

Hydrogen Sulfide (H2S) Mitigates Sepsis-Induced Adrenal Dysfunction via Inhibition of TNFα-Mediated Necroptosis
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cell Treatment
2.3. Animals and Experimental Protocols
2.4. Cecal Ligation and Puncture Methods
2.5. Inflammation Detection
2.6. Western Blot Analysis
2.7. Immunofluorescence Staining
2.8. H&E Staining
2.9. Cell Counting Kit-8 (CCK-8)
2.10. Genetic Identification
2.11. Statistical Analysis
3. Results
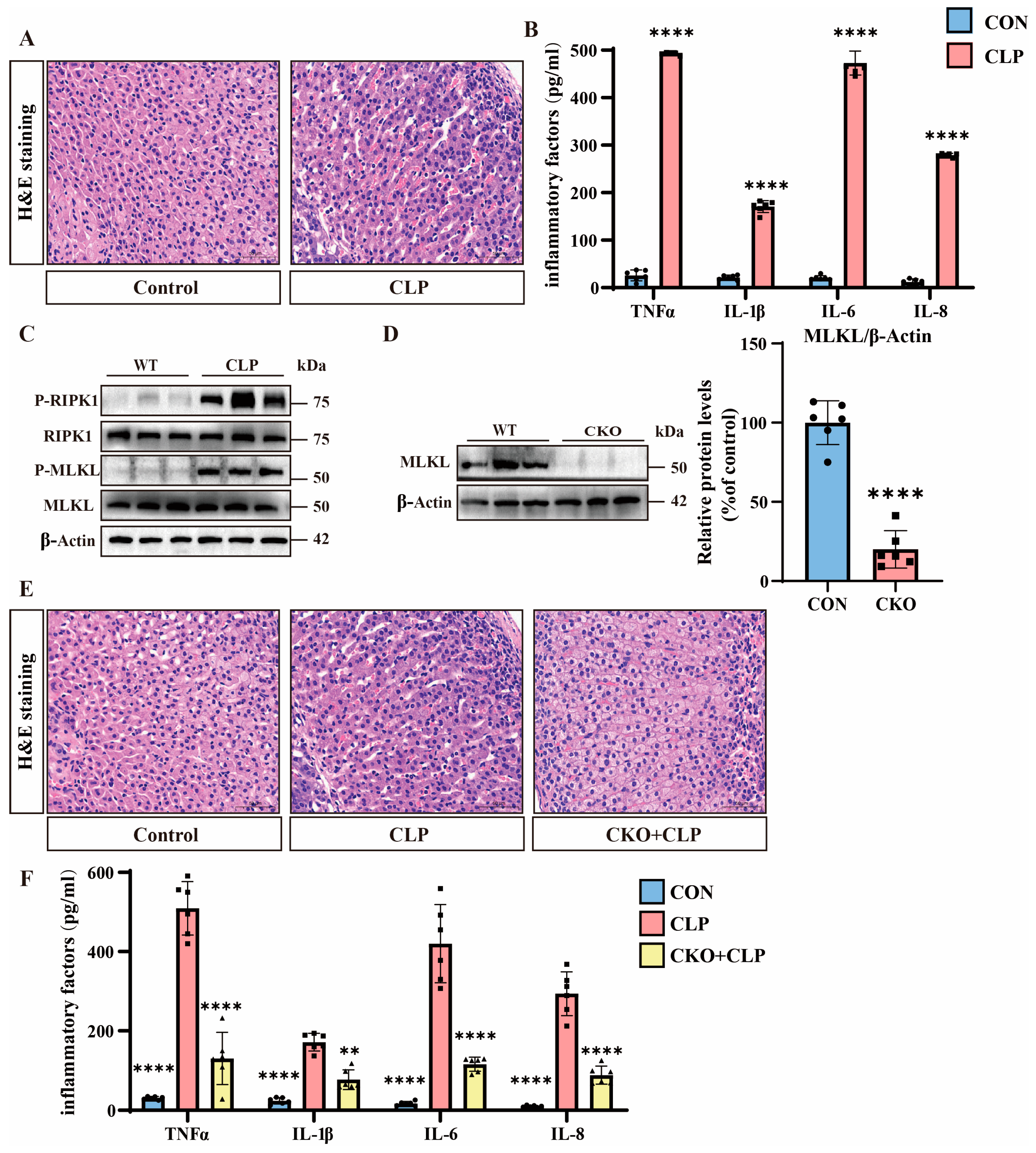
3.1. Septic Adrenal Dysfunction and Necroptosis Upregulation in Mice
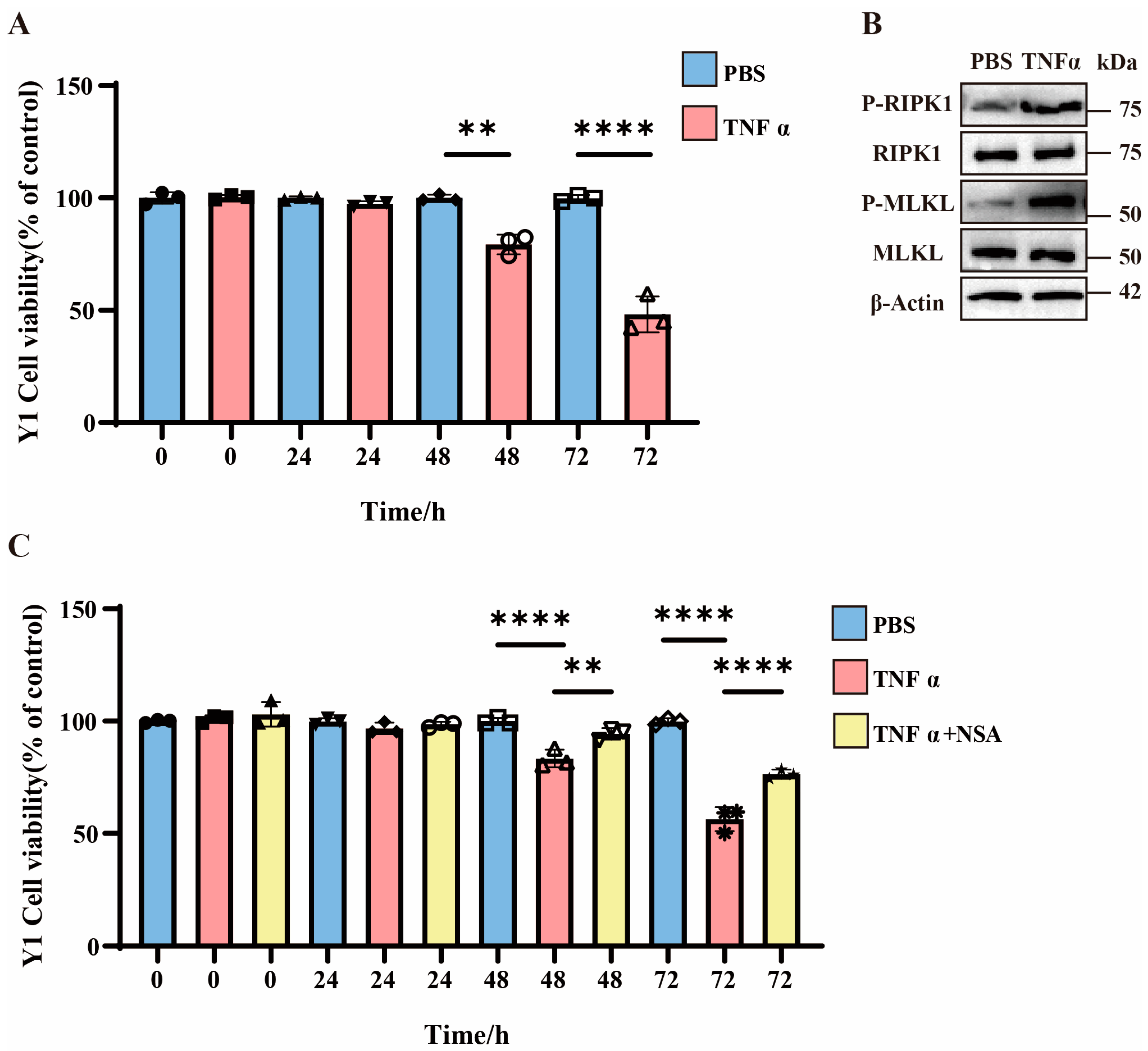
3.2. TNFα-Induced Necroptosis in Y1 Cells and Enhanced Cell Mortality
3.3. H2S Suppresses Necroptosis and Alleviates Septic Adrenal Dysfunction
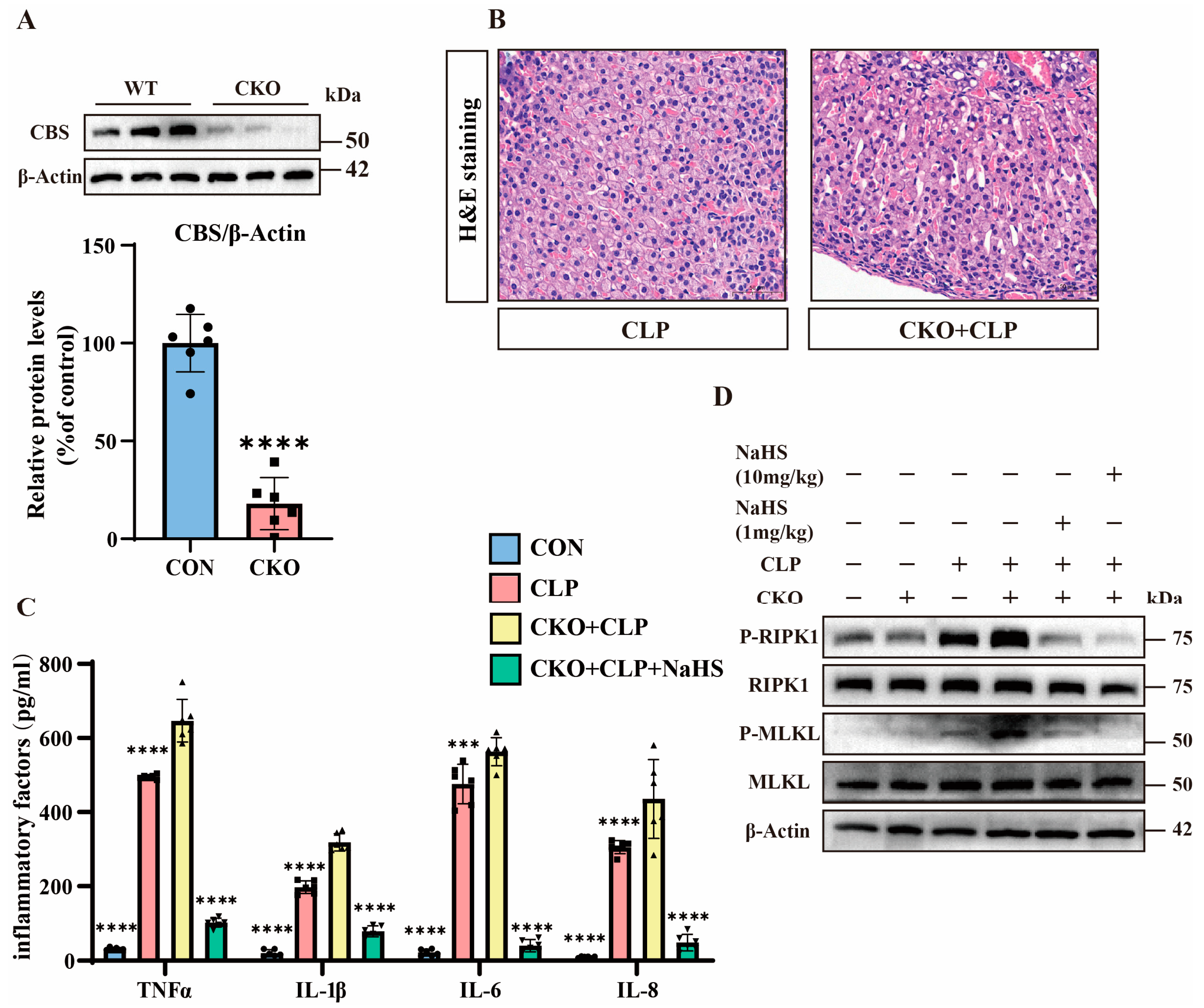
3.4. H2S Attenuates TNFα-Induced Y1 Cell Necroptosis Through CBS Activation
3.5. H2S Modulates TNFα-Mediated Inflammation and Necroptosis in Septic Mice
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- van der Poll, T.; Shankar-Hari, M.; Wiersinga, W.J. The immunology of sepsis. Immunity 2021, 54, 2450–2464. [Google Scholar] [CrossRef]
- Kyriacou, D.N. Government Regulation of Sepsis Care. JAMA 2019, 322, 250–251. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Zhang, C.; Wang, T.; Wang, J.; Fu, F. Effects of natural products on skin inflammation caused by abnormal hormones secreted by the adrenal gland. Front. Pharmacol. 2023, 14, 1156271. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Zhou, Z.; Huang, Y.; Wang, G.; Tang, Z.; Lu, J.; Wang, C.; Ni, X. Reduced hydrogen sulfide production contributes to adrenal insufficiency induced by hypoxia via modulation of NLRP3 inflammasome activation. Redox Rep. 2023, 28, 2163354. [Google Scholar] [CrossRef] [PubMed]
- Angus, D.C.; Wax, R.S. Epidemiology of sepsis: An update. Crit. Care Med. 2001, 29 (Suppl. 7), S109–S116. [Google Scholar] [CrossRef]
- Kertai, M.D.; Fontes, M.L. Predicting Adrenal Insufficiency in Severe Sepsis. Crit. Care Med. 2015, 43, 715–716. [Google Scholar] [CrossRef]
- Venkatesh, B.; Cohen, J. Pathophysiology and management of adrenal disorders in the critically ill. In Oxford Medicine Online; Oxford University Press: Oxford, UK, 2016. [Google Scholar]
- Marik, P.E. Mechanisms and clinical consequences of critical illness associated adrenal Insufficiency. Curr. Opin. Crit. Care 2007, 13, 363–369. [Google Scholar] [CrossRef]
- Wu, Y.Y.; Li, C.C.; Lin, X.; Xu, F.; Shan, S.K.; Guo, B.; Li, F.X.; Zheng, M.H.; Xu, Q.S.; Lei, L.M.; et al. Global publication trends and research trends of necroptosis application in tumor: A bibliometric analysis. Front. Pharmacol. 2023, 14, 1112484. [Google Scholar] [CrossRef]
- Garnish, S.E.; Tovey Crutchfield, E.C.; Murphy, J.M.; Hildebrand, J.M. Add necroptosis to your asthma action Plan. Immunol. Cell Biol. 2021, 99, 800–802. [Google Scholar] [CrossRef]
- Della Torre, L.; Nebbioso, A.; Stunnenberg, H.G.; Martens, J.H.A.; Carafa, V.; Altucci, L. The Role of Necroptosis: Biological Relevance and Its Involvement in Cancer. Cancers 2021, 13, 684. [Google Scholar] [CrossRef]
- Zhuang, C.; Chen, F. Small-Molecule Inhibitors of Necroptosis: Current Status and Perspectives. J. Med. Chem. 2019, 63, 1490–1510. [Google Scholar] [CrossRef] [PubMed]
- Tonnus, W.; Gembardt, F.; Latk, M.; Parmentier, S.; Hugo, C.; Bornstein, S.R.; Linkermann, A. The clinical relevance of necroinflammation—Highlighting the importance of acute kidney injury and the adrenal glands. Cell Death Differ. 2018, 26, 68–82. [Google Scholar] [CrossRef]
- Yoo, H.; Im, Y.; Ko, R.E.; Lee, J.Y.; Park, J.; Jeon, K. Association of plasma level of High-mobility group box-1 with necroptosis and sepsis Outcomes. Sci. Rep. 2021, 11, 9512. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Du, J.; Du, S.; Liu, Y.; Li, D.; Zhu, X.; Ni, X. Endogenous H(2)S resists mitochondria-mediated apoptosis in the adrenal glands via ATP5A1 S-sulfhydration in male mice. Mol. Cell Endocrinol. 2018, 474, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Pang, Q.; Huo, F.; Yin, C. Research Progress in the Field of Hydrogen Sulfide Donors in the Last Five Years. ChemBioChem 2024, 26, e202400817. [Google Scholar] [CrossRef]
- Sen, N. Functional and Molecular Insights of Hydrogen Sulfide Signaling and Protein Sulfhydration. J. Mol. Biol. 2017, 429, 543–561. [Google Scholar] [CrossRef]
- Xia, M.; Chen, L.; Muh, R.W.; Li, P.L.; Li, N. Production and Actions of Hydrogen Sulfide, a Novel Gaseous Bioactive Substance, in the Kidneys. J. Pharmacol. Exp. Ther. 2009, 329, 1056–1062. [Google Scholar] [CrossRef]
- Liu, Y.H.; Lu, M.; Xie, Z.Z.; Hua, F.; Xie, L.; Gao, J.H.; Koh, Y.H.; Bian, J.S. Hydrogen Sulfide Prevents Heart Failure Development via Inhibition of Renin Release from Mast Cells in Isoproterenol-Treated Rats. Antioxid. Redox Signal. 2013, 20, 759–769. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, C. Application and value of hydrogen sulfide modulated autophagy in sepsis. Int. Immunopharmacol. 2023, 122, 110662. [Google Scholar] [CrossRef]
- Li, H.; Wang, S.; An, S.; Gao, B.; Wu, D.; Li, Y. Hydrogen Sulfide Reduces Renal Ischemia-Reperfusion Injury by Enhancing Autophagy and Reducing Oxidative Stress. Nephrology 2024, 29, 645–654. [Google Scholar] [CrossRef]
- Wang, C.N.; Liu, Y.J.; Duan, G.L.; Zhao, W.; Li, X.H.; Zhu, X.Y.; Ni, X. CBS and CSE are critical for maintenance of mitochondrial function and glucocorticoid production in adrenal cortex. Antioxid. Redox Signal 2014, 21, 2192–2207. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhou, Y.; Yu, D.; Hu, W.; Wu, X.; Wang, J.; Huang, S.; Zhao, S.; Fan, X.; Chu, Z.; et al. The Autophagy Signaling Pathway in Necroptosis-Dependent Cerebral Ischemia/Reperfusion Injury. Neurochem. J. 2021, 15, 247–253. [Google Scholar] [CrossRef]
- Xie, Y.; Zhao, Y.; Shi, L.; Li, W.; Chen, K.; Li, M.; Chen, X.; Zhang, H.; Li, T.; Matsuzawa-Ishimoto, Y.; et al. Gut epithelial TSC1/mTOR controls RIPK3-dependent necroptosis in intestinal inflammation and Cancer. J. Clin. Investig. 2020, 130, 2111–2128. [Google Scholar] [CrossRef]
- Zhang, N.; Wang, C.; Ni, X. Construction of transgenic mice with specific Cre recombinase expression in the zona fasciculata in adrenal cortex. Acta Physiol. Sin. 2020, 72, 148–156. [Google Scholar] [CrossRef]
- Kaya-Yasar, Y.; Karaman, Y.; Bozkurt, T.E.; Onder, S.C.; Sahin-Erdemli, I. Effects of intranasal treatment with slow (GYY4137) and rapid (NaHS) donors of hydrogen sulfide in lipopolysaccharide-induced airway inflammation in Mice. Pulm. Pharmacol. Ther. 2017, 45, 170–180. [Google Scholar] [CrossRef]
- Aslami, H.; Pulskens, W.P.; Kuipers, M.T.; Bos, A.P.; van Kuilenburg, A.B.; Wanders, R.J.; Roelofsen, J.; Roelofs, J.J.; Kerindongo, R.P.; Beurskens, C.J.; et al. Hydrogen Sulfide Donor NaHS Reduces Organ Injury in a Rat Model of Pneumococcal Pneumosepsis, Associated with Improved Bio-Energetic Status. PLoS ONE 2013, 8, e63497. [Google Scholar] [CrossRef]
- Tonnus, W.; Belavgeni, A.; Beuschlein, F.; Eisenhofer, G.; Fassnacht, M.; Kroiss, M.; Krone, N.P.; Reincke, M.; Bornstein, S.R.; Linkermann, A. The role of regulated necrosis in endocrine diseases. Nat. Rev. Endocrinol. 2021, 17, 497–510. [Google Scholar] [CrossRef]
- Deng, X.X.; Li, S.S.; Sun, F.Y. Necrostatin-1 Prevents Necroptosis in Brains after Ischemic Stroke via Inhibition of RIPK1-Mediated RIPK3/MLKL Signaling. Aging Dis. 2019, 10, 807. [Google Scholar] [CrossRef]
- Wang, Z.; Feng, J.; Yu, J.; Chen, G. FKBP12 mediates necroptosis by initiating RIPK1–RIPK3–MLKL signal transduction in response to TNF receptor 1 Ligation. J. Cell Sci. 2019, 132, jcs227777. [Google Scholar] [CrossRef]
- Gu, C.; Hou, C.; Zhang, S. miR-425-5p improves inflammation and septic liver damage through negatively regulating the RIP1-mediated Necroptosis. Inflamm. Res. 2020, 69, 299–308. [Google Scholar] [CrossRef]
- Nakamura, H.; Kinjo, T.; Arakaki, W.; Miyagi, K.; Tateyama, M.; Fujita, J. Serum levels of receptor-interacting protein kinase-3 in patients with COVID-19. Crit Care 2020, 318, L215–L225. [Google Scholar] [CrossRef]
- Chisholm, L.O.; Jaeger, N.M.; Murawsky, H.E.; Harms, M.J. S100A9 interacts with a dynamic region on CD14 to activate Toll-like receptor 4. bioRxiv 2024. [Google Scholar] [CrossRef]
- Du, Y.; Zhong, Y.; Ding, R.; Wang, X.; Xia, F.; Zhang, Q.; Peng, Q. New insights of necroptosis and immune infiltration in sepsis-induced myocardial dysfunction from bioinformatics analysis through RNA-seq in mice. Front. Cell. Infect. Microbiol. 2022, 12, 1068324. [Google Scholar] [CrossRef]
- Kanczkowski, W.; Evert, K.; Stadtmüller, M.; Haberecker, M.; Laks, L.; Chen, L.-S.; Frontzek, K.; Pablik, J.; Hantel, C.; Beuschlein, F.; et al. COVID-19 targets human adrenal glands. Lancet Diabetes Endocrinol. 2022, 10, 13–16. [Google Scholar] [CrossRef]
- Chen, X.; Xu, W.; Wang, Y.; Luo, H.; Quan, S.; Zhou, J.; Yang, N.; Zhang, T.; Wu, L.; Liu, J.; et al. Hydrogen sulfide reduces kidney injury due to urinary-derived sepsis by inhibiting NF-κB expression, decreasing TNF-α levels and increasing IL-10 levels. Exp. Ther. Med. 2014, 8, 464–470. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Zhao, J.; Miao, S.; Chen, Y.; Xu, Y.; Liu, Y. Protective effect of H2S on LPS-induced AKI by promoting autophagy. Mol. Med. Rep. 2022, 25, 96. [Google Scholar] [CrossRef] [PubMed]
- Sohda, M.; Misumi, Y.; Oda, K. TNFα triggers release of extracellular vesicles containing TNFR1 and TRADD, which can modulate TNFα responses of the parental Cells. Arch. Biochem. Biophys. 2015, 587, 31–37. [Google Scholar] [CrossRef]
- Saddala, M.S.; Huang, H. Identification of novel inhibitors for TNFR1 and TNFα-TNFR1 complex using pharmacophore-based Approaches. J. Transl. Med. 2019, 17, 215. [Google Scholar] [CrossRef]
- Kuai, J.; Wooters, J.; Hall, J.P.; Rao, V.R.; Nickbarg, E.; Li, B.; Chatterjee-Kishore, M.; Qiu, Y.; Lin, L.L. NAK Is Recruited to the TNFR1 Complex in a TNFα-dependent Manner and Mediates the Production of RANTES. J. Biol. Chem. 2004, 279, 53266–53271. [Google Scholar] [CrossRef]
- Chen, S.W.; Zhu, J.; Zuo, S.; Zhang, J.L.; Chen, Z.Y.; Chen, G.W.; Wang, X.; Pan, Y.S.; Liu, Y.C.; Wang, P.Y. Protective effect of hydrogen sulfide on TNF-α and IFN-γ-induced injury of intestinal epithelial barrier function in Caco-2 Monolayers. Inflamm. Res. 2015, 64, 789–797. [Google Scholar] [CrossRef]
- Toro, G.; Szaniszlo, P.; Almenas, F.A.; Thanki, K.; Maskey, M.; Chao, C.; Hellmich, M.; Yochum, G.; Pinchuk, I.; Modis, K. The function of the cystathionine-gamma-lyase/hydrogen sulfide axis in the pathogenesis of ulcerative colitis. Gastroenterology 2024, 166, S84–S85. [Google Scholar] [CrossRef]
- Lohninger, L.; Tomasova, L.; Praschberger, M.; Hintersteininger, M.; Erker, T.; Gmeiner, B.M.; Laggner, H. Hydrogen sulphide induces HIF-1α and Nrf2 in THP-1 Macrophages. Biochimie 2015, 112, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Li, X.; Lin, Y.; Huang, Z.; Wu, W. Exogenous sodium hydrosulfide protects against high glucose-induced injury and inflammation in human umbilical vein endothelial cells by inhibiting necroptosis via the p38 MAPK signaling pathway. Mol. Med. Rep. 2020, 23, 67. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Lei, H.; Hu, X.; Dong, W. Hesperetin ameliorates DSS-induced colitis by maintaining the epithelial barrier via blocking RIPK3/MLKL necroptosis Signaling. Eur. J. Pharmacol. 2020, 873, 172992. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, K.; Huang, J.; Zhang, J.; Tian, Y.; Hu, J.; Ma, L.; Wang, C. Hydrogen Sulfide (H2S) Mitigates Sepsis-Induced Adrenal Dysfunction via Inhibition of TNFα-Mediated Necroptosis. Pathogens 2025, 14, 439. https://doi.org/10.3390/pathogens14050439
Ma K, Huang J, Zhang J, Tian Y, Hu J, Ma L, Wang C. Hydrogen Sulfide (H2S) Mitigates Sepsis-Induced Adrenal Dysfunction via Inhibition of TNFα-Mediated Necroptosis. Pathogens. 2025; 14(5):439. https://doi.org/10.3390/pathogens14050439
Chicago/Turabian StyleMa, Kai, Jingwen Huang, Jin Zhang, Yuan Tian, Jing Hu, Linhao Ma, and Changnan Wang. 2025. "Hydrogen Sulfide (H2S) Mitigates Sepsis-Induced Adrenal Dysfunction via Inhibition of TNFα-Mediated Necroptosis" Pathogens 14, no. 5: 439. https://doi.org/10.3390/pathogens14050439
APA StyleMa, K., Huang, J., Zhang, J., Tian, Y., Hu, J., Ma, L., & Wang, C. (2025). Hydrogen Sulfide (H2S) Mitigates Sepsis-Induced Adrenal Dysfunction via Inhibition of TNFα-Mediated Necroptosis. Pathogens, 14(5), 439. https://doi.org/10.3390/pathogens14050439