
Cell Death Mechanisms in Mycobacterium abscessus Infection: A Double-Edged Sword



{kind=link}
{kind=link}
Abstract
1. Introduction
2. Cell Death in the Immune Response
2.1. Apoptosis: Mechanisms, Immune Regulation, and Disease Implications
2.2. Necrosis and Necroptosis: Mechanisms, Immune Response and Pathological Implications
2.3. Autophagy: Mechanisms, Immune Functions, and Role in Cell Death
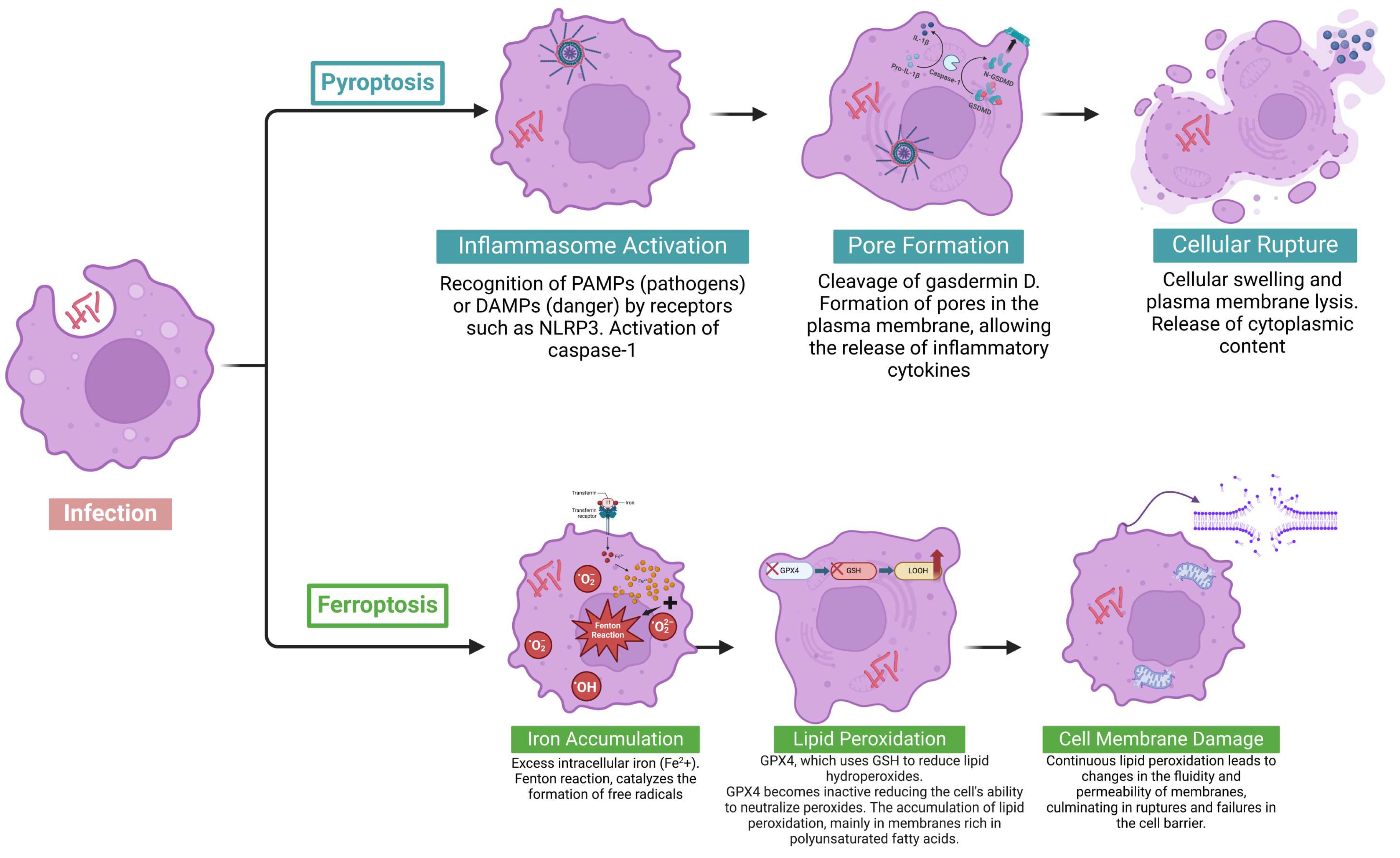
2.4. Pyroptosis: Mechanisms, Immune Functions, and Role in Cell Death
2.5. Ferroptosis: Mechanisms and Role in Cell Death
3. Cell Death in M. abscessus Infection
3.1. Modulation of Apoptotic Pathways by M. abscessus
3.2. Mechanisms of Necrosis Induced by M. abscessus
3.3. Modulation of Autophagy by M. abscessus and Its Role in Host–Pathogen Interaction
3.4. Modulation of Pyroptosis and Inflammasome Activation by M. abscessus in Infection
3.5. Modulation of Ferroptosis and Inflammasome Activation by M. abscessus in Infection
4. Conclusions
5. Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Falkinham, J.O. Environmental Sources of Nontuberculous Mycobacteria. Clin. Chest. Med. 2015, 36, 35–41. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.Y.; Chen, W.C.; Chen, Y.Y.; Su, W.J. Clinical Relevance and Diagnosis of Nontuberculous Mycobacterial Pulmonary Disease in Populations at Risk. J. Formos. Med. Assoc. 2020, 119 (Suppl. S1), S23–S31. [Google Scholar] [CrossRef] [PubMed]
- Cowman, S.; Van Ingen, J.; Griffith, D.E.; Loebinger, M.R. Non-Tuberculous Mycobacterial Pulmonary Disease. Eur. Respir. J. 2019, 54, 1900250. [Google Scholar] [CrossRef]
- Lake, M.A.; Ambrose, L.R.; Lipman, M.C.I.; Lowe, D.M. “Why Me, Why Now?” Using Clinical Immunology and Epidemiology to Explain Who Gets Nontuberculous Mycobacterial Infection. BMC Med. 2016, 14, 54. [Google Scholar] [CrossRef]
- de Mello, K.G.C.; Queiroz Mello, F.C.; Borga, L.; Rolla, V.; Duarte, R.S.; Sampaio, E.P.; Holland, S.M.; Prevots, D.R.; Dalcolmo, M.P. Clinical and Therapeutic Features of Pulmonary Nontuberculous Mycobacterial Disease, Brazil, 1993–2011. Emerg. Infect. Dis. 2013, 19, 393–399. [Google Scholar]
- Gochi, M.; Takayanagi, N.; Kanauchi, T.; Ishiguro, T.; Yanagisawa, T.; Sugita, Y. Retrospective Study of the Predictors of Mortality and Radiographic Deterioration in 782 Patients with Nodular/Bronchiectatic Mycobacterium avium Complex Lung Disease. BMJ Open 2015, 5, 8. [Google Scholar] [CrossRef]
- Qu, H.Q.; Fisher-Hoch, S.P.; McCormick, J.B. Molecular Immunity to Mycobacteria: Knowledge from the Mutation and Phenotype Spectrum Analysis of Mendelian Susceptibility to Mycobacterial Diseases. Int. J. Infect. Dis. 2011, 15, e305–e313. [Google Scholar] [CrossRef]
- Haverkamp, M.H.; van Dissel, J.T.; Holland, S.M. Human Host Genetic Factors in Nontuberculous Mycobacterial Infection: Lessons from Single Gene Disorders Affecting Innate and Adaptive Immunity and Lessons from Molecular Defects in Interferon-Gamma-Dependent Signaling. Microbes Infect. 2006, 8, 1157–1166. [Google Scholar] [CrossRef]
- Rosenzweig, S.D.; Holland, S.M. Defects in the Interferon-γ and Interleukin-12 Pathways. Immunol. Rev. 2005, 203, 38–47. [Google Scholar] [CrossRef]
- Holland, S.M. Treatment of Infections in the Patient with Mendelian Susceptibility to Mycobacterial Infection. Microbes Infect. 2000, 2, 1579–1590. [Google Scholar] [CrossRef]
- Guide, S.V.; Holland, S.M. Host Susceptibility Factors in Mycobacterial Infection. Genetics and Body Morphotype. Infect. Dis. Clin. N. Am. 2002, 16, 163–186. [Google Scholar] [CrossRef] [PubMed]
- Furuya, E.Y.; Paez, A.; Srinivasan, A.; Cooksey, R.; Augenbraun, M.; Baron, M.; Brudney, K.; Della-Latta, P.; Estivariz, C.; Fischer, S.; et al. Outbreak of Mycobacterium abscessus Wound Infections among “Lipotourists” from the United States Who Underwent Abdominoplasty in the Dominican Republic. Clin. Infect. Dis. 2008, 46, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Eustace, K.; Jolliffe, V.; Sahota, A.; Gholam, K. Cutaneous Mycobacterium abscessus Infection Following Hair Transplant. Clin. Exp. Dermatol. 2016, 41, 768–770. [Google Scholar] [CrossRef] [PubMed]
- Tuan, H.T.; Ngoc, N.A.; Ai, L.D.; Van Luat, N. Complicated Surgical Site Infection with Mycobacterium abscessus After Liposuction and Affections of Corticosteroids in the Treatment Regimen: Three Cases Report and a Systematic Review. Aesthet. Plast. Surg. 2024, 48, 1365–1385. [Google Scholar] [CrossRef]
- Ou, Y.; Liu, D.; Feng, J.; Xu, X.; Lin, T.; Zhang, Y.; Luo, L.; Wu, M.; Cui, Y. Subcutaneous Infection Caused by Mycobacterium abscessus Following Botulinum Toxin Injections: A Case Report and Literature Review. J. Cosmet. Dermatol. 2024, 23, 1527–1532. [Google Scholar] [CrossRef]
- Pace, V.; Antinolfi, P.; Borroni, E.; Cirillo, D.M.; Cenci, E.; Piersimoni, C.; Cardaccia, A.; Nofri, M.; Papalini, C.; Petruccelli, R.; et al. Treating Primary Arthroprosthesis Infection Caused by Mycobacterium abscessus Subsp. abscessus. Case Rep. Infect. Dis. 2019, 2019, 5892913. [Google Scholar] [CrossRef]
- Appelgren, P.; Farnebo, F.; Dotevall, L.; Studahl, M.; Jönsson, B.; Petrini, B. Late-Onset Posttraumatic Skin and Soft-Tissue Infections Caused by Rapid-Growing Mycobacteria in Tsunami Survivors. Clin. Infect. Dis. 2008, 47, e11–e16. [Google Scholar] [CrossRef]
- Dahl, V.N.; Mølhave, M.; Fløe, A.; van Ingen, J.; Schön, T.; Lillebaek, T.; Andersen, A.B.; Wejse, C. Global Trends of Pulmonary Infections with Nontuberculous Mycobacteria: A Systematic Review. Int. J. Infect. Dis. 2022, 125, 120–131. [Google Scholar] [CrossRef]
- Prieto, M.D.; Alam, M.E.; Franciosi, A.N.; Quon, B.S. Global Burden of Nontuberculous Mycobacteria in the Cystic Fibrosis Population: A Systematic Review and Meta-Analysis. ERJ Open Res. 2023, 9, 00336–02022. [Google Scholar] [CrossRef]
- Moore, M.; Frerichs, J.B. An Unusual Acid-Fast Infection of the Knee with Subcutaneous, Abscess-like Lesions of the Gluteal Region; Report of a Case with a Study of the Organism, Mycobacterium abscessus, n. Sp. J. Investig. Dermatol. 1953, 20, 133–169. [Google Scholar] [CrossRef]
- Bryant, J.M.; Grogono, D.M.; Greaves, D.; Foweraker, J.; Roddick, I.; Inns, T.; Reacher, M.; Haworth, C.S.; Curran, M.D.; Harris, S.R.; et al. Whole-Genome Sequencing to Identify Transmission of Mycobacterium abscessus between Patients with Cystic Fibrosis: A Retrospective Cohort Study. Lancet 2013, 381, 1551–1560. [Google Scholar] [CrossRef] [PubMed]
- Tortoli, E.; Kohl, T.A.; Brown-Elliott, B.A.; Trovato, A.; Leão, S.C.; Garcia, M.J.; Vasireddy, S.; Turenne, C.Y.; Griffith, D.E.; Philley, J.V.; et al. Emended Description of Mycobacterium abscessus, Mycobacterium abscessus subsp. abscessus and Mycobacterium abscessus subsp. bolletii and Designation of Mycobacterium abscessus subsp. massiliense Comb. Nov. Int. J. Syst. Evol. Microbiol. 2016, 66, 4471–4479. [Google Scholar] [CrossRef] [PubMed]
- Leao, S.C.; Tortoli, E.; Paul Euzé, J.; Garcia, M.J. Proposal That Mycobacterium massiliense and Mycobacterium bolletii Be United and Reclassified as Mycobacterium abscessus subsp. bolletii Comb. Nov., Designation of Mycobacterium abscessus subsp. abscessus Subsp. Nov. and Emended Description of Mycobacterium abscessus. Int. J. Syst. Evol. Microbiol. 2011, 61, 2311–2313. [Google Scholar] [CrossRef]
- Jones, R.S.; Shier, K.L.; Master, R.N.; Bao, J.R.; Clark, R.B. Current Significance of the Mycobacterium chelonae-abscessus Group. Diagn. Microbiol. Infect. Dis. 2019, 94, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Simmon, K.E.; Brown-Elliott, B.A.; Ridge, P.G.; Durtschi, J.D.; Mann, L.B.; Slechta, E.S.; Steigerwalt, A.G.; Moser, B.D.; Whitney, A.M.; Brown, J.M.; et al. Mycobacterium chelonae-abscessus Complex Associated with Sinopulmonary Disease, Northeastern USA. Emerg. Infect. Dis. 2011, 17, 1692–1700. [Google Scholar] [CrossRef]
- Abdelaal, H.F.M.; Chan, E.D.; Young, L.; Baldwin, S.L.; Coler, R.N. Mycobacterium abscessus: It’s Complex. Microorganisms 2022, 10, 1454. [Google Scholar] [CrossRef]
- Clary, G.; Sasindran, S.J.; Nesbitt, N.; Mason, L.; Cole, S.; Azad, A.; McCoy, K.; Schlesinger, L.S.; Hall-Stoodley, L. Mycobacterium abscessus Smooth and Rough Morphotypes Form Antimicrobial-Tolerant Biofilm Phenotypes but Are Killed by Acetic Acid. Antimicrob. Agents Chemother. 2018, 62, 3. [Google Scholar] [CrossRef]
- Bernut, A.; Herrmann, J.-L.; Kissa, K.; Dubremetz, J.-F.; Gaillard, J.-L.; Lutfalla, G.; Kremer, L. Mycobacterium abscessus Cording Prevents Phagocytosis and Promotes Abscess Formation. Proc. Natl. Acad. Sci. USA 2014, 111, E943–E952. [Google Scholar] [CrossRef]
- Helguera-Repetto, A.C.; Chacon-Salinas, R.; Cerna-Cortes, J.F.; Rivera-Gutierrez, S.; Ortiz-Navarrete, V.; Estrada-Garcia, I.; Gonzalez-Y-Merchand, J.A. Differential Macrophage Response to Slow- and Fast-Growing Pathogenic Mycobacteria. BioMed Res. Int. 2014, 2014, 916521. [Google Scholar] [CrossRef]
- Newton, K.; Strasser, A.; Kayagaki, N.; Dixit, V.M. Cell Death. Cell 2024, 187, 235–256. [Google Scholar] [CrossRef]
- Bertheloot, D.; Latz, E.; Franklin, B.S. Necroptosis, Pyroptosis and Apoptosis: An Intricate Game of Cell Death. Cell. Mol. Immunol. 2021, 18, 1106–1121. [Google Scholar] [CrossRef] [PubMed]
- Wanford, J.J.; Hachani, A.; Odendall, C. Reprogramming of Cell Death Pathways by Bacterial Effectors as a Widespread Virulence Strategy. Infect. Immun. 2022, 90, 5. [Google Scholar] [CrossRef] [PubMed]
- Place, D.E.; Lee, S.; Kanneganti, T.-D. PANoptosis in Microbial Infection. Curr. Opin. Microbiol. 2021, 59, 42–49. [Google Scholar] [CrossRef]
- Bittencourt, T.L.; da Silva Prata, R.B.; de Andrade Silva, B.J.; de Mattos Barbosa, M.G.; Dalcolmo, M.P.; Pinheiro, R.O. Autophagy as a Target for Drug Development of Skin Infection Caused by Mycobacteria. Front. Immunol. 2021, 12, 1785. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- D’Arcy, M.S. Cell Death: A Review of the Major Forms of Apoptosis, Necrosis and Autophagy. Cell. Biol. Int. 2019, 43, 582–592. [Google Scholar] [CrossRef]
- Igney, F.H.; Krammer, P.H. Death and Anti-Death: Tumour Resistance to Apoptosis. Nat. Rev. Cancer 2002, 2, 277–288. [Google Scholar] [CrossRef]
- Yuan, J.; Ofengeim, D. A Guide to Cell Death Pathways. Nat. Rev. Mol. Cell. Biol. 2024, 25, 379–395. [Google Scholar] [CrossRef]
- Green, D.R. The Mitochondrial Pathway of Apoptosis Part II: The BCL-2 Protein Family. Cold Spring Harb. Perspect. Biol. 2022, 14, a041046. [Google Scholar] [CrossRef]
- Lakhani, S.A.; Masud, A.; Kuida, K.; Porter, G.A.; Booth, C.J.; Mehal, W.Z.; Inayat, I.; Flavell, R.A. Caspases 3 and 7: Key Mediators of Mitochondrial Events of Apoptosis. Science (1979) 2006, 311, 847–851. [Google Scholar] [CrossRef]
- Glover, H.L.; Schreiner, A.; Dewson, G.; Tait, S.W.G. Mitochondria and Cell Death. Nat. Cell. Biol. 2024, 26, 1434–1446. [Google Scholar] [CrossRef] [PubMed]
- Rudd-Schmidt, J.A.; Trapani, J.A.; Voskoboinik, I. Distinguishing Perforin-Mediated Lysis and Granzyme-Dependent Apoptosis. Methods Enzymol. 2019, 629, 291–306. [Google Scholar] [PubMed]
- Kiselevsky, D.B. Granzymes and Mitochondria. Biochemistry 2020, 85, 131–139. [Google Scholar] [CrossRef] [PubMed]
- Martinvalet, D.; Zhu, P.; Lieberman, J. Granzyme A Induces Caspase-Independent Mitochondrial Damage, a Required First Step for Apoptosis. Immunity 2005, 22, 355–370. [Google Scholar] [CrossRef]
- Ben-Abdallah, M.; Sturny-Leclère, A.; Avé, P.; Louise, A.; Moyrand, F.; Weih, F.; Janbon, G.; Mémet, S. Fungal-Induced Cell Cycle Impairment, Chromosome Instability and Apoptosis via Differential Activation of NF-ΚB. PLoS. Pathog. 2012, 8, e1002555. [Google Scholar] [CrossRef]
- Jie, W.; Rui-Fen, Z.; Zhong-Xiang, H.; Yan, W.; Wei-Na, L.; Yong-Ping, M.; Jing, S.; Jing-Yi, C.; Wan-Hong, L.; Xiao-Hua, H.; et al. Inhibition of Cell Proliferation by Tas of Foamy Viruses through Cell Cycle Arrest or Apoptosis Underlines the Different Mechanisms of Virus-Host Interactions. Virulence 2022, 13, 342–354. [Google Scholar] [CrossRef]
- Zhou, X.; Jiang, W.; Liu, Z.; Liu, S.; Liang, X. Virus Infection and Death Receptor-Mediated Apoptosis. Viruses 2017, 9, 316. [Google Scholar] [CrossRef]
- Selvaraj, C.; Vierra, M.; Dinesh, D.C.; Abhirami, R.; Singh, S.K. Structural Insights of Macromolecules Involved in Bacteria-Induced Apoptosis in the Pathogenesis of Human Diseases. Adv. Protein Chem. Struct. Biol. 2021, 126, 1–38. [Google Scholar] [CrossRef]
- Krammer, P.H.; Behrmann, I.; Daniel, P.; Dhein, J.; Debatin, K.-M. Regulation of Apoptosis in the Immune System. Curr. Opin. Immunol. 1994, 6, 279–289. [Google Scholar] [CrossRef]
- Ji, W.; Xin, Y.; Zhang, L.; Liu, X. ALG2 Influences T Cell Apoptosis by Regulating FASLG Intracellular Transportation. Biochem. J. 2020, 477, 3105–3121. [Google Scholar] [CrossRef]
- Pol, J.G.; Caudana, P.; Paillet, J.; Piaggio, E.; Kroemer, G. Effects of Interleukin-2 in Immunostimulation and Immunosuppression. J. Exp. Med. 2020, 217, e20191247. [Google Scholar] [CrossRef] [PubMed]
- Katzman, S.D.; Hoyer, K.K.; Dooms, H.; Gratz, I.K.; Rosenblum, M.D.; Paw, J.S.; Isakson, S.H.; Abbas, A.K. Opposing Functions of IL-2 and IL-7 in the Regulation of Immune Responses. Cytokine 2011, 56, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Morana, O.; Wood, W.; Gregory, C.D. The Apoptosis Paradox in Cancer. Int. J. Mol. Sci. 2022, 23, 1328. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; He, H.; Wang, K.; Shi, X.; Wang, Y.; Su, Y.; Wang, Y.; Li, D.; Liu, W.; Zhang, Y.; et al. Granzyme A from Cytotoxic Lymphocytes Cleaves GSDMB to Trigger Pyroptosis in Target Cells. Science (1979) 2020, 368, 6494. [Google Scholar] [CrossRef]
- Bellone, M. Apoptosis, Cross-Presentation, and the Fate of the Antigen Specific Immune Response. Apoptosis 2000, 5, 307–314. [Google Scholar] [CrossRef]
- Van Zanten, J.; Hospers, G.A.P.; Harmsen, M.C.; The, T.H.; Mulder, N.H.; De Leij, L.F.M.H. Dendritic Cells Present an Intracellular Viral Antigen Derived from Apoptotic Cells and Induce a T-Cell Response. Scand. J. Immunol. 2002, 56, 254–259. [Google Scholar] [CrossRef]
- Mahoney, J.A.; Rosen, A. Apoptosis and Autoimmunity. Curr. Opin. Immunol. 2005, 17, 583–588. [Google Scholar] [CrossRef]
- Mountz, J.D.; Wu, J.; Cheng, J.; Zhou, T. Autoimmune Disease. a Problem of Defective Apoptosis. Arthritis Rheum. 1994, 37, 1415–1420. [Google Scholar] [CrossRef]
- Ravirajan, C.T.; Pittoni, V.; Isenberg, D.A. Apoptosis in Human Autoimmune Diseases. Int. Rev. Immunol. 1999, 18, 563–589. [Google Scholar] [CrossRef]
- Wong, R.S. Apoptosis in Cancer: From Pathogenesis to Treatment. J. Exp. Clin. Cancer Res. 2011, 30, 87. [Google Scholar] [CrossRef]
- Conrad, M.; Angeli, J.P.F.; Vandenabeele, P.; Stockwell, B.R. Regulated Necrosis: Disease Relevance and Therapeutic Opportunities. Nat. Rev. Drug Discov. 2016, 15, 348–366. [Google Scholar] [CrossRef] [PubMed]
- Festjens, N.; Vanden Berghe, T.; Vandenabeele, P. Necrosis, a Well-Orchestrated Form of Cell Demise: Signalling Cascades, Important Mediators and Concomitant Immune Response. Biochim. Biophys. Acta. (BBA)-Bioenerg. 2006, 1757, 1371–1387. [Google Scholar] [CrossRef] [PubMed]
- Linkermann, A.; Green, D.R. Necroptosis. N. Engl. J. Med. 2014, 370, 455–465. [Google Scholar] [CrossRef]
- Berghe, T.V.; Linkermann, A.; Jouan-Lanhouet, S.; Walczak, H.; Vandenabeele, P. Regulated Necrosis: The Expanding Network of Non-Apoptotic Cell Death Pathways. Nat. Rev. Mol. Cell Biol. 2014, 15, 135–147. [Google Scholar] [CrossRef]
- Tonnus, W.; Meyer, C.; Paliege, A.; Belavgeni, A.; von Mässenhausen, A.; Bornstein, S.R.; Hugo, C.; Becker, J.U.; Linkermann, A. The Pathological Features of Regulated Necrosis. J. Pathol. 2019, 247, 697–707. [Google Scholar] [CrossRef]
- Pasparakis, M.; Vandenabeele, P. Necroptosis and Its Role in Inflammation. Nature 2015, 517, 311–320. [Google Scholar] [CrossRef]
- Grootjans, S.; Vanden Berghe, T.; Vandenabeele, P. Initiation and Execution Mechanisms of Necroptosis: An Overview. Cell Death Differ. 2017, 24, 1184–1195. [Google Scholar] [CrossRef]
- Vakkila, J.; Lotze, M.T. Inflammation and Necrosis Promote Tumour Growth. Nat. Rev. Immunol. 2004, 4, 641–648. [Google Scholar] [CrossRef]
- Mocarski, E.S. Programmed Necrosis in Host Defense. Curr Trop Microbiol Immunol. 2023, 442, 1–40. [Google Scholar] [CrossRef]
- Murao, A.; Aziz, M.; Wang, H.; Brenner, M.; Wang, P. Release Mechanisms of Major DAMPs. Apoptosis 2021, 26, 152–162. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Chan, F.K.-M.; Kroemer, G. Necroptosis: Mechanisms and Relevance to Disease. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 103–130. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and Molecular Mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Yang, L.; Zhang, B.; Lv, X.; Gong, F.; Yang, W. Hydrogen Inhalation Enhances Autophagy via the AMPK/MTOR Pathway, Thereby Attenuating Doxorubicin-Induced Cardiac Injury. Int. Immunopharmacol. 2023, 119, 110071. [Google Scholar] [CrossRef] [PubMed]
- Khalil, M.I.; Ali, M.M.; Holail, J.; Houssein, M. Growth or Death? Control of Cell Destiny by MTOR and Autophagy Pathways. Prog. Biophys. Mol. Biol. 2023, 185, 39–55. [Google Scholar] [CrossRef]
- Alers, S.; Löffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-MTOR-Ulk1/2 in the Regulation of Autophagy: Cross Talk, Shortcuts, and Feedbacks. Mol. Cell Biol. 2012, 32, 2–11. [Google Scholar] [CrossRef]
- Hamaoui, D.; Subtil, A. ATG16L1 Functions in Cell Homeostasis beyond Autophagy. FEBS J. 2022, 289, 1779–1800. [Google Scholar] [CrossRef]
- Changotra, H.; Kaur, S.; Yadav, S.S.; Gupta, G.L.; Parkash, J.; Duseja, A. ATG5: A Central Autophagy Regulator Implicated in Various Human Diseases. Cell Biochem. Funct. 2022, 40, 650–667. [Google Scholar] [CrossRef]
- Metlagel, Z.; Otomo, C.; Ohashi, K.; Takaesu, G.; Otomo, T. Structural Insights into E2–E3 Interaction for LC3 Lipidation. Autophagy 2014, 10, 522–523. [Google Scholar] [CrossRef]
- Bortoluci, K.R.; Medzhitov, R. Control of Infection by Pyroptosis and Autophagy: Role of TLR and NLR. Cell. Mol. Life Sci. 2010, 67, 1643–1651. [Google Scholar] [CrossRef]
- Huyghe, J.; Priem, D.; Bertrand, M.J.M. Cell Death Checkpoints in the TNF Pathway. Trends Immunol. 2023, 44, 628–643. [Google Scholar] [CrossRef]
- Wu, W.; Wang, X.; Sun, Y.; Berleth, N.; Deitersen, J.; Schlütermann, D.; Stuhldreier, F.; Wallot-Hieke, N.; José Mendiburo, M.; Cox, J.; et al. TNF-Induced Necroptosis Initiates Early Autophagy Events via RIPK3-Dependent AMPK Activation, but Inhibits Late Autophagy. Autophagy 2021, 17, 3992–4009. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.-M.; Jeon, B.-Y.; Lee, H.-M.; Jin, H.S.; Yuk, J.-M.; Song, C.-H.; Lee, S.-H.; Lee, Z.-W.; Cho, S.-N.; Kim, J.-M.; et al. Mycobacterium tuberculosis Eis Regulates Autophagy, Inflammation, and Cell Death through Redox-Dependent Signaling. PLoS Pathog. 2010, 6, e1001230. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yao, S.; Yang, H.; Liu, S.; Wang, Y. Autophagy: Regulator of Cell Death. Cell Death Dis. 2023, 14, 648. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, M.; Hirata, N.; Tanaka, T.; Suizu, F.; Nakajima, H.; Chiorini, J.A. Autophagy as a Modulator of Cell Death Machinery. Cell Death Dis. 2020, 11, 517. [Google Scholar] [CrossRef]
- Mahapatra, K.K.; Mishra, S.R.; Behera, B.P.; Patil, S.; Gewirtz, D.A.; Bhutia, S.K. The Lysosome as an Imperative Regulator of Autophagy and Cell Death. Cell. Mol. Life Sci. 2021, 78, 7435–7449. [Google Scholar] [CrossRef]
- Arumugam, P.; Shankaran, D.; Bothra, A.; Gandotra, S.; Rao, V. The MmpS6-MmpL6 Operon Is an Oxidative Stress Response System Providing Selective Advantage to Mycobacterium tuberculosis in Stress. J. Infect. Dis. 2019, 219, 459–469. [Google Scholar] [CrossRef]
- Shankaran, D.; Arumugam, P.; Vasanthakumar, R.P.; Singh, A.; Bothra, A.; Gandotra, S.; Rao, V. Modern Clinical Mycobacterium tuberculosis Strains Leverage Type I IFN Pathway for a Proinflammatory Response in the Host. J. Immunol. 2022, 209, 1736–1745. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of Assembly, Regulation and Signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef]
- Li, L.; Dickinson, M.S.; Coers, J.; Miao, E.A. Pyroptosis in Defense against Intracellular Bacteria. Semin. Immunol. 2023, 69, 101805. [Google Scholar] [CrossRef]
- Xiao, C.; Cao, S.; Li, Y.; Luo, Y.; Liu, J.; Chen, Y.; Bai, Q.; Chen, L. Pyroptosis in Microbial Infectious Diseases. Mol. Biol. Rep. 2024, 51, 42. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, S.B.; Miao, E.A. Gasdermins: Effectors of Pyroptosis. Trends Cell Biol. 2017, 27, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, S.O.; Behl, B.; Rathinam, V.A. Pyroptosis-Induced Inflammation and Tissue Damage. Semin. Immunol. 2023, 69, 101781. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; Fulda, S.; Gascón, S.; Hatzios, S.K.; Kagan, V.E.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Ursini, F.; Maiorino, M. Lipid Peroxidation and Ferroptosis: The Role of GSH and GPx4. Free Radic. Biol. Med. 2020, 152, 175–185. [Google Scholar] [CrossRef]
- Henning, Y.; Blind, U.S.; Larafa, S.; Matschke, J.; Fandrey, J. Hypoxia Aggravates Ferroptosis in RPE Cells by Promoting the Fenton Reaction. Cell Death Dis. 2022, 13, 662. [Google Scholar] [CrossRef]
- Feng, H.; Schorpp, K.; Jin, J.; Yozwiak, C.E.; Hoffstrom, B.G.; Decker, A.M.; Rajbhandari, P.; Stokes, M.E.; Bender, H.G.; Csuka, J.M.; et al. Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep. 2020, 30, 3411–3423.e7. [Google Scholar] [CrossRef]
- Park, E.; Chung, S.W. ROS-Mediated Autophagy Increases Intracellular Iron Levels and Ferroptosis by Ferritin and Transferrin Receptor Regulation. Cell Death Dis. 2019, 10, 822. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, M.; Bi, R.; Su, Y.; Quan, F.; Lin, Y.; Yue, C.; Cui, X.; Zhao, Q.; Liu, S.; et al. ACSL4 Deficiency Confers Protection against Ferroptosis-Mediated Acute Kidney Injury. Redox Biol. 2022, 51, 102262. [Google Scholar] [CrossRef]
- Cui, J.; Wang, Y.; Tian, X.; Miao, Y.; Ma, L.; Zhang, C.; Xu, X.; Wang, J.; Fang, W.; Zhang, X. LPCAT3 Is Transcriptionally Regulated by YAP/ZEB/EP300 and Collaborates with ACSL4 and YAP to Determine Ferroptosis Sensitivity. Antioxid. Redox Signal. 2023, 39, 491–511. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Kang, R.; Tang, D. Signaling Pathways and Defense Mechanisms of Ferroptosis. FEBS J. 2022, 289, 7038–7050. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Kim, K.J.; Gaschler, M.M.; Patel, M.; Shchepinov, M.S.; Stockwell, B.R. Peroxidation of Polyunsaturated Fatty Acids by Lipoxygenases Drives Ferroptosis. Proc. Natl. Acad. Sci. USA 2016, 113, E4966–E4975. [Google Scholar] [CrossRef]
- Whang, J.; Back, Y.W.; Lee, K.-I.; Fujiwara, N.; Paik, S.; Choi, C.H.; Park, J.-K.; Kim, H.-J. Mycobacterium abscessus Glycopeptidolipids Inhibit Macrophage Apoptosis and Bacterial Spreading by Targeting Mitochondrial Cyclophilin D. Cell Death Dis. 2017, 8, e3012. [Google Scholar] [CrossRef]
- Shin, D.M.; Yang, C.S.; Yuk, J.M.; Lee, J.Y.; Kim, K.H.; Shin, S.J.; Takahara, K.; Lee, S.J.; Jo, E.K. Mycobacterium abscessus Activates the Macrophage Innate Immune Response via a Physical and Functional Interaction between TLR2 and Dectin-1. Cell. Microbiol. 2008, 10, 1608–1621. [Google Scholar] [CrossRef]
- Blanco-Conde, S.; González-Cortés, C.; López-Medrano, R.; Carazo-Fernández, L.; Diez-Tascón, C.; Marcos-Benavides, M.F.; Rivero-Lezcano, O.M. Mycobacterium abscessus Infected Neutrophils as an In Vitro Model for Bronchiectasis. Neutrophils Prevent Mycobacterial Aggregation. Arch. Bronconeumol. 2022, 58, 578–581. [Google Scholar] [CrossRef]
- Ahn, J.-H.; Jung, D.-H.; Kim, D.-Y.; Lee, T.-S.; Kim, Y.-J.; Lee, Y.-J.; Seo, I.-S.; Kim, W.-G.; Cho, Y.J.; Shin, S.J.; et al. Impact of IL-1β on Lung Pathology Caused by Mycobacterium abscessus Infection and Its Association with IL-17 Production. Microbes Infect. 2024, 26, 105351. [Google Scholar] [CrossRef]
- Sheng, S.; Xin, L.; Yam, J.K.H.; Salido, M.M.; Khong, N.Z.J.; Liu, Q.; Chea, R.A.; Li, H.Y.; Yang, L.; Liang, Z.-X.; et al. The MapZ-Mediated Methylation of Chemoreceptors Contributes to Pathogenicity of Pseudomonas aeruginosa. Front. Microbiol. 2019, 10, 67. [Google Scholar] [CrossRef]
- Bates, N.A.; Rodriguez, R.; Drwich, R.; Ray, A.; Stanley, S.A.; Penn, B.H. Reactive Oxygen Detoxification Contributes to Mycobacterium abscessus Antibiotic Survival. bioRxiv 2024, preprint. [Google Scholar] [CrossRef]
- Kim, B.R.; Kim, B.J.; Kook, Y.H.; Kim, B.J. Phagosome Escape of Rough Mycobacterium abscessus Strains in Murine Macrophage via Phagosomal Rupture Can Lead to Type I Interferon Production and Their Cell-to-Cell Spread. Front. Immunol. 2019, 10, 125. [Google Scholar] [CrossRef]
- Bonay, M.; Roux, A.-L.; Floquet, J.; Retory, Y.; Herrmann, J.-L.; Lofaso, F.; Deramaudt, T. Caspase-Independent Apoptosis in Infected Macrophages Triggered by Sulforaphane via Nrf2/P38 Signaling Pathways. Cell Death Discov. 2015, 1, 15022. [Google Scholar] [CrossRef] [PubMed]
- Parmar, S.; Tocheva, E.I. The Cell Envelope of Mycobacterium abscessus and Its Role in Pathogenesis. PLoS Pathog. 2023, 19, e1011318. [Google Scholar] [CrossRef] [PubMed]
- Roux, A.L.; Viljoen, A.; Bah, A.; Simeone, R.; Bernut, A.; Laencina, L.; Deramaudt, T.; Rottman, M.; Gaillard, J.L.; Majlessi, L.; et al. The Distinct Fate of Smooth and Rough Mycobacterium abscessus Variants inside Macrophages. Open Biol. 2016, 6, 160185. [Google Scholar] [CrossRef] [PubMed]
- Catherinot, E.; Clarissou, J.; Etienne, G.; Ripoll, F.; Emile, J.-F.; Daffé, M.; Perronne, C.; Soudais, C.; Gaillard, J.-L.; Rottman, M. Hypervirulence of a Rough Variant of the Mycobacterium abscessus Type Strain. Infect. Immun. 2007, 75, 1055–1058. [Google Scholar] [CrossRef]
- PETRINI, B. Mycobacterium abscessus: An Emerging Rapid-growing Potential Pathogen. Apmis 2006, 114, 319–328. [Google Scholar] [CrossRef]
- Leestemaker-Palmer, A.L.; Bermudez, L.E. Mycobacterium abscessus Infection Results in Decrease of Oxidative Metabolism of Lung Airways Cells and Relaxation of the Epithelial Mucosal Tight Junctions. Tuberculosis 2023, 138, 102303. [Google Scholar] [CrossRef]
- Feng, Z.; Bai, X.; Wang, T.; Garcia, C.; Bai, A.; Li, L.; Honda, J.R.; Nie, X.; Chan, E.D. Differential Responses by Human Macrophages to Infection With Mycobacterium tuberculosis and Non-Tuberculous Mycobacteria. Front. Microbiol. 2020, 11, 116. [Google Scholar] [CrossRef]
- Quan, H.; Chung, H.; Je, S.; Hong, J.J.; Kim, B.-J.; Na, Y.R.; Seok, S.H. Pyruvate Dehydrogenase Kinase Inhibitor Dichloroacetate Augments Autophagy Mediated Constraining the Replication of Mycobacteroides massiliense in Macrophages. Microbes Infect. 2023, 25, 105139. [Google Scholar] [CrossRef]
- Kim, S.W.; Subhadra, B.; Whang, J.; Back, Y.W.; Bae, H.S.; Kim, H.J.; Choi, C.H. Clinical Mycobacterium abscessus Strain Inhibits Autophagy Flux and Promotes Its Growth in Murine Macrophages. Pathog. Dis. 2017, 75, 8. [Google Scholar] [CrossRef]
- Viljoen, A.; Herrmann, J.L.; Onajole, O.K.; Stec, J.; Kozikowski, A.P.; Kremer, L. Controlling Extra- and Intramacrophagic Mycobacterium abscessus by Targeting Mycolic Acid Transport. Front. Cell. Infect. Microbiol. 2017, 7, 388. [Google Scholar] [CrossRef]
- Shamaei, M.; Mirsaeidi, M. Nontuberculous Mycobacteria, Macrophages, and Host Innate Immune Response. Infect. Immun. 2021, 89, e0081220. [Google Scholar] [CrossRef] [PubMed]
- Silwal, P.; Kim, I.S.; Jo, E.-K. Autophagy and Host Defense in Nontuberculous Mycobacterial Infection. Front. Immunol. 2021, 12, 728742. [Google Scholar] [CrossRef]
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed Cell Death as a Defence against Infection. Nat. Rev. Immunol. 2017, 17, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Ratnatunga, C.N.; Tungatt, K.; Proietti, C.; Halstrom, S.; Holt, M.R.; Lutzky, V.P.; Price, P.; Doolan, D.L.; Bell, S.C.; Field, M.A.; et al. Characterizing and Correcting Immune Dysfunction in Non-Tuberculous Mycobacterial Disease. Front. Immunol. 2022, 13, 1047781. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Koch, B.E.V.; Lamers, G.E.M.; Forn-Cuní, G.; Spaink, H.P. Specificity of the Innate Immune Responses to Different Classes of Non-Tuberculous Mycobacteria. Front. Immunol. 2022, 13, 1075473. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Z.; Ruan, J.; Pan, Y.; Magupalli, V.G.; Wu, H.; Lieberman, J. Inflammasome-Activated Gasdermin D Causes Pyroptosis by Forming Membrane Pores. Nature 2016, 535, 153–158. [Google Scholar] [CrossRef]
- Coll, R.C.; Robertson, A.A.B.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A Small-Molecule Inhibitor of the NLRP3 Inflammasome for the Treatment of Inflammatory Diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef]
- Bagayoko, S.; Meunier, E. Emerging Roles of Ferroptosis in Infectious Diseases. FEBS J. 2022, 289, 7869–7890. [Google Scholar] [CrossRef]
- Sun, S.; Shen, J.; Jiang, J.; Wang, F.; Min, J. Targeting Ferroptosis Opens New Avenues for the Development of Novel Therapeutics. Signal Transduct. Target. Ther. 2023, 8, 372. [Google Scholar] [CrossRef]
- Bar-Oz, M.; Meir, M.; Barkan, D. Virulence-Associated Secretion in Mycobacterium abscessus. Front. Immunol. 2022, 13, 938895. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in Major Human Diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef] [PubMed]
- Nisa, A.; Kipper, F.C.; Panigrahy, D.; Tiwari, S.; Kupz, A.; Subbian, S. Different Modalities of Host Cell Death and Their Impact on Mycobacterium tuberculosis Infection. Am. J. Physiol. Cell Physiol. 2022, 323, C1444–C1474. [Google Scholar] [CrossRef] [PubMed]
- Awuh, J.A.; Flo, T.H. Molecular Basis of Mycobacterial Survival in Macrophages. Cell. Mol. Life Sci. 2017, 74, 1625–1648. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, A.E.; Congel, J.H.; Corley, J.M.; Janssen, W.J.; Nick, J.A.; Malcolm, K.C.; Hisert, K.B. Dectin-1-Independent Macrophage Phagocytosis of Mycobacterium abscessus. Int. J. Mol. Sci. 2023, 24, 11062. [Google Scholar] [CrossRef]
- Behar, S.M.; Martin, C.J.; Booty, M.G.; Nishimura, T.; Zhao, X.; Gan, H.-X.; Divangahi, M.; Remold, H.G. Apoptosis Is an Innate Defense Function of Macrophages against Mycobacterium tuberculosis. Mucosal. Immunol. 2011, 4, 279–287. [Google Scholar] [CrossRef]
- Kale, J.; Osterlund, E.J.; Andrews, D.W. BCL-2 Family Proteins: Changing Partners in the Dance towards Death. Cell Death Differ. 2018, 25, 65–80. [Google Scholar] [CrossRef]
- Gutierrez, M.G.; Master, S.S.; Singh, S.B.; Taylor, G.A.; Colombo, M.I.; Deretic, V. Autophagy Is a Defense Mechanism Inhibiting BCG and Mycobacterium tuberculosis Survival in Infected Macrophages. Cell 2004, 119, 753–766. [Google Scholar] [CrossRef]
- Xiao, Z.; Kong, B.; Fang, J.; Qin, T.; Dai, C.; Shuai, W.; Huang, H. Ferrostatin-1 Alleviates Lipopolysaccharide-Induced Cardiac Dysfunction. Bioengineered 2021, 12, 9367–9376. [Google Scholar] [CrossRef]
- Tong, J.; Lan, X.; Zhang, Z.; Liu, Y.; Sun, D.; Wang, X.; Ou-Yang, S.; Zhuang, C.; Shen, F.; Wang, P.; et al. Ferroptosis Inhibitor Liproxstatin-1 Alleviates Metabolic Dysfunction-Associated Fatty Liver Disease in Mice: Potential Involvement of PANoptosis. Acta Pharmacol. Sin. 2023, 44, 1014–1028. [Google Scholar] [CrossRef]
- Fan, B.-Y.; Pang, Y.-L.; Li, W.-X.; Zhao, C.-X.; Zhang, Y.; Wang, X.; Ning, G.-Z.; Kong, X.-H.; Liu, C.; Yao, X.; et al. Liproxstatin-1 Is an Effective Inhibitor of Oligodendrocyte Ferroptosis Induced by Inhibition of Glutathione Peroxidase 4. Neural Regen. Res. 2021, 16, 561. [Google Scholar] [CrossRef]
- Tanouchi, Y.; Lee, A.J.; Meredith, H.; You, L. Programmed Cell Death in Bacteria and Implications for Antibiotic Therapy. Trends Microbiol. 2013, 21, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, Y.; Zhang, J.; Hu, C.; Jiang, J.; Li, Y.; Peng, Z. ROS-Induced Lipid Peroxidation Modulates Cell Death Outcome: Mechanisms behind Apoptosis, Autophagy, and Ferroptosis. Arch. Toxicol. 2023, 97, 1439–1451. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Liu, J.; Kang, R.; Klionsky, D.J.; Kroemer, G.; Tang, D. Ferroptosis Is a Type of Autophagy-Dependent Cell Death. Semin. Cancer Biol. 2020, 66, 89–100. [Google Scholar] [CrossRef] [PubMed]
- Pang, Q.; Wang, P.; Pan, Y.; Dong, X.; Zhou, T.; Song, X.; Zhang, A. Irisin Protects against Vascular Calcification by Activating Autophagy and Inhibiting NLRP3-Mediated Vascular Smooth Muscle Cell Pyroptosis in Chronic Kidney Disease. Cell Death Dis. 2022, 13, 283. [Google Scholar] [CrossRef]
- Tang, Q.; Liu, W.; Yang, X.; Tian, Y.; Chen, J.; Hu, Y.; Fu, N. ATG5-Mediated Autophagy May Inhibit Pyroptosis to Ameliorate Oleic Acid-Induced Hepatocyte Steatosis. DNA Cell. Biol. 2022, 41, 1038–1052. [Google Scholar] [CrossRef]
- Deng, Z.; Wang, Y.; Liu, J.; Zhang, H.; Zhou, L.; Zhao, H.; Han, Y.; Yan, S.; Dong, Z.; Wang, Y.; et al. WBP2 Restrains the Lysosomal Degradation of GPX4 to Inhibit Ferroptosis in Cisplatin-Induced Acute Kidney Injury. Redox Biol. 2023, 65, 102826. [Google Scholar] [CrossRef]
- Cai, X.; Hua, S.; Deng, J.; Du, Z.; Zhang, D.; Liu, Z.; Khan, N.U.; Zhou, M.; Chen, Z. Astaxanthin Activated the Nrf2/HO-1 Pathway to Enhance Autophagy and Inhibit Ferroptosis, Ameliorating Acetaminophen-Induced Liver Injury. ACS Appl. Mater. Interfaces J. 2022, 14, 42887–42903. [Google Scholar] [CrossRef]
- Bhagwat, A.C.; Patil, A.M.; Saroj, S.D. CRISPR/Cas 9-Based Editing in the Production of Bioactive Molecules. Mol. Biotechnol. 2022, 64, 245–251. [Google Scholar] [CrossRef]
- de Maat, V.; Stege, P.B.; Dedden, M.; Hamer, M.; van Pijkeren, J.-P.; Willems, R.J.L.; van Schaik, W. CRISPR-Cas9-mediated genome editing in vancomycin-resistant Enterococcus faecium. FEMS Microbiol. Lett. 2019, 366, fnz256. [Google Scholar] [CrossRef]
- Bhattacharjee, G.; Gohil, N.; Khambhati, K.; Mani, I.; Maurya, R.; Karapurkar, J.K.; Gohil, J.; Chu, D.-T.; Vu-Thi, H.; Alzahrani, K.J.; et al. Current Approaches in CRISPR-Cas9 Mediated Gene Editing for Biomedical and Therapeutic Applications. J. Control. Release 2022, 343, 703–723. [Google Scholar] [CrossRef]
- Yang, X. Applications of CRISPR-Cas9 Mediated Genome Engineering. Mil. Med. Res. 2015, 2, 11. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prata, R.B.d.S.; Pinheiro, R.O. Cell Death Mechanisms in Mycobacterium abscessus Infection: A Double-Edged Sword. Pathogens 2025, 14, 391. https://doi.org/10.3390/pathogens14040391
Prata RBdS, Pinheiro RO. Cell Death Mechanisms in Mycobacterium abscessus Infection: A Double-Edged Sword. Pathogens. 2025; 14(4):391. https://doi.org/10.3390/pathogens14040391
Chicago/Turabian StylePrata, Rhana Berto da Silva, and Roberta Olmo Pinheiro. 2025. "Cell Death Mechanisms in Mycobacterium abscessus Infection: A Double-Edged Sword" Pathogens 14, no. 4: 391. https://doi.org/10.3390/pathogens14040391
APA StylePrata, R. B. d. S., & Pinheiro, R. O. (2025). Cell Death Mechanisms in Mycobacterium abscessus Infection: A Double-Edged Sword. Pathogens, 14(4), 391. https://doi.org/10.3390/pathogens14040391