH tssI, tssJ, tssK, tssL, tssM, imp, impG, impH, impJ vasG, icmF/tssM, dotU/tssL, vasE/tssK, clpV/tssH, vgrG/tssI hcp/tssD, impA/tssD, vasA/impG, vipA/tssB vipB/tssC, sciN/tssJ, tli1, tle1, sci, dotU, impF, impN. |
| Type II secretion system (T2SS) | 25 | 0.45% | gspC, gspD, gspE, gspG, gspH, gspI, gspJ, gspL gspM, gspN, pulB, pulC, pulD, pulE, pulF, pulG pulH, pulI, pulJ, pulK, pulL, pulO, pulP, pulS, PulN. |
| Toxins and genotoxins colibactin | 23 | 3.37% | clb, clb1, clb2, clb3, clbA, clbB, clbC, clbD clbE, clbF, clbG, clbH, clbI, clbJ, clbK, clbL clbM, clbN, clbO, clbP, clpQ, clbR, clbS. |
| Other toxins and cytotoxic factors | 9 | 0.18% | hlyA, EAST-1, CNF-1, khe, SAT, usp, astA, hlyE, VAT. |
| Regulators, stress response and outer membrane | 12 | 0.84% | OmpA, pagO, ompX, ompR, ompW, ompN ompC, traT, shiF, lon, htrA, zapA. |
| Toxin–antitoxin systems and persistence | 5 | 0.07% | hipA, hipB, phd, doc, ratA. |
| Nutrient acquisition and metabolism (excluding iron) | 7 | 0.81% | ureC, ureD, ureE, ureF, ureG, ureB, ureA. |
| Allantoin, purine and glycosylate metabolism | 16 | 2.01% | all, allA, allB, allC, allD, allR, allS, ybbW ybbY, ylbE, glxK, glxR, fdrA, hyi, ylbF, arcC. |
| Gene | cKp n/N (%) | hvKp n/N (%) | Absolute Difference (%) | 95% CI | p Value (Fisher) |
|---|---|---|---|---|---|
| rmpA | 238/450 (52.89) | 346/436 (79.36) | 26.47 | (20.50–32.73) | <0.0001 |
| rmpA2 | 162/450 (36.00) | 256/436 (58.72) | 22.72 | (16.32–29.39) | <0.0001 |
| mrkD | 166/450 (36.89) | 144/436 (33.03) | 3.86 | (−2.58–10.25) | 0.2320 |
| iucA | 150/450 (33.33) | 247/436 (56.65) | 23.32 | (16.96–29.97) | <0.0001 |
| iutA | 154/450 (34.22) | 205/436 (47.02) | 12.8 | (6.32–19.42) | 0.0001 |
| Gene | PCR-Based Methods n/N (%) | Sequencing-Based Methods n/N (%) | Absolute Difference (%) | 95% CI | p Value (Fisher) |
|---|---|---|---|---|---|
| rmpA | 205/305 (67.21) | 215/371 (57.95) | 9.26 | (1.71 to 16.61) | 0.0137 |
| rmpA2 | 105/305 (34.43) | 190/371 (51.21) | −16.79 | (−24.48 to −9.40) | <0.0001 |
| mrkD | 133/305 (43.61) | 115/371 (31.00) | 12.61 | (5.08 to 20.00) | 0.0008 |
| iucA | 106/305 (34.75) | 183/371 (49.33) | −14.58 | (−22.26 to −7.16) | 0.0002 |
| iutA | 92/305 (30.16) | 186/371 (50.13) | −19.97 | (−27.57 to −12.74) | <0.0001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2026 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license.
Share and Cite
Aldana-Ortega, C.A.; Pérez-Villadiego, A.J.; Monterrosa-Taborda, Y.; Angulo-Ortíz, A.; Contreras-Martínez, O.I. Genetic Fingerprint of Klebsiella pneumoniae Virulence: A Systematic Review. Pathogens 2026, 15, 556. https://doi.org/10.3390/pathogens15050556
Aldana-Ortega CA, Pérez-Villadiego AJ, Monterrosa-Taborda Y, Angulo-Ortíz A, Contreras-Martínez OI. Genetic Fingerprint of Klebsiella pneumoniae Virulence: A Systematic Review. Pathogens. 2026; 15(5):556. https://doi.org/10.3390/pathogens15050556
Chicago/Turabian StyleAldana-Ortega, Carlos Andrés, Alexander José Pérez-Villadiego, Yohelys Monterrosa-Taborda, Alberto Angulo-Ortíz, and Orfa Inés Contreras-Martínez. 2026. "Genetic Fingerprint of Klebsiella pneumoniae Virulence: A Systematic Review" Pathogens 15, no. 5: 556. https://doi.org/10.3390/pathogens15050556
APA StyleAldana-Ortega, C. A., Pérez-Villadiego, A. J., Monterrosa-Taborda, Y., Angulo-Ortíz, A., & Contreras-Martínez, O. I. (2026). Genetic Fingerprint of Klebsiella pneumoniae Virulence: A Systematic Review. Pathogens, 15(5), 556. https://doi.org/10.3390/pathogens15050556