Abstract
We hypothesize that a unified mitochondrial perspective on aging, HIV, and long COVID reveals shared pathogenic mechanisms and specific therapeutic vulnerabilities that are overlooked when these conditions are treated independently. Mitochondrial dysfunction is increasingly recognized as a common factor driving aging, HIV, and long COVID. Shared mechanisms—including oxidative stress, impaired mitophagy and dynamics, mtDNA damage, and metabolic reprogramming—contribute to ongoing energy failure and chronic inflammation. Recent advancements highlight new therapeutic strategies such as mitochondrial transfer, transplantation, and genome-level correction of mtDNA variants, with early preclinical and clinical studies providing proof-of-concept. This review summarizes current evidence on mitochondrial changes across aging and post-viral syndromes, examines emerging organelle-based therapies, and discusses key challenges related to safety, durability, and translation.
1. Introduction
Mitochondria are central hubs for cellular metabolism, redox signaling, and innate immunity. Their dysfunction thus impacts more than just energy production, affecting tissue health and overall systemic well-being [1,2]. Viral infections often disrupt mitochondrial stability, either directly through interactions between viral proteins and organelles or indirectly via ongoing inflammation and oxidative stress. This damage can persist well after the virus is cleared, especially when mitochondrial DNA (mtDNA) is harmed, resulting in lasting bioenergetic problems and immune system imbalance [3,4].
We propose that a unified mitochondrial perspective across aging, HIV, and long COVID will reveal shared pathogenic mechanisms and specific therapeutic vulnerabilities that are amenable to organelle-targeted interventions.
This phenomenon is well illustrated in HIV and, more recently, in long COVID. Both conditions are associated with structural and functional mitochondrial alterations that resemble features of accelerated aging [5,6]. These include impaired respiratory chain activity, increased reactive oxygen species (ROS), altered metabolic programming, and defective quality-control mechanisms such as mitophagy [7,8]. Such processes contribute not only to chronic inflammation but also to the development of comorbidities typically associated with advanced age [9].
Although aging, HIV, and long COVID have each been separately linked to mitochondrial dysfunction; to our knowledge, a systematic comparison emphasizing how shared mitochondrial alterations inform organelle-based therapies is lacking. In this review we (1) compare mitochondrial changes across aging, HIV, and long COVID to identify convergent mechanisms; (2) dissect how these shared pathways contribute to bioenergetic failure and chronic inflammation; (3) evaluate emerging organelle-based therapeutic strategies; and (4) discuss key translational challenges—safety, durability, and feasibility—and propose research priorities to advance clinical translation.
By outlining this roadmap, we guide the reader through a cohesive argument that links mitochondrial pathogenesis to specific therapeutic solutions, highlighting vulnerabilities that are overlooked when these conditions are considered in isolation.
2. Mitochondrial Dynamics in Energy Production and Cellular Regulation
Mitochondria are multifunctional organelles whose roles extend well beyond ATP synthesis and whose dysfunction underlies a wide spectrum of human disease. In the matrix, tricarboxylic acid (TCA) enzymes produce reducing equivalents (NADH, FADH2) that deliver electrons to the inner-membrane electron transport chain (ETC, complexes I–IV), driving proton translocation and establishing the electrochemical gradient that powers ATP synthase (complex V) and supports protein import and organelle quality control [10,11,12,13,14,15].
Dysfunction of ETC components—particularly complexes I and III—raises electron leak and ROS production. Low, regulated ROS levels are signaling mediators that influence differentiation, immune responses, and adaptive stress resistance (mitohormesis). In contrast, excessive ROS causes oxidative damage, inflammasome activation, and pro-inflammatory signaling implicated in aging, HIV, and severe/long COVID [8,16,17,18,19]. Thus, the balance between bioenergetic competence and redox homeostasis is a central mechanistic node linking metabolic, infectious, and degenerative pathologies.
Beyond energy conversion, mitochondria supply precursors and cofactors for biosynthetic processes (pyrimidine and lipid synthesis), conduct fatty-acid β-oxidation, and assemble heme and iron–sulfur (Fe-S) clusters essential for respiration and DNA repair [20,21]. They also act as dynamic Ca2+ buffers that shape cytosolic and endoplasmic reticulum signaling, thereby influencing secretion, contractility, cell fate decisions, and programmed cell death [22]. Mitochondria determine cellular capacity to respond to stress, infection, and metabolic demand by integrating these biochemical and signaling roles.
Importantly for infectious disease, mitochondria are central platforms for innate immunity: mitochondrial antiviral signaling (MAVS), regulated fission–fusion dynamics, and mitophagy coordinate type I interferon responses, inflammasome activation, and cell survival programs. Viruses commonly target these interfaces to subvert host defenses—modulating fusion/fission, impairing mitophagy, degrading MAVS, or altering mitochondrial metabolism—to promote replication and persistence. Given this pathogen–host interplay, focused discussion of viral strategies that remodel mitochondrial form and function is essential (Figure 1).
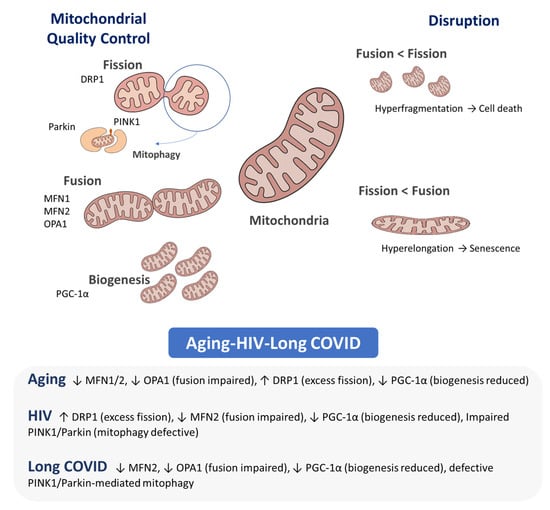
Figure 1.
Mitochondrial dynamics. Mitochondria cycle through biogenesis, fusion, fission and mitophagy; balance among these processes preserves mitochondrial integrity and function. Key mediators—Fusion: MFN1/2 (outer-membrane fusion, network integrity), OPA1 (inner-membrane fusion, cristae maintenance); Fission: DRP1 (scission); Mitophagy: PINK1 (damage sensor), Parkin (E3 ligase); Biogenesis: PGC-1α (master coactivator of mitochondrial biogenesis). Alterations: Aging: ↓ MFN1/2, ↓ OPA1, ↑ DRP1, ↓ PGC-1α. HIV: ↑ DRP1, ↓ MFN2, ↓ PGC-1α, impaired PINK1/Parkin. Long COVID: ↓ MFN2, ↓ OPA1, ↓ PGC-1α, defective PINK1/Parkin.
Similarly, the therapeutic potential of mitochondrial transfer—through isolated mitochondria, organelle-bearing extracellular vesicles, or cell-mediated delivery—deserves recognition. Restoring organelle function provides a translational pathway to address bioenergetic failure in damaged tissues, but clinical application faces specific hurdles: ensuring long-lasting engraftment and functional recovery, preventing immune activation and harmful mtDNA heteroplasmy, standardizing isolation, storage, and potency testing, optimizing delivery methods (such as local injection, systemic mito-EVs, or cell carriers), and establishing meaningful endpoints and regulatory procedures.
3. Mitochondrial Aging
Aging has long been regarded as a degenerative process driven by the accumulation of damage at the cellular level, ultimately leading to tissue dysfunction and organismal decline. Among the numerous theories proposed to explain the mechanisms underlying aging, the mitochondrial free radical theory of aging (MFRTA) has dominated the field for several decades [23]. This theory posits that ROS, as byproducts of aerobic metabolism, are highly reactive molecules that induce oxidative damage to cellular macromolecules, including lipids, proteins, and DNA.
For years, the MFRTA has suggested that ROS-induced damage is the main cause of aging. However, recent studies have offered new insights into mitochondrial function in aging and challenged this long-held theory. It is clear that mitochondria have developed mechanisms to control physiological levels of oxidative stress, and that ROS alone cannot fully explain the aging process. For example, genetic mouse models have shown that somatic mtDNA mutations can cause progeroid phenotypes without a corresponding increase in oxidative stress [24,25,26]. This highlights the importance of mtDNA integrity in aging. Although the overall levels of mtDNA mutations in aging tissues stay relatively low, the clonal expansion of these mutations creates a mosaic pattern of ETC dysfunction, which contributes to functional decline in aging cells and tissues [27,28,29,30,31].
The connection between mitochondrial dysfunction, signaling pathways, and lifespan regulation has become clearer. Mitochondria are not just passive energy producers; they actively influence cellular signaling and metabolic processes that affect aging. For instance, caloric restriction (CR), a well-known way to extend lifespan, has been shown to improve mitochondrial function, indicating that better mitochondrial health may delay the onset of age-related diseases [32,33,34,35].
Despite these advances, many questions remain unanswered, including how mtDNA mutations accumulate during aging and the extent to which reducing these mutations can improve health and longevity. These challenges underscore the need for therapeutic strategies aimed at enhancing mitochondrial function as a means to combat age-related degeneration [36].
While the MFRTA has largely been refuted, the role of mitochondria in aging is undeniable [37]. Rather than viewing ROS as purely detrimental, it is crucial to recognize that these molecules also play signaling roles essential for adaptive stress responses and cellular homeostasis [38]. Understanding how mitochondrial dysfunction integrates with other hallmarks of aging will be key to developing interventions that promote healthy aging and potentially extend human lifespan.
4. Comparative Analysis of Mitochondrial Dysfunction in Aging, HIV, and COVID-19
Mitochondria play a key role as metabolic hubs, influencing not only the aging process but also the development of chronic viral diseases such as COVID-19 and HIV [7,39]. At first glance, aging and chronic viral infections may seem unrelated; however, they share important underlying mechanisms, including mitochondrial dysfunction, oxidative stress, metabolic disturbances, and immune system activation [40,41,42]. Importantly, these shared mechanisms also encompass impaired mitochondrial quality control (mitophagy) and disruptions of mitochondrial dynamics (fusion and fission), which together determine organelle integrity and cellular resilience.
These parallels highlight the intimate link between mitochondrial health, disease, and aging, while emphasizing the distinct ways in which these processes evolve. Nevertheless, these associations remain controversial in parts of the literature: aging, HIV and long COVID are biologically and temporally distinct conditions, and the causal link between viral infection and sustained mitochondrial dysfunction is not uniformly established across studies.
With age, mitochondrial function commonly declines because of accumulating mtDNA mutations, impaired activity of the ETC, and a diminished capacity for mitochondrial biogenesis. Together, these alterations lead to excessive production of ROS, which amplify oxidative damage to lipids, proteins, and nucleic acids, ultimately driving cellular dysfunction and tissue degeneration [43,44]. Chronic viral infections, including HIV and SARS-CoV-2, place considerable stress on mitochondria. These viruses hijack host mitochondrial functions to support their replication and to dampen antiviral immune responses. In doing so, they often generate oxidative stress and inflammation—processes that closely resemble the biological hallmarks of aging [45,46,47]. Viral perturbations commonly affect pathways that regulate mitophagy and mitochondrial dynamics, shifting the balance of fusion and fission and altering post-translational modification and turnover of the fusion/fission machinery (e.g., phosphorylation, ubiquitination, SUMOylation). These effects can impair PINK1–Parkin–dependent mitophagy and other quality-control pathways [48]. Specific viral factors have been implicated: SARS-CoV-2 ORF9b interacts with TOM70 and interferes with MAVS/TOM70-dependent antiviral signaling, modifying mitochondrial protein import and innate immune signaling [49].
HIV proteins such as Tat, Nef and Vrp induce mitochondrial membrane depolarization, disrupt respiration, and trigger mitochondrial damage or mitophagy in infected and bystander cells [50,51,52,53,54,55].
Both viruses can also suppress mitochondrial biogenesis by perturbing PGC-1α/NRF1/TFAM signaling, thereby limiting the generation of new mitochondria and impairing organelle turnover [8,56,57,58].
Collectively, these disruptions promote accumulation of dysfunctional mitochondria with mtDNA and respiratory-chain damage, excessive ROS production, bioenergetic collapse and amplified inflammatory signaling that are linked to the pathophysiology of HIV disease and the chronic sequelae observed in long COVID.
Besides the acute phase of SARS-CoV-2 infection, long COVID—a condition marked by ongoing symptoms months after the initial infection—has been linked to mitochondrial dysfunction [59,60,61,62,63]. New evidence indicates that aging mitochondria might play a key role in developing long COVID, especially in people with existing metabolic disorders or older age [64]. During long COVID, mitochondrial functions are disrupted, with less mitochondrial biogenesis, reduced respiratory chain activity, and increased ROS production [65]. These changes are similar to those seen in normal aging, where mitochondrial dysfunction is a sign of cell aging and inflammation. Constant mitochondrial stress in long COVID can increase systemic inflammation, resembling the “inflammaging” pattern observed with aging, leading to a cycle of immune activation and tissue damage [66,67].
A central shared hallmark of aging and chronic viral diseases is oxidative stress. In aging, ROS produced by the mitochondrial electron transport chain—especially at complexes I and III—oxidize cellular macromolecules, impair function, and trigger inflammation [68]. Similarly, in COVID-19, excessive ROS production has been implicated in the cytokine storm observed in severe cases [69,70,71,72,73]. Excessive ROS generation has likewise been implicated in the cytokine storm of severe COVID-19 [69,70,71,72,73] and in long COVID, persistent mitochondrial dysfunction prolongs this injury, contributing to fatigue, myalgia, and cognitive deficits that resemble mitochondrial disease phenotypes [6,65,74,75,76,77,78,79,80,81]. In HIV, chronic oxidative stress drives systemic inflammation and immunosenescence, further reinforcing parallels with aging [82,83,84,85,86].
Clinically, people living with HIV (PLWH)—even those on long-term suppressive antiretroviral therapy (ART)—show a higher prevalence and earlier onset of age-related comorbidities such as cardiovascular disease, metabolic syndrome, bone problems, and neurocognitive disorders compared to matched uninfected populations [87,88,89,90]. This faster accumulation of multiple conditions reflects chronic immune activation, residual viral reservoirs, lifestyle factors, and cumulative ART exposure. Mechanistically, mitochondrial disturbances in PLWH result from several interacting sources: direct effects of HIV proteins on mitochondrial dynamics and function, inflammation-driven oxidative stress, and drug-related mitochondrial toxicity [50,83,91,92,93,94]. Importantly, drug-induced mitochondrial toxicity—particularly from older nucleoside reverse transcriptase inhibitors (NRTIs)—is among the best-established mechanisms linking HIV treatment to mitochondrial dysfunction. Mechanistically, many NRTIs inhibit mitochondrial DNA polymerase γ (POLG), causing mtDNA depletion, impaired ETC assembly and activity, and consequent bioenergetic failure; classic examples include zidovudine (AZT) and stavudine (d4T), which were associated with pronounced mitochondrial side effects [95,96,97,98,99,100].
Although older nucleoside reverse transcriptase inhibitors (NRTIs) were strongly associated with mtDNA depletion, newer ART regimens—including INSTI-based combinations and updated NRTI backbones—have lessened visible mitochondrial toxicity; however, subtle mitochondrial impairments and metabolic side effects still appear in some treated individuals and likely add to long-term comorbidity risk [93,94]. Consequently, in PLWH it is critical to disentangle mitochondrial alterations driven by the virus itself from those that are treatment-related—and to consider that in many cohorts the cumulative exposure to older, more toxic ART compounds may confound comparisons with aging or with untreated viral infection.
Therefore, “aging with HIV” reflects the interaction of infection-driven mitochondrial stress with treatment- and host-related factors that speed up age-related diseases [82,83,84,85,86,93].
Metabolic disruption is another key intersection. Aged cells often shift from oxidative phosphorylation (OXPHOS) to glycolysis—a Warburg-like reprogramming that reduces energetic efficiency and promotes metabolic exhaustion [101]. Both HIV and SARS-CoV-2 similarly rewire host metabolism toward glycolysis, with consequences for immune exhaustion and impaired tissue repair [8,102,103,104,105,106,107]. In SARS-CoV-2, this metabolic shift supports rapid viral replication and impairs the immune response, while in HIV, the reprogramming of immune cell metabolism contributes to their functional exhaustion [108,109,110,111,112]. SARS-CoV-2 alters host folate and one-carbon metabolism post-transcriptionally, enabling de novo purine synthesis despite shutting down host translation. Infected cells show glucose and folate depletion, and viral replication is highly sensitive to folate metabolism inhibitors like methotrexate [108]. High-content screens in human airway organoids identify Hypoxia-Inducible Factor (HIF)-1α–glycolysis and fatty-acid biosynthesis as actionable vulnerabilities for blocking SARS-CoV-2 [108], and HIF-1α–driven metabolic reprogramming in monocytes/macrophages enhances glycolysis while impairing T cell responses and epithelial survival, positioning HIF-1α as a therapeutic target for COVID-19 [110]. Long COVID exacerbates these disruptions, since aged or dysfunctional mitochondria cannot meet recovery energy demands, worsening fatigue and repair deficits.
In HIV, CD8+ T cells show elevated oxygen consumption, reduced glycolytic capacity, and dysregulated mTOR signaling; impaired glycolysis contributes to T-cell exhaustion, whereas early infection or viral controllers display less metabolic derangement—suggesting metabolic interventions might restore CD8+ function [111,113,114,115]. Conversely, increased OXPHOS and glycolysis in CD4+ T cells heighten HIV-1 susceptibility, and partial glycolysis inhibition reduces infection, viability, and viral amplification. It reveals metabolic vulnerabilities that could be exploited to target HIV reservoirs mtDNA mutations provide an additional overlap [112]. Somatic mtDNA mutations accumulate with age and, through clonal expansion, generate mosaic ETC dysfunction across tissues, contributing to decline [116,117]. Chronic viral infections (HIV, SARS-CoV-2) further drive mtDNA damage via oxidative stress and inflammation [7,118], a burden that may be compounded by ART-related mitochondrial toxicity [93]. Although mtDNA mutations are relatively low in many aging tissues, their amplification in chronic viral contexts highlights convergent aging–viral mechanisms.
Inflammation unites these processes. Aging is marked by “inflammaging,” a persistent low-grade inflammatory state partly driven by mitochondrial dysfunction [119]; chronic inflammation is likewise central to COVID-19 and HIV. Hyperinflammatory acute SARS-CoV-2 responses can persist as long COVID, and chronic immune activation in HIV accelerates aging and promotes age-associated comorbidities [120,121,122].
A mechanistic synthesis links HIV-associated mitochondrial dysfunction to immunosenescence: impaired mitochondria increase ROS and release mitochondrial DAMPs (e.g., oxidized mtDNA) that activate innate sensors (cGAS–STING, inflammasomes), sustaining type I interferon and proinflammatory signaling [123,124,125]. This milieu expands senescent and exhausted T cell populations with diminished proliferative and effector capacity; senescent cells secrete a pro-inflammatory SASP that impairs tissue homeostasis and repair. Concurrent metabolic rewiring (impaired OXPHOS, altered NAD+/NADH balance, dysregulated mitophagy) further undermines immune resilience, linking mitochondrial stress to systemic comorbidity risk in PLWH [82,83,84,85,86,119]. Taken together, the evidence supports some common mechanistic themes (oxidative stress, metabolic reprogramming, chronic inflammation) but also underscores that the relative contribution of direct viral effects, host age-related decline, and drug toxicity varies markedly between conditions and cohorts.
In conclusion, although aging, HIV and COVID-19 converge on ROS overproduction, they differ in predominant etiology and affected pathways. In aging, ROS generation is mainly associated with dysfunction of the ETC (complexes I and III) due to accumulated mtDNA mutations and progressive declines in biogenesis and mitophagy. In HIV, mitochondrial dysfunction arises from a combination of direct viral protein effects, chronic inflammation and, in some cases, antiretroviral-associated toxicity; this introduces membrane depolarization–mediated damage in addition to oxidative stress. In SARS-CoV-2 and long COVID, beyond ETC damage, viral proteins (such as ORF9b) interfere with mitochondrial import and signaling (TOM70/MAVS), and defective mitochondrial quality control (mitophagy) promotes accumulation of dysfunctional organelles that continuously produce ROS. These etiological differences have clinical and therapeutic implications.
Mitochondria are a nexus connecting aging and chronic/post-infectious syndromes such as HIV and long COVID. These conditions share oxidative stress, metabolic reprogramming, and chronic inflammation, yet differ in timing, progression, and underlying triggers. Dissecting both common and distinct pathways offers routes to therapies that preserve mitochondrial health across this spectrum of disorders.
5. Mitochondrial Dynamics in Aging and Viral Infection
Mitochondrial shape and function are constantly remodeled through cycles of fission, fusion, selective autophagy (mitophagy), and intracellular transport. Together, these processes define the morphology, number, positioning, and overall quality of the organelles within the cell. Since mitochondria carry their own genome, they also rely on continuous repair, biogenesis, and protein turnover to maintain proper function [126].
Fission allows the separation of damaged mitochondrial regions, making it easier to eliminate dysfunctional fragments, while fusion enables the exchange of matrix and membrane components between organelles, helping to dilute damage and sustain respiratory efficiency. However, under intense stress, excessive fission can trigger apoptotic pathways. Mitophagy acts as a quality-control mechanism by selectively removing mitochondria that are beyond repair, thereby limiting the release of pro-apoptotic factors and reducing oxidative stress. In addition, transport along microtubules ensures that mitochondria are delivered to regions of the cell with high energy or Ca2+ demands, securing local ATP availability and maintaining homeostasis. The fine-tuned regulation of these mechanisms is therefore crucial not only for mitochondrial health but also for broader cell-fate decisions [127,128,129] (Figure 1).
Aging, chronic HIV infection and long COVID all converge on impaired mitochondrial quality control—mitophagy, biogenesis and mitochondrial dynamics—leading to accumulation of dysfunctional mitochondria, ROS overproduction and chronic inflammation [37,47,65,130,131,132,133]. In aging, ROS generation is largely attributable to progressive ETC dysfunction (notably complexes I and III) driven by accumulated mtDNA mutations and reduced proteostasis, changes that promote chronic oxidative damage and “inflammaging” [43,68,116,117]. Age-related declines in mitophagy reflect reduced expression and activity of PINK1 and Parkin, favoring retention of damaged organelles and increased ROS and inflammation [134,135,136,137,138,139,140,141], while diminished biogenesis via downregulation of PGC-1α/NRF1/TFAM limits replacement of dysfunctional mitochondria [57,142,143].
In chronic HIV, mitochondrial injury results from a combination of direct viral protein effects that perturb membrane potential and autophagic flux, persistent immune activation that amplifies oxidative stress, and, historically, antiretroviral-associated mitochondrial toxicity; these factors disrupt mitophagy, alter fusion–fission dynamics through post-translational modification of MFN/OPA1/DRP1, and blunt biogenesis, promoting bioenergetic collapse and accelerated comorbidity [46,50,51,52,53,83,91,92,93,94,144,145,146].
In long COVID, beyond acute ETC injury, viral interference with mitochondrial import/signaling (for example, ORF9b–TOM70/MAVS) and sustained inflammatory/metabolic reprogramming impair mitophagy (PINK1–Parkin and other routes) and dysregulate fusion–fission balance (reduced effective fusion; inadequate segregation/fission), leading to persistence of ROS-producing mitochondria and prolonged symptoms such as fatigue and exertional intolerance [8,49,53,59,60,61,62,63,64,65,144,145].
These distinctions—mtDNA accumulation and declining turnover in aging, viral-protein plus inflammation and drug effects in HIV, and defective import/quality-control with persistent metabolic stress in long COVID—have therapeutic implications (e.g., prioritizing mitophagy and biogenesis enhancement in aging and long COVID, while minimizing ART mitochondrial toxicity and targeting inflammation/mitoprotection in HIV).
Therefore, aging, HIV and long COVID each rewire mitochondrial metabolism (reduced pyruvate dehydrogenase (PDH)/pyruvate oxidation, greater reliance on glycolysis, NAD+/NADH shifts) but differ in proximate drivers (intrinsic aging-related mtDNA accumulation vs. viral proteins + ART vs. post-infectious persistent signaling). Integrating pyruvate-centric assays into comparative studies will clarify which interventions (PDH-centric, mitophagy/biogenesis, anti-inflammatory metabolic modulators) are most likely to restore mitochondrial health in each setting [147].
Overall, the convergence of impaired mitophagy, mitochondrial biogenesis, and dynamics in aging, HIV, and long COVID underscores their role as critical drivers of cellular dysfunction, energy deficits, and chronic inflammation (Figure 1 and Figure 2, Table 1).
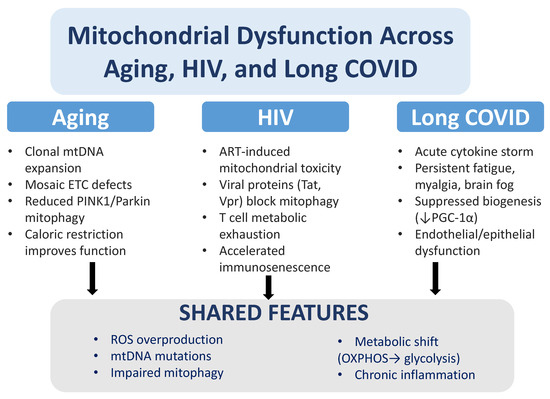
Figure 2.
Mitochondrial dysfunction in aging, HIV, and long COVID. While aging, HIV infection, and long COVID affect mitochondria through distinct mechanisms, they converge on similar pathological outcomes. In aging, mitochondrial dysfunction is linked to clonal expansion of mtDNA mutations, mosaic defects in the electron transport chain (ETC), reduced PINK1/Parkin-dependent mitophagy, and improvements observed with caloric restriction. In HIV, antiretroviral therapy (ART) can cause mitochondrial toxicity, and viral proteins such as Tat and Vpr suppress mitophagy, driving T cell metabolic exhaustion and accelerating immunosenescence. In long COVID, cytokine storms, persistent fatigue, muscle pain, and brain fog are associated with reduced mitochondrial biogenesis (↓ PGC-1α) and endothelial/epithelial dysfunction. Across all three conditions, common features include excess reactive oxygen species (ROS) production, mtDNA mutations, impaired mitophagy, metabolic reprogramming from oxidative phosphorylation (OXPHOS) to glycolysis, and chronic inflammation.

Table 1.
Comparative summary of mitochondrial changes across Aging, Long COVID, and HIV. This table outlines representative proteins and viral factors, the status of key mitophagy pathways, fusion–fission dynamics, predominant sources of ROS, mtDNA integrity, and characteristic patterns of metabolic reprogramming across aging, long COVID, and HIV.
6. Mitochondrial Transplantation
Mitochondrial transplantation is a therapeutic procedure involving the isolation of functional mitochondria, which are then introduced into a living organism to integrate into target cells and restore or enhance cellular function [148].
An early groundbreaking study exploring the therapeutic applications of isolated mitochondria centered on transplanting healthy cardiac mitochondria into the rabbits’ hearts subjected to ischemia followed by reperfusion [149]. This pioneering approach demonstrated a notable reduction in ischemia–reperfusion injury and significantly improved heart function. Encouragingly, similar outcomes were later observed in studies involving pigs, further supporting the intervention’s potential [150]. Building on these findings, clinical trials explored the technique in pediatric patients requiring extracorporeal membrane oxygenation. By isolating skeletal muscle mitochondria from the patients and transplanting them back autologously, researchers accelerate recovery, allowing most patients to be successfully weaned off extracorporeal membrane oxygenation in a shorter timeframe [151,152]. More recently, mitochondrial transplantation promoted faster wound healing by reducing wound size, enhancing granulation tissue formation, and accelerating the process of epithelialization [153]. The effectiveness of platelet-derived mitochondrial transplantation in the treatment of ischemic heart disease was demonstrated in a randomized controlled clinical trial [154]. In addition, a mechanism underlying the effects of mitochondrial transfer between mesenchymal and endothelial cells is revealed when it is demonstrated that mesenchymal stromal cells (MSCs) use tunneling nanotubes to transfer mitochondria to endothelial cells (ECs) during cellular stress, and that preventing this transfer hinders EC engraftment [155]. Intercellular nanotube-mediated mitochondrial transfer was also used as a new immunotherapy approach, where healthy mitochondria are transferred from donor cells to T cells via nanotubes. This process boosts the T cells’ ability to produce energy, improving their function and reducing exhaustion. When applied in cancer therapy, these “supercharged” T cells become more effective at infiltrating and destroying tumors, leading to better tumor control and improved survival in patients [156].
Mitochondrial transplantation is classified into three primary types: autologous, heterologous (or allogeneic), and xenogenic transplants.
Autologous mitochondrial transplants involve isolating mitochondria from the same individual who will later receive them. This approach carries minimal risk since the mitochondria are entirely self-derived, ensuring compatibility and avoiding immune rejection [149,150,152].
In contrast, heterologous or allogeneic mitochondrial transplants rely on mitochondria isolated from another individual or a cell line. In some cases, mitochondria may be sourced from a biological mother or a relative within the same maternal lineage, as mtDNA is maternally inherited. Although clinical trials using heterologous mitochondria are still in preclinical stages [157]. However, in cases where donor mitochondria persist, the potential impact of mtDNA heteroplasmy on the recipient’s cells requires careful consideration. While extreme heteroplasmy has been linked to dysfunction [158], achieving such levels through transplantation appears unlikely based on current research [155]. Viral- and therapy-associated stresses accelerate mtDNA damage or alter heteroplasmy, which can produce focal respiratory chain defects and functional mosaicism in tissues; these lesions can persist and drive organ dysfunction [159].
Xenogenic mitochondrial transplantation, on the other hand, involves transferring mitochondria between species, such as from humans to mice. While this method serves as a valuable experimental model, its translational potential is limited due to the likelihood of immune reactions and incompatibility between mitochondrial and nuclear genomes.
A critical aspect of mitochondrial transplantation is its durability and mechanisms of action. For transplanted mitochondria to have a therapeutic effect, they must be taken up by recipient cells and integrated into their endogenous mitochondrial pool [160]. Studies suggest that metabolically compromised cells are more efficient at retaining and utilizing transplanted mitochondria [161]. Animal models of ischemia–reperfusion injury have demonstrated that donor mitochondria can persist in recipient tissues for up to 28 days [162]. However, recent findings reveal that transplanted mitochondria are often rapidly degraded by certain cell types, such as endothelial cells and macrophages [163,164]. While engraftment does occur in some instances, the therapeutic benefits of mitochondrial transplantation may also stem from indirect effects, such as the stimulation of mitophagy or mitochondrial biogenesis [155,165,166,167].
Not all mitochondrial transplants are identical, and their outcomes can vary significantly depending on multiple factors, including the source of mitochondria, the isolation process, and the route of administration [168]. Moreover, mitochondria can be engineered to target specific tissues [169]. These variations highlight the need for standardized protocols to improve the reproducibility of results.
To address this variability, it is recommended that mitochondrial transplantation studies provide detailed descriptions of the source material, isolation methods, mitochondrial size, and the composition of the product, whether it consists of free mitochondria or mitochondrial extracellular vesicles (Mito-EVs) [170,171].
In this context, MoDL, a deep learning-based software package, offers a transformative approach by bridging the gap between mitochondrial morphology and function [172]. Its deep learning algorithm facilitates precise segmentation of mitochondrial structures and accurate prediction of their functions using live-cell imaging data. Trained on a comprehensive dataset of super-resolution images annotated with functional biochemical data, MoDL empowers researchers to analyze heterogeneous mitochondria across various cell types. This software is particularly valuable in mitochondrial transplantation, where identifying healthy, functional mitochondria and predicting their therapeutic potential are critical. MoDL’s ability to generalize across diverse biological contexts makes it an indispensable resource for optimizing transplantation strategies, evaluating mitochondrial integration, and assessing therapeutic outcomes. By doing so, it drives progress in both fundamental research and clinical applications.
Moreover, any modifications made to the mitochondria after isolation should be transparently reported. Rather than aiming for absolute purity, researchers are encouraged to focus on enrichment, defined as the proportion of particles containing mitochondria, which can be quantified using techniques such as flow cytometry or immunoblotting [173,174,175].
As research in this field advances, a deeper understanding of the pharmacokinetics and pharmacodynamics of transplanted mitochondria will be essential. These unique biological therapies behave fundamentally differently from traditional small or large molecule treatments, necessitating tailored approaches for their characterization and clinical application. With these considerations, mitochondrial transplantation holds the potential to become a transformative therapeutic tool across a range of medical conditions.
7. Chronic Viral Infection and Mitochondrial Transplantation
Mitochondrial transplantation has emerged as an exciting and innovative therapeutic approach that offers a way to directly address cellular dysfunction by introducing healthy mitochondria into cells with impaired function. Over the past few years, this strategy has drawn considerable attention, particularly in conditions where mitochondrial health is profoundly compromised, as occurs in PLWH and long COVID [47,65,92,133]. Both conditions are characterized by shared underlying problems—chronic inflammation, oxidative stress, and disrupted energy metabolism—that make mitochondrial transplantation a potentially groundbreaking intervention.
However, the proposal to apply mitochondrial transplantation to chronic viral syndromes requires a much more explicit cell-biological justification, and it must be considered in the context of recent community recommendations [160]. The mechanistic rationale is currently incomplete: transplantation replaces organelles but does not necessarily correct upstream causes such as viral-driven mitophagy blockade or drug-induced mtDNA replication defects.
One of the most compelling advantages of mitochondrial transplantation is its ability to restore energy production within cells [176,177,178,179]. For PLWH or those stressed with the persistent effects of long COVID, mitochondrial dysfunction plays a central role in many of the symptoms they experience. Reduced ATP production in these conditions leads to lower energy availability in vital organs such as the brain, liver, and muscles, often resulting in debilitating fatigue and other systemic issues [63,78,79,92,180,181,182,183,184,185]. By transplanting healthy mitochondria, it might be possible to replenish energy stores and improve cellular function, offering hope for reversing these symptoms. Additionally, mitochondrial transplantation could help address the overproduction of ROS, a key driver of oxidative stress, which further damages cells and contributes to disease progression [186].
Membrane integrity and permeability are central, unresolved technical issues. Isolated donor mitochondria are highly sensitive to mechanical and oxidative damage during isolation and handling; loss of outer- or inner-membrane integrity or dissipation of the membrane potential (Δψm) will markedly reduce ATP-generating capacity and can provoke release of mitochondrial DAMPs (mtDNA, cardiolipin, formyl peptides) that trigger robust innate immune responses. The mitochondrial permeability transition pore (mPTP) and other membrane-permeabilizing events must be monitored because permeabilized organelles cannot provide the intended bioenergetic benefit and may instead increase inflammation or cell stress. These concerns are emphasized in the recent consensus statement, which recommends rigorous quality control of donor organelles (including Δψm and membrane intactness) before any translational application [160].
Importantly, mitochondrial transplantation could also influence the burden of pathogenic mtDNA variants that accumulate with age. By introducing functional mitochondria, the procedure may dilute cells’ load of mutated mtDNA and/or promote a shift in heteroplasmy towards wild-type genomes, thereby counteracting clonal expansion of deleterious mtDNA. However, this potential benefit must be balanced against the risk of creating new heteroplasmic mixtures; careful donor selection and longitudinal monitoring of mtDNA heteroplasmy will be required. Conversely, transplantation may create new heteroplasmic mixtures or introduce donor mtDNA variants; therefore, donor screening and longitudinal heteroplasmy monitoring (single-cell and bulk assays) are necessary safeguards.
Beyond energy restoration, mitochondrial transplantation may have profound effects on the immune system [187,188,189]. Chronic immune activation, a hallmark of HIV—even in individuals on effective antiretroviral therapy—plays a major role in disease progression [190]. Similarly, long COVID has been associated with persistent systemic inflammation, which affects various tissues and organs [191,192]. By improving mitochondrial health, mitochondrial transplantation could help modulate the immune system, dampening inflammation and restoring balance [193]. For example, CD4+ T cells, critical to immune defense but often dysfunctional in HIV, could benefit from the energy boost provided by healthier mitochondria, leading to better immune responses [194,195]. Likewise, in long COVID, improved mitochondrial function in endothelial and epithelial cells could reduce the widespread inflammation and vascular dysfunction that many patients experience [196,197].
Uptake routes and intracellular fate remain poorly defined and are decisive for efficacy. Key mechanistic gaps include: (i) how intact mitochondria cross the plasma membrane of different cell types (direct fusion, endocytosis, tunnelling nanotubes, or EV-mediated transfer); (ii) whether internalized mitochondria escape endolysosomal degradation; (iii) whether they retain Δψm and integrate (fuse) with host mitochondrial networks or are rapidly targeted for mitophagy; and (iv) the kinetics of persistence versus clearance in vivo. The consensus paper recommends minimal characterization assays (membrane potential, ROS generation, mtDNA release, endolysosomal co-localization) to address these unknowns prior to claiming therapeutic benefit.
In the context of HIV, where viral proteins can directly impair mitophagy and mitochondrial quality control, mitochondrial transplantation might be combined with strategies that restore mitophagy or inhibit the viral factors that block it. Thus, transplantation could both replenish functional organelles and work synergistically with targeted therapies to overcome virus-driven defects in mitochondrial turnover. However, unless upstream defects (e.g., POLG inhibition from prior NRTI exposure, chronic blocking of mitophagy by viral products) are corrected, transplanted mitochondria risk being subjected to the same dysfunctional quality-control pathways and cleared, limiting long-term benefit.
Mitochondrial transplantation could also offer targeted benefits for specific tissues. Neurocognitive issues, such as HIV-associated neurocognitive disorders and the brain fog reported by many long COVID patients, often stem from mitochondrial dysfunction in neurons [65,198]. The transplantation of healthy mitochondria into these cells could enhance their energy production, reduce inflammation in the brain, and potentially improve cognitive function. Moreover, mitochondrial transplantation might address cellular senescence—a process in which cells lose their ability to function properly but continue to contribute to inflammation [199,200]. Senescent cells are common in aging, HIV, and long COVID, and rejuvenating these cells through mitochondrial transplantation could restore tissue health and reduce chronic inflammation [201]. Yet CNS application faces major delivery barriers: the blood–brain barrier limits systemic access, and invasive routes (intrathecal or intracerebral) carry safety and scalability challenges. Carrier strategies (EVs/exosome-like particles, receptor-mediated shuttles, nasal delivery, transient BBB modulation) are being explored preclinically but require demonstration of safe biodistribution and lack of neuroinflammatory sequelae.
Targeting the central nervous system (CNS) presents significant practical challenges that are highly relevant to “brain fog” in long COVID and HIV. The blood–brain barrier limits access to systemically delivered mitochondria, so CNS-directed strategies (for example, intrathecal or intracerebral delivery, carrier systems such as extracellular vesicles or exosome-like particles, receptor-mediated transport, nasal administration, or transient BBB-permeabilizing approaches) are being explored preclinically. Each approach has distinct safety and scalability considerations, and successful CNS application will require rigorous assessment of delivery efficiency, off-target distribution, and neuroinflammatory risk.
Despite its promising potential, mitochondrial transplantation is not without challenges. One major concern is the risk of immune rejection [202,203]. Mitochondria from a donor, even if closely matched, may be perceived as foreign by the recipient’s immune system, leading to inflammation and negating the therapeutic benefits. Furthermore, the risk of introducing mitochondria with genetic mutations (in mitochondrial DNA) must be carefully managed, as this could worsen cellular function instead of improving it [204,205,206].
Another issue is the possibility of off-target effects. Transplanted mitochondria may integrate into unintended cells or tissues, potentially disrupting normal cellular processes [207]. This risk is particularly relevant for individuals with underlying inflammatory conditions, such as those seen in HIV and long COVID, where an overactive immune response could be further exacerbated. In HIV, an additional concern is the potential effect on viral reservoirs—latently infected cells that harbor the virus. ROS could act as promoters or reversers of latency, depending on the cell type. Therefore, ROS reverse HIV latency in macrophages but promote latency in T cells [208]. Improved mitochondrial function could theoretically activate these reservoirs, complicating disease management for patients already on antiretroviral therapy.
The persistence of transplanted mitochondria also raises important questions. Early studies suggest that the introduced mitochondria can integrate into recipient cells and remain functional, but it is unclear how long these benefits last. In vivo studies have shown that donor mitochondria can persist in recipient tissues for up to 28 days [150,209,210]. These uncertainties must be addressed in future research. Furthermore, ethical and regulatory considerations—such as donor selection, mitochondrial compatibility, and the long-term safety of the procedure—require careful planning and oversight to ensure the technology is applied responsibly (Figure 3).
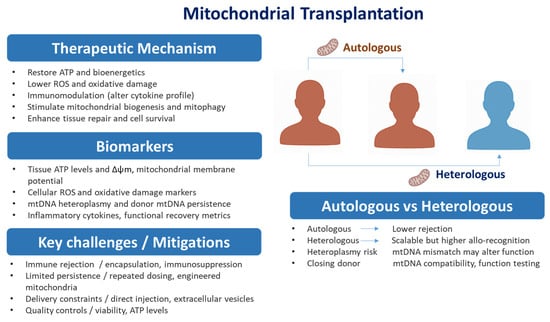
Figure 3.
Mitochondrial Transplantation. Mitochondrial transplantation overview. Right: schematic of autologous (patient-derived—lower rejection, limited supply) versus heterologous (allogeneic—scalable, higher allo-recognition/heteroplasmy) approaches. Left: concise panels showing Therapeutic mechanisms, Biomarkers/options, and Key challenges/mitigations. Arrows link the routes to the panels; most strategies remain preclinical and require rigorous assessment of donor compatibility, safety, and persistence. Abbreviations: ROS, reactive oxygen species; mtDNA, mitochondrial DNA; Δψm, mitochondrial membrane potential.
Recommended experimental and reporting roadmap (cell-biology-driven): (1) standardized quality control of donor mitochondria (Δψm, membrane intactness, absence of damaging mtDNA variants, redox state); (2) mechanistic uptake studies in primary human target cells (T cells, macrophages, endothelial cells, neurons) with assays for endolysosomal escape, fusion with host mitochondria, and mitophagy targeting; (3) immunogenicity testing in inflammatory milieu models (measure mtDNA release, cytokine induction, innate sensor activation); (4) heteroplasmy tracking and donor mtDNA fate over time at single-cell resolution; (5) biodistribution and persistence studies for each delivery modality (systemic, intrathecal, EV-assisted); and (6) adherence to the standardized nomenclature, minimal reporting criteria and safety endpoints recommended in the recent consensus [160]. These steps will help determine whether transplantation is mechanistically plausible and clinically safe for chronic viral conditions.
In summary, mitochondrial transplantation holds remarkable promise for addressing the mitochondrial dysfunction that underlies HIV and long COVID. By restoring cellular energy production, reducing oxidative stress, and modulating immune responses, this therapy could alleviate many of the systemic and tissue-specific problems that plague individuals with these conditions. Nevertheless, to address age-associated clonal mtDNA expansion, virus-mediated mitophagy blockade, and CNS-targeted symptoms such as brain fog, combined approaches (optimized donor selection, heteroplasmy monitoring, mitophagy-restoring agents, and targeted delivery systems) will likely be necessary. However, challenges such as immune rejection, off-target effects, and ethical concerns need to be resolved before this approach can be widely adopted. To fully harness the potential of mitochondrial transplantation, rigorous clinical trials and continued research into its mechanisms, safety, and long-term efficacy are essential. With these efforts, mitochondrial transplantation could emerge as a transformative therapy, offering hope to those living with the long-term effects of HIV and COVID-19.
8. Limitations and Future Directions for Mitochondrial Therapies in HIV, Aging, and Long COVID
Several important limitations shape our current understanding and point to priorities for future research. Much of the evidence on long COVID is still early and inconsistent, with wide variation in case definitions and cohort sizes. This makes it hard to draw firm causal conclusions or generalize findings. In people living with HIV (PLWH), the picture is even more complex: mitochondrial changes are influenced not only by the infection itself but also by aging, coexisting health conditions, and diverse histories of antiretroviral therapy (ART). These overlapping factors complicate efforts to separate drug-related mitochondrial injury from virus-driven effects.
Age-related clonal expansion of mtDNA mutations and heteroplasmy adds yet another layer of difficulty. While organelle transfer may dilute harmful mitochondrial genomes in some cells, it can also create new heteroplasmic mixtures with unknown long-term consequences.
On the technical side, several key issues remain unresolved. These include identifying the most suitable source of mitochondria, refining isolation methods, optimizing delivery routes, improving engraftment efficiency, managing immune recognition, limiting off-target uptake, and ensuring durable benefits. These challenges are particularly daunting when targeting the central nervous system, given the blood–brain barrier and the risks tied to intraparenchymal delivery.
Methodological inconsistencies also limit progress. Studies differ in their assays for viability and potency, their outcome measures, and even their reporting standards. Ethical and regulatory questions—such as how to select donors and how these therapies might affect viral reservoirs—remain unsettled as well.
Overcoming these challenges will require carefully designed studies. Only through such work can we determine whether mitochondrial therapies can safely and effectively correct the specific defects seen in aging, HIV, and long COVID, and guide their responsible translation into clinical practice.
Author Contributions
J.Q. and M.V.D.: Both have contributed equally to conceptualization, methodology, writing, review, and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| mtDNA | Mitochondrial DNA |
| ROS | Reactive oxygen species |
| TCA | Tricarboxylic acid |
| ETC | Electron transport chain |
| MAVS | Mitochondrial antiviral signaling |
| MFRTA | Mitochondrial free radical theory of aging |
| CR | Caloric restriction |
| PLWH | People living with HIV |
| ART | Antiretroviral therapy |
| NRTIs | Nucleoside reverse transcriptase inhibitors |
| OXPHOS | oxidative phosphorylation |
| HIF | Hypoxia-Inducible Factor |
| DRP1 | Dynamin-related protein 1 |
| MSCs | Mesenchymal stromal cells |
| EC | Endothelial cells |
References
- Martijn, J.; Vosseberg, J.; Guy, L.; Offre, P.; Ettema, T.J.G. Deep mitochondrial origin outside the sampled alphaproteobacteria. Nature 2018, 557, 101–105. [Google Scholar] [CrossRef]
- Ott, M.; Gogvadze, V.; Orrenius, S.; Zhivotovsky, B. Mitochondria, oxidative stress and cell death. Apoptosis 2007, 12, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Sorouri, M.; Chang, T.; Hancks, D.C. Mitochondria and Viral Infection: Advances and Emerging Battlefronts. mBio 2022, 13, e0209621. [Google Scholar] [CrossRef]
- Shokolenko, I.N.; Wilson, G.L.; Alexeyev, M.F. Persistent damage induces mitochondrial DNA degradation. DNA Repair 2013, 12, 488–499. [Google Scholar] [CrossRef]
- Sun, J.; Brown, T.T.; Samuels, D.C.; Hulgan, T.; D’Souza, G.; Jamieson, B.D.; Erlandson, K.M.; Martinson, J.; Palella, F.J., Jr.; Margolick, J.B.; et al. The Role of Mitochondrial DNA Variation in Age-Related Decline in Gait Speed Among Older Men Living with Human Immunodeficiency Virus. Clin. Infect. Dis. 2018, 67, 778–784. [Google Scholar] [CrossRef]
- Szogi, T.; Borsos, B.N.; Masic, D.; Radics, B.; Bella, Z.; Banfi, A.; Ordog, N.; Zsiros, C.; Kiricsi, A.; Pankotai-Bodo, G.; et al. Novel biomarkers of mitochondrial dysfunction in Long COVID patients. Geroscience 2024, 47, 2245–2261. [Google Scholar] [CrossRef]
- Gay, L.; Desquiret-Dumas, V.; Nagot, N.; Rapenne, C.; Van de Perre, P.; Reynier, P.; Moles, J.P. Long-term persistence of mitochondrial dysfunctions after viral infections and antiviral therapies: A review of mechanisms involved. J. Med. Virol. 2024, 96, e29886. [Google Scholar] [CrossRef]
- Macnaughtan, J.; Chau, K.Y.; Brennan, E.; Toffoli, M.; Spinazzola, A.; Hillman, T.; Heightman, M.; Schapira, A.H.V. Mitochondrial function is impaired in long COVID patients. Ann. Med. 2025, 57, 2528167. [Google Scholar] [CrossRef]
- Savedchuk, S.; Raslan, R.; Nystrom, S.; Sparks, M.A. Emerging Viral Infections and the Potential Impact on Hypertension, Cardiovascular Disease, and Kidney Disease. Circ. Res. 2022, 130, 1618–1641. [Google Scholar] [CrossRef]
- Tzameli, I. The evolving role of mitochondria in metabolism. Trends Endocrinol. Metab. 2012, 23, 417–419. [Google Scholar] [CrossRef]
- Formosa, L.E.; Dibley, M.G.; Stroud, D.A.; Ryan, M.T. Building a complex complex: Assembly of mitochondrial respiratory chain complex I. Semin. Cell Dev. Biol. 2018, 76, 154–162. [Google Scholar] [CrossRef]
- Vonck, J.; Schafer, E. Supramolecular organization of protein complexes in the mitochondrial inner membrane. Biochim. Biophys. Acta 2009, 1793, 117–124. [Google Scholar] [CrossRef]
- Racker, E.; Horstman, L.L. Partial resolution of the enzymes catalyzing oxidative phosphorylation. 13. Structure and function of submitochondrial particles completely resolved with respect to coupling factor. J. Biol. Chem. 1967, 242, 2547–2551. [Google Scholar] [CrossRef]
- Mitchell, P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 1961, 191, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Zorova, L.D.; Popkov, V.A.; Plotnikov, E.Y.; Silachev, D.N.; Pevzner, I.B.; Jankauskas, S.S.; Babenko, V.A.; Zorov, S.D.; Balakireva, A.V.; Juhaszova, M.; et al. Mitochondrial membrane potential. Anal. Biochem. 2018, 552, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Labunskyy, V.M.; Gladyshev, V.N. Role of reactive oxygen species-mediated signaling in aging. Antioxid. Redox Signal. 2013, 19, 1362–1372. [Google Scholar] [CrossRef]
- Ristow, M.; Schmeisser, K. Mitohormesis: Promoting Health and Lifespan by Increased Levels of Reactive Oxygen Species (ROS). Dose Response 2014, 12, 288–341. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef]
- Spinelli, J.B.; Haigis, M.C. The multifaceted contributions of mitochondria to cellular metabolism. Nat. Cell Biol. 2018, 20, 745–754. [Google Scholar] [CrossRef]
- Read, A.D.; Bentley, R.E.; Archer, S.L.; Dunham-Snary, K.J. Mitochondrial iron-sulfur clusters: Structure, function, and an emerging role in vascular biology. Redox Biol. 2021, 47, 102164. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Wang, F.; Li, P.; Gao, Y. Mitochondrial Ca(2+) Homeostasis: Emerging Roles and Clinical Significance in Cardiac Remodeling. Int. J. Mol. Sci. 2022, 23, 3025. [Google Scholar] [CrossRef]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly, Y.M.; Gidlof, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef]
- Ross, J.M.; Stewart, J.B.; Hagstrom, E.; Brene, S.; Mourier, A.; Coppotelli, G.; Freyer, C.; Lagouge, M.; Hoffer, B.J.; Olson, L.; et al. Germline mitochondrial DNA mutations aggravate ageing and can impair brain development. Nature 2013, 501, 412–415. [Google Scholar] [CrossRef]
- Kujoth, G.C.; Prolla, T.A. Evolving insight into the role of mitochondrial DNA mutations in aging. Exp. Gerontol. 2008, 43, 20–23. [Google Scholar] [CrossRef]
- Somasundaram, I.; Jain, S.M.; Blot-Chabaud, M.; Pathak, S.; Banerjee, A.; Rawat, S.; Sharma, N.R.; Duttaroy, A.K. Mitochondrial dysfunction and its association with age-related disorders. Front. Physiol. 2024, 15, 1384966. [Google Scholar] [CrossRef]
- Corral-Debrinski, M.; Horton, T.; Lott, M.T.; Shoffner, J.M.; Beal, M.F.; Wallace, D.C. Mitochondrial DNA deletions in human brain: Regional variability and increase with advanced age. Nat. Genet. 1992, 2, 324–329. [Google Scholar] [CrossRef]
- Yen, T.C.; Su, J.H.; King, K.L.; Wei, Y.H. Ageing-associated 5 kb deletion in human liver mitochondrial DNA. Biochem. Biophys. Res. Commun. 1991, 178, 124–131. [Google Scholar] [CrossRef]
- Fayet, G.; Jansson, M.; Sternberg, D.; Moslemi, A.R.; Blondy, P.; Lombes, A.; Fardeau, M.; Oldfors, A. Ageing muscle: Clonal expansions of mitochondrial DNA point mutations and deletions cause focal impairment of mitochondrial function. Neuromuscul. Disord. 2002, 12, 484–493. [Google Scholar] [CrossRef]
- Larsson, N.G. Somatic mitochondrial DNA mutations in mammalian aging. Annu. Rev. Biochem. 2010, 79, 683–706. [Google Scholar] [CrossRef]
- Balasubramanian, P.; Howell, P.R.; Anderson, R.M. Aging and Caloric Restriction Research: A Biological Perspective with Translational Potential. eBioMedicine 2017, 21, 37–44. [Google Scholar] [CrossRef]
- Peterson, C.M.; Johannsen, D.L.; Ravussin, E. Skeletal muscle mitochondria and aging: A review. J. Aging Res. 2012, 2012, 194821. [Google Scholar] [CrossRef]
- Civitarese, A.E.; Carling, S.; Heilbronn, L.K.; Hulver, M.H.; Ukropcova, B.; Deutsch, W.A.; Smith, S.R.; Ravussin, E.; the CALERIE Pennnigton Team. Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med. 2007, 4, e76. [Google Scholar] [CrossRef]
- Civitarese, A.E.; Smith, S.R.; Ravussin, E. Diet, energy metabolism and mitochondrial biogenesis. Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Sprason, C.; Tucker, T.; Clancy, D. MtDNA deletions and aging. Front. Aging 2024, 5, 1359638. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yuan, Y.; Liu, J.; Li, C.; Jiang, X. The role of mitochondria in aging, cell death, and tumor immunity. Front. Immunol. 2024, 15, 1520072. [Google Scholar] [CrossRef] [PubMed]
- Sinenko, S.A.; Starkova, T.Y.; Kuzmin, A.A.; Tomilin, A.N. Physiological Signaling Functions of Reactive Oxygen Species in Stem Cells: From Flies to Man. Front. Cell Dev. Biol. 2021, 9, 714370. [Google Scholar] [CrossRef]
- Moreno Fernandez-Ayala, D.J.; Navas, P.; Lopez-Lluch, G. Age-related mitochondrial dysfunction as a key factor in COVID-19 disease. Exp. Gerontol. 2020, 142, 111147. [Google Scholar] [CrossRef]
- Elesela, S.; Lukacs, N.W. Role of Mitochondria in Viral Infections. Life 2021, 11, 232. [Google Scholar] [CrossRef]
- Srivastava, S. The Mitochondrial Basis of Aging and Age-Related Disorders. Genes 2017, 8, 398. [Google Scholar] [CrossRef]
- Guo, J.; Huang, X.; Dou, L.; Yan, M.; Shen, T.; Tang, W.; Li, J. Aging and aging-related diseases: From molecular mechanisms to interventions and treatments. Signal Transduct. Target. Ther. 2022, 7, 391. [Google Scholar] [CrossRef]
- Amorim, J.A.; Coppotelli, G.; Rolo, A.P.; Palmeira, C.M.; Ross, J.M.; Sinclair, D.A. Mitochondrial and metabolic dysfunction in ageing and age-related diseases. Nat. Rev. Endocrinol. 2022, 18, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Ladha, J.S.; Tripathy, M.K.; Mitra, D. Mitochondrial complex I activity is impaired during HIV-1-induced T-cell apoptosis. Cell Death Differ. 2005, 12, 1417–1428. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.J.; Lee, W.; Ku, K.B.; Yoon, G.Y.; Moon, H.W.; Kim, C.; Kim, M.H.; Yi, Y.S.; Jun, S.; Kim, B.T.; et al. SARS-CoV-2 aberrantly elevates mitochondrial bioenergetics to induce robust virus propagation. Signal Transduct. Target. Ther. 2024, 9, 125. [Google Scholar] [CrossRef]
- Hinton, A.O., Jr.; N’Jai, A.U.; Vue, Z.; Wanjalla, C. Connection Between HIV and Mitochondria in Cardiovascular Disease and Implications for Treatments. Circ. Res. 2024, 134, 1581–1606. [Google Scholar] [CrossRef]
- Cho, D.H.; Kim, J.K.; Jo, E.K. Mitophagy and Innate Immunity in Infection. Mol. Cell 2020, 43, 10–22. [Google Scholar] [CrossRef]
- Jiang, H.W.; Zhang, H.N.; Meng, Q.F.; Xie, J.; Li, Y.; Chen, H.; Zheng, Y.X.; Wang, X.N.; Qi, H.; Zhang, J.; et al. SARS-CoV-2 Orf9b suppresses type I interferon responses by targeting TOM70. Cell. Mol. Immunol. 2020, 17, 998–1000. [Google Scholar] [CrossRef]
- Lecoeur, H.; Borgne-Sanchez, A.; Chaloin, O.; El-Khoury, R.; Brabant, M.; Langonne, A.; Porceddu, M.; Briere, J.J.; Buron, N.; Rebouillat, D.; et al. HIV-1 Tat protein directly induces mitochondrial membrane permeabilization and inactivates cytochrome c oxidase. Cell Death Dis. 2012, 3, e282. [Google Scholar] [CrossRef]
- Thangaraj, A.; Periyasamy, P.; Liao, K.; Bendi, V.S.; Callen, S.; Pendyala, G.; Buch, S. HIV-1 TAT-mediated microglial activation: Role of mitochondrial dysfunction and defective mitophagy. Autophagy 2018, 14, 1596–1619. [Google Scholar] [CrossRef]
- Huang, C.Y.; Chiang, S.F.; Lin, T.Y.; Chiou, S.H.; Chow, K.C. HIV-1 Vpr triggers mitochondrial destruction by impairing Mfn2-mediated ER-mitochondria interaction. PLoS ONE 2012, 7, e33657. [Google Scholar] [CrossRef] [PubMed]
- Rozzi, S.J.; Avdoshina, V.; Fields, J.A.; Mocchetti, I. Human immunodeficiency virus Tat impairs mitochondrial fission in neurons. Cell Death Discov. 2018, 4, 8. [Google Scholar] [CrossRef] [PubMed]
- Fields, J.A.; Ellis, R.J. HIV in the cART era and the mitochondrial: Immune interface in the CNS. Int. Rev. Neurobiol. 2019, 145, 29–65. [Google Scholar] [CrossRef] [PubMed]
- Villeneuve, L.M.; Purnell, P.R.; Stauch, K.L.; Callen, S.E.; Buch, S.J.; Fox, H.S. HIV-1 transgenic rats display mitochondrial abnormalities consistent with abnormal energy generation and distribution. J. Neurovirol. 2016, 22, 564–574. [Google Scholar] [CrossRef]
- Figarola-Centurion, I.; Escoto-Delgadillo, M.; Gonzalez-Enriquez, G.V.; Gutierrez-Sevilla, J.E.; Vazquez-Valls, E.; Cardenas-Bedoya, J.; Torres-Mendoza, B.M. HIV-1 Tat Induces Dysregulation of PGC1-Alpha and Sirtuin 3 Expression in Neurons: The Role of Mitochondrial Biogenesis in HIV-Associated Neurocognitive Disorder (HAND). Int. J. Mol. Sci. 2023, 24, 17566. [Google Scholar] [CrossRef]
- Swinton, M.K.; Carson, A.; Telese, F.; Sanchez, A.B.; Soontornniyomkij, B.; Rad, L.; Batki, I.; Quintanilla, B.; Perez-Santiago, J.; Achim, C.L.; et al. Mitochondrial biogenesis is altered in HIV+ brains exposed to ART: Implications for therapeutic targeting of astroglia. Neurobiol. Dis. 2019, 130, 104502. [Google Scholar] [CrossRef]
- Zhao, T.; Zhang, J.; Lei, H.; Meng, Y.; Cheng, H.; Zhao, Y.; Geng, G.; Mu, C.; Chen, L.; Liu, Q.; et al. NRF1-mediated mitochondrial biogenesis antagonizes innate antiviral immunity. EMBO J. 2023, 42, e113258. [Google Scholar] [CrossRef]
- Akanchise, T.; Angelova, A. Potential of Nano-Antioxidants and Nanomedicine for Recovery from Neurological Disorders Linked to Long COVID Syndrome. Antioxidants 2023, 12, 393. [Google Scholar] [CrossRef]
- Prasada Kabekkodu, S.; Chakrabarty, S.; Jayaram, P.; Mallya, S.; Thangaraj, K.; Singh, K.K.; Satyamoorthy, K. Severe acute respiratory syndrome coronaviruses contributing to mitochondrial dysfunction: Implications for post-COVID complications. Mitochondrion 2023, 69, 43–56. [Google Scholar] [CrossRef]
- Saito, S.; Shahbaz, S.; Luo, X.; Osman, M.; Redmond, D.; Cohen Tervaert, J.W.; Li, L.; Elahi, S. Metabolomic and immune alterations in long COVID patients with chronic fatigue syndrome. Front. Immunol. 2024, 15, 1341843. [Google Scholar] [CrossRef]
- Mikuteit, M.; Baskal, S.; Klawitter, S.; Dopfer-Jablonka, A.; Behrens, G.M.N.; Muller, F.; Schroder, D.; Klawonn, F.; Steffens, S.; Tsikas, D. Amino acids, post-translational modifications, nitric oxide, and oxidative stress in serum and urine of long COVID and ex COVID human subjects. Amino Acids 2023, 55, 1173–1188. [Google Scholar] [CrossRef] [PubMed]
- Noonong, K.; Chatatikun, M.; Surinkaew, S.; Kotepui, M.; Hossain, R.; Bunluepuech, K.; Noothong, C.; Tedasen, A.; Klangbud, W.K.; Imai, M.; et al. Mitochondrial oxidative stress, mitochondrial ROS storms in long COVID pathogenesis. Front. Immunol. 2023, 14, 1275001. [Google Scholar] [CrossRef] [PubMed]
- Russell, S.J.; Parker, K.; Lehoczki, A.; Lieberman, D.; Partha, I.S.; Scott, S.J.; Phillips, L.R.; Fain, M.J.; Nikolich, J.Z. Post-acute sequelae of SARS-CoV-2 infection (Long COVID) in older adults. Geroscience 2024, 46, 6563–6581. [Google Scholar] [CrossRef]
- Molnar, T.; Lehoczki, A.; Fekete, M.; Varnai, R.; Zavori, L.; Erdo-Bonyar, S.; Simon, D.; Berki, T.; Csecsei, P.; Ezer, E. Mitochondrial dysfunction in long COVID: Mechanisms, consequences, and potential therapeutic approaches. Geroscience 2024, 46, 5267–5286. [Google Scholar] [CrossRef]
- Santoro, A.; Martucci, M.; Conte, M.; Capri, M.; Franceschi, C.; Salvioli, S. Inflammaging, hormesis and the rationale for anti-aging strategies. Ageing Res. Rev. 2020, 64, 101142. [Google Scholar] [CrossRef]
- Queiroz, M.A.F.; Brito, W.; Pereira, K.A.S.; Pereira, L.M.S.; Amoras, E.; Lima, S.S.; Santos, E.F.D.; Costa, F.P.D.; Sarges, K.M.L.; Cantanhede, M.H.D.; et al. Severe COVID-19 and long COVID are associated with high expression of STING, cGAS and IFN-alpha. Sci. Rep. 2024, 14, 4974. [Google Scholar] [CrossRef]
- Alfadda, A.A.; Sallam, R.M. Reactive oxygen species in health and disease. J. Biotechnol. Biomed. 2012, 2012, 936486. [Google Scholar] [CrossRef]
- Akbal, A.; Dernst, A.; Lovotti, M.; Mangan, M.S.J.; McManus, R.M.; Latz, E. How location and cellular signaling combine to activate the NLRP3 inflammasome. Cell. Mol. Immunol. 2022, 19, 1201–1214. [Google Scholar] [CrossRef]
- Chen, I.Y.; Moriyama, M.; Chang, M.F.; Ichinohe, T. Severe Acute Respiratory Syndrome Coronavirus Viroporin 3a Activates the NLRP3 Inflammasome. Front. Microbiol. 2019, 10, 50. [Google Scholar] [CrossRef]
- Xu, H.; Akinyemi, I.A.; Chitre, S.A.; Loeb, J.C.; Lednicky, J.A.; McIntosh, M.T.; Bhaduri-McIntosh, S. SARS-CoV-2 viroporin encoded by ORF3a triggers the NLRP3 inflammatory pathway. Virology 2022, 568, 13–22. [Google Scholar] [CrossRef]
- Planes, R.; Pinilla, M.; Santoni, K.; Hessel, A.; Passemar, C.; Lay, K.; Paillette, P.; Valadao, A.C.; Robinson, K.S.; Bastard, P.; et al. Human NLRP1 is a sensor of pathogenic coronavirus 3CL proteases in lung epithelial cells. Mol. Cell 2022, 82, 2385–2400.e9. [Google Scholar] [CrossRef]
- de Almeida, L.; da Silva, A.L.N.; Rodrigues, T.S.; Oliveira, S.; Ishimoto, A.Y.; Seribelli, A.A.; Becerra, A.; Andrade, W.A.; Ataide, M.A.; Caetano, C.C.S.; et al. Identification of immunomodulatory drugs that inhibit multiple inflammasomes and impair SARS-CoV-2 infection. Sci. Adv. 2022, 8, eabo5400. [Google Scholar] [CrossRef]
- Guarnieri, J.W.; Dybas, J.M.; Fazelinia, H.; Kim, M.S.; Frere, J.; Zhang, Y.; Soto Albrecht, Y.; Murdock, D.G.; Angelin, A.; Singh, L.N.; et al. Core mitochondrial genes are down-regulated during SARS-CoV-2 infection of rodent and human hosts. Sci. Transl. Med. 2023, 15, eabq1533. [Google Scholar] [CrossRef] [PubMed]
- Appelman, B.; Charlton, B.T.; Goulding, R.P.; Kerkhoff, T.J.; Breedveld, E.A.; Noort, W.; Offringa, C.; Bloemers, F.W.; van Weeghel, M.; Schomakers, B.V.; et al. Muscle abnormalities worsen after post-exertional malaise in long COVID. Nat. Commun. 2024, 15, 17. [Google Scholar] [CrossRef] [PubMed]
- Tryfonos, A.; Pourhamidi, K.; Jornaker, G.; Engvall, M.; Eriksson, L.; Elhallos, S.; Asplund, N.; Mandic, M.; Sundblad, P.; Sepic, A.; et al. Functional Limitations and Exercise Intolerance in Patients with Post-COVID Condition: A Randomized Crossover Clinical Trial. JAMA Netw. Open 2024, 7, e244386. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.Y.; Wei, A.C. Transcriptome and machine learning analysis of the impact of COVID-19 on mitochondria and multiorgan damage. PLoS ONE 2024, 19, e0297664. [Google Scholar] [CrossRef]
- Grossini, E.; Concina, D.; Rinaldi, C.; Russotto, S.; Garhwal, D.; Zeppegno, P.; Gramaglia, C.; Kul, S.; Panella, M. Association Between Plasma Redox State/Mitochondria Function and a Flu-Like Syndrome/COVID-19 in the Elderly Admitted to a Long-Term Care Unit. Front. Physiol. 2021, 12, 707587. [Google Scholar] [CrossRef]
- Pintos, I.; Soriano, V. Mitochondrial damage as cause of long COVID. AIDS Rev. 2023, 26, 145–149. [Google Scholar] [CrossRef]
- Bizjak, D.A.; Ohmayer, B.; Buhl, J.L.; Schneider, E.M.; Walther, P.; Calzia, E.; Jerg, A.; Matits, L.; Steinacker, J.M. Functional and Morphological Differences of Muscle Mitochondria in Chronic Fatigue Syndrome and Post-COVID Syndrome. Int. J. Mol. Sci. 2024, 25, 1675. [Google Scholar] [CrossRef]
- Chen, Z.Z.; Johnson, L.; Trahtemberg, U.; Baker, A.; Huq, S.; Dufresne, J.; Bowden, P.; Miao, M.; Ho, J.A.; Hsu, C.C.; et al. Mitochondria and cytochrome components released into the plasma of severe COVID-19 and ICU acute respiratory distress syndrome patients. Clin. Proteom. 2023, 20, 17. [Google Scholar] [CrossRef] [PubMed]
- Buckley, S.; Byrnes, S.; Cochrane, C.; Roche, M.; Estes, J.D.; Selemidis, S.; Angelovich, T.A.; Churchill, M.J. The role of oxidative stress in HIV-associated neurocognitive disorders. Brain Behav. Immun. Health 2021, 13, 100235. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Ivanova, O.N.; Kochetkov, S.N.; Starodubova, E.S.; Bartosch, B.; Isaguliants, M.G. Oxidative Stress during HIV Infection: Mechanisms and Consequences. Oxidative Med. Cell. Longev. 2016, 2016, 8910396. [Google Scholar] [CrossRef] [PubMed]
- Hileman, C.O.; Turner, R.; Funderburg, N.T.; Semba, R.D.; McComsey, G.A. Changes in oxidized lipids drive the improvement in monocyte activation and vascular disease after statin therapy in HIV. AIDS 2016, 30, 65–73. [Google Scholar] [CrossRef]
- Harshithkumar, R.; Shah, P.; Jadaun, P.; Mukherjee, A. ROS Chronicles in HIV Infection: Genesis of Oxidative Stress, Associated Pathologies, and Therapeutic Strategies. Curr. Issues Mol. Biol. 2024, 46, 8852–8873. [Google Scholar] [CrossRef]
- Montano, M.; Oursler, K.K.; Xu, K.; Sun, Y.V.; Marconi, V.C. Biological ageing with HIV infection: Evaluating the geroscience hypothesis. Lancet Healthy Longev. 2022, 3, e194–e205. [Google Scholar] [CrossRef]
- Rodes, B.; Cadinanos, J.; Esteban-Cantos, A.; Rodriguez-Centeno, J.; Arribas, J.R. Ageing with HIV: Challenges and biomarkers. eBioMedicine 2022, 77, 103896. [Google Scholar] [CrossRef]
- Zhu, S.; Wang, W.; He, J.; Duan, W.; Ma, X.; Guan, H.; Wu, Y.; Li, S.; Li, Y.; Tian, T.; et al. Higher cardiovascular disease risks in people living with HIV: A systematic review and meta-analysis. J. Glob. Health 2024, 14, 04078. [Google Scholar] [CrossRef]
- Pramukti, I.; Lindayani, L.; Chen, Y.C.; Yeh, C.Y.; Tai, T.W.; Fetzer, S.; Ko, N.Y. Bone fracture among people living with HIV: A systematic review and meta-regression of prevalence, incidence, and risk factors. PLoS ONE 2020, 15, e0233501. [Google Scholar] [CrossRef]
- Vastag, Z.; Fira-Mladinescu, O.; Rosca, E.C. HIV-Associated Neurocognitive Disorder (HAND): Obstacles to Early Neuropsychological Diagnosis. Int. J. Gen. Med. 2022, 15, 4079–4090. [Google Scholar] [CrossRef]
- Wang, Y.; Santerre, M.; Tempera, I.; Martin, K.; Mukerjee, R.; Sawaya, B.E. HIV-1 Vpr disrupts mitochondria axonal transport and accelerates neuronal aging. Neuropharmacology 2017, 117, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Schank, M.; Zhao, J.; Moorman, J.P.; Yao, Z.Q. The Impact of HIV- and ART-Induced Mitochondrial Dysfunction in Cellular Senescence and Aging. Cells 2021, 10, 174. [Google Scholar] [CrossRef] [PubMed]
- Selvaraj, S.; Ghebremichael, M.; Li, M.; Foli, Y.; Langs-Barlow, A.; Ogbuagu, A.; Barakat, L.; Tubridy, E.; Edifor, R.; Lam, W.; et al. Antiretroviral therapy-induced mitochondrial toxicity: Potential mechanisms beyond polymerase-gamma inhibition. Clin. Pharmacol. Ther. 2014, 96, 110–120. [Google Scholar] [CrossRef] [PubMed]
- Margolis, A.M.; Heverling, H.; Pham, P.A.; Stolbach, A. A review of the toxicity of HIV medications. J. Med. Toxicol. 2014, 10, 26–39. [Google Scholar] [CrossRef]
- Brinkman, K.; Smeitink, J.A.; Romijn, J.A.; Reiss, P. Mitochondrial toxicity induced by nucleoside-analogue reverse-transcriptase inhibitors is a key factor in the pathogenesis of antiretroviral-therapy-related lipodystrophy. Lancet 1999, 354, 1112–1115. [Google Scholar] [CrossRef]
- Hung, K.M.; Chen, P.C.; Hsieh, H.C.; Calkins, M.J. Mitochondrial defects arise from nucleoside/nucleotide reverse transcriptase inhibitors in neurons: Potential contribution to HIV-associated neurocognitive disorders. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 406–413. [Google Scholar] [CrossRef]
- Note, R.; Maisonneuve, C.; Letteron, P.; Peytavin, G.; Djouadi, F.; Igoudjil, A.; Guimont, M.C.; Biour, M.; Pessayre, D.; Fromenty, B. Mitochondrial and metabolic effects of nucleoside reverse transcriptase inhibitors (NRTIs) in mice receiving one of five single- and three dual-NRTI treatments. Antimicrob. Agents Chemother. 2003, 47, 3384–3392. [Google Scholar] [CrossRef]
- Haynes, J.; Joshi, A.; Larue, R.C.; Eisenmann, E.D.; Govindarajan, R. Nucleoside Reverse Transcriptase Inhibitor (NRTI)-Induced Neuropathy and Mitochondrial Toxicity: Limitations of the Poly-gamma Hypothesis and the Potential Roles of Autophagy and Drug Transport. Pharmaceutics 2024, 16, 1592. [Google Scholar] [CrossRef]
- Saitoh, A.; Fenton, T.; Alvero, C.; Fletcher, C.V.; Spector, S.A. Impact of nucleoside reverse transcriptase inhibitors on mitochondria in human immunodeficiency virus type 1-infected children receiving highly active antiretroviral therapy. Antimicrob. Agents Chemother. 2007, 51, 4236–4242. [Google Scholar] [CrossRef]
- Santos-Llamas, A.; Monte, M.J.; Marin, J.J.G.; Perez, M.J. Dysregulation of autophagy in rat liver with mitochondrial DNA depletion induced by the nucleoside analogue zidovudine. Arch. Toxicol. 2018, 92, 2109–2118. [Google Scholar] [CrossRef]
- Zhang, Y.; Oliveira, A.N.; Hood, D.A. The intersection of exercise and aging on mitochondrial protein quality control. Exp. Gerontol. 2020, 131, 110824. [Google Scholar] [CrossRef]
- Zhao, Q.; Yu, Z.; Zhang, S.; Shen, X.R.; Yang, H.; Xu, Y.; Liu, Y.; Yang, L.; Zhang, Q.; Chen, J.; et al. Metabolic modeling of single bronchoalveolar macrophages reveals regulators of hyperinflammation in COVID-19. iScience 2022, 25, 105319. [Google Scholar] [CrossRef] [PubMed]
- Icard, P.; Lincet, H.; Wu, Z.; Coquerel, A.; Forgez, P.; Alifano, M.; Fournel, L. The key role of Warburg effect in SARS-CoV-2 replication and associated inflammatory response. Biochimie 2021, 180, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Demmer, R.T. Impaired glucose regulation, SARS-CoV-2 infections and adverse COVID-19 outcomes. Transl. Res. 2022, 241, 52–69. [Google Scholar] [CrossRef] [PubMed]
- Montefusco, L.; Ben Nasr, M.; D’Addio, F.; Loretelli, C.; Rossi, A.; Pastore, I.; Daniele, G.; Abdelsalam, A.; Maestroni, A.; Dell’Acqua, M.; et al. Acute and long-term disruption of glycometabolic control after SARS-CoV-2 infection. Nat. Metab. 2021, 3, 774–785. [Google Scholar] [CrossRef]
- Deme, P.; Rubin, L.H.; Yu, D.; Xu, Y.; Nakigozi, G.; Nakasujja, N.; Anok, A.; Kisakye, A.; Quinn, T.C.; Reynolds, S.J.; et al. Immunometabolic Reprogramming in Response to HIV Infection Is Not Fully Normalized by Suppressive Antiretroviral Therapy. Viruses 2022, 14, 1313. [Google Scholar] [CrossRef]
- Palmer, C.S.; Ostrowski, M.; Gouillou, M.; Tsai, L.; Yu, D.; Zhou, J.; Henstridge, D.C.; Maisa, A.; Hearps, A.C.; Lewin, S.R.; et al. Increased glucose metabolic activity is associated with CD4+ T-cell activation and depletion during chronic HIV infection. AIDS 2014, 28, 297–309. [Google Scholar] [CrossRef]
- Duan, X.; Tang, X.; Nair, M.S.; Zhang, T.; Qiu, Y.; Zhang, W.; Wang, P.; Huang, Y.; Xiang, J.; Wang, H.; et al. An airway organoid-based screen identifies a role for the HIF1alpha-glycolysis axis in SARS-CoV-2 infection. Cell Rep. 2021, 37, 109920. [Google Scholar] [CrossRef]
- Bojkova, D.; Klann, K.; Koch, B.; Widera, M.; Krause, D.; Ciesek, S.; Cinatl, J.; Munch, C. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature 2020, 583, 469–472. [Google Scholar] [CrossRef]
- Codo, A.C.; Davanzo, G.G.; Monteiro, L.B.; de Souza, G.F.; Muraro, S.P.; Virgilio-da-Silva, J.V.; Prodonoff, J.S.; Carregari, V.C.; de Biagi, C.A.O., Jr.; Crunfli, F.; et al. Elevated Glucose Levels Favor SARS-CoV-2 Infection and Monocyte Response through a HIF-1alpha/Glycolysis-Dependent Axis. Cell Metab. 2020, 32, 437–446.e5. [Google Scholar] [CrossRef]
- Rahman, A.N.; Liu, J.; Mujib, S.; Kidane, S.; Ali, A.; Szep, S.; Han, C.; Bonner, P.; Parsons, M.; Benko, E.; et al. Elevated glycolysis imparts functional ability to CD8(+) T cells in HIV infection. Life Sci. Alliance 2021, 4, e202101081. [Google Scholar] [CrossRef]
- Valle-Casuso, J.C.; Angin, M.; Volant, S.; Passaes, C.; Monceaux, V.; Mikhailova, A.; Bourdic, K.; Avettand-Fenoel, V.; Boufassa, F.; Sitbon, M.; et al. Cellular Metabolism Is a Major Determinant of HIV-1 Reservoir Seeding in CD4(+) T Cells and Offers an Opportunity to Tackle Infection. Cell Metab. 2019, 29, 611–626.e5. [Google Scholar] [CrossRef]
- Bengsch, B.; Johnson, A.L.; Kurachi, M.; Odorizzi, P.M.; Pauken, K.E.; Attanasio, J.; Stelekati, E.; McLane, L.M.; Paley, M.A.; Delgoffe, G.M.; et al. Bioenergetic Insufficiencies Due to Metabolic Alterations Regulated by the Inhibitory Receptor PD-1 Are an Early Driver of CD8(+) T Cell Exhaustion. Immunity 2016, 45, 358–373. [Google Scholar] [CrossRef]
- Cao, J.; Liao, S.; Zeng, F.; Liao, Q.; Luo, G.; Zhou, Y. Effects of altered glycolysis levels on CD8(+) T cell activation and function. Cell Death Dis. 2023, 14, 407. [Google Scholar] [CrossRef]
- Loucif, H.; Dagenais-Lussier, X.; Beji, C.; Telittchenko, R.; Routy, J.P.; van Grevenynghe, J. Plasticity in T-cell mitochondrial metabolism: A necessary peacekeeper during the troubled times of persistent HIV-1 infection. Cytokine Growth Factor Rev. 2020, 55, 26–36. [Google Scholar] [CrossRef]
- Wolf, A.M. MtDNA mutations and aging-not a closed case after all? Signal Transduct. Target. Ther. 2021, 6, 56. [Google Scholar] [CrossRef]
- Kauppila, T.E.S.; Kauppila, J.H.K.; Larsson, N.G. Mammalian Mitochondria and Aging: An Update. Cell Metab. 2017, 25, 57–71. [Google Scholar] [CrossRef] [PubMed]
- Foo, J.; Bellot, G.; Pervaiz, S.; Alonso, S. Mitochondria-mediated oxidative stress during viral infection. Trends Microbiol. 2022, 30, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Saavedra, D.; Ane-Kouri, A.L.; Barzilai, N.; Caruso, C.; Cho, K.H.; Fontana, L.; Franceschi, C.; Frasca, D.; Ledon, N.; Niedernhofer, L.J.; et al. Aging and chronic inflammation: Highlights from a multidisciplinary workshop. Immun. Ageing 2023, 20, 25. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G.; Lewin, S.R.; Havlir, D.V. The end of AIDS: HIV infection as a chronic disease. Lancet 2013, 382, 1525–1533. [Google Scholar] [CrossRef]
- Deeks, S.G.; Tracy, R.; Douek, D.C. Systemic effects of inflammation on health during chronic HIV infection. Immunity 2013, 39, 633–645. [Google Scholar] [CrossRef]
- Bohmwald, K.; Diethelm-Varela, B.; Rodriguez-Guilarte, L.; Rivera, T.; Riedel, C.A.; Gonzalez, P.A.; Kalergis, A.M. Pathophysiological, immunological, and inflammatory features of long COVID. Front. Immunol. 2024, 15, 1341600. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef]
- McArthur, K.; Whitehead, L.W.; Heddleston, J.M.; Li, L.; Padman, B.S.; Oorschot, V.; Geoghegan, N.D.; Chappaz, S.; Davidson, S.; San Chin, H.; et al. BAK/BAX macropores facilitate mitochondrial herniation and mtDNA efflux during apoptosis. Science 2018, 359, eaao6047. [Google Scholar] [CrossRef]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef]
- Tilokani, L.; Nagashima, S.; Paupe, V.; Prudent, J. Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 2018, 62, 341–360. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018, 20, 1013–1022. [Google Scholar] [CrossRef]
- Passarella, S.; Atlante, A.; Valenti, D.; de Bari, L. The role of mitochondrial transport in energy metabolism. Mitochondrion 2003, 2, 319–343. [Google Scholar] [CrossRef]
- Ojeda, D.S.; Grasso, D.; Urquiza, J.; Till, A.; Vaccaro, M.I.; Quarleri, J. Cell Death Is Counteracted by Mitophagy in HIV-Productively Infected Astrocytes but Is Promoted by Inflammasome Activation Among Non-productively Infected Cells. Front. Immunol. 2018, 9, 2633. [Google Scholar] [CrossRef]
- Onishi, M.; Yamano, K.; Sato, M.; Matsuda, N.; Okamoto, K. Molecular mechanisms and physiological functions of mitophagy. EMBO J. 2021, 40, e104705. [Google Scholar] [CrossRef]
- Picca, A.; Faitg, J.; Auwerx, J.; Ferrucci, L.; D’Amico, D. Mitophagy in human health, ageing and disease. Nat. Metab. 2023, 5, 2047–2061. [Google Scholar] [CrossRef] [PubMed]
- van der Heijden, W.A.; van de Wijer, L.; Jaeger, M.; Grintjes, K.; Netea, M.G.; Urbanus, R.T.; van Crevel, R.; van den Heuvel, L.P.; Brink, M.; Rodenburg, R.J.; et al. Long-term treated HIV infection is associated with platelet mitochondrial dysfunction. Sci. Rep. 2021, 11, 6246. [Google Scholar] [CrossRef] [PubMed]
- Cesari, R.; Martin, E.S.; Calin, G.A.; Pentimalli, F.; Bichi, R.; McAdams, H.; Trapasso, F.; Drusco, A.; Shimizu, M.; Masciullo, V.; et al. Parkin, a gene implicated in autosomal recessive juvenile parkinsonism, is a candidate tumor suppressor gene on chromosome 6q25-q27. Proc. Natl. Acad. Sci. USA 2003, 100, 5956–5961. [Google Scholar] [CrossRef]
- Drummond, M.J.; Addison, O.; Brunker, L.; Hopkins, P.N.; McClain, D.A.; LaStayo, P.C.; Marcus, R.L. Downregulation of E3 ubiquitin ligases and mitophagy-related genes in skeletal muscle of physically inactive, frail older women: A cross-sectional comparison. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 1040–1048. [Google Scholar] [CrossRef] [PubMed]
- Veeriah, S.; Taylor, B.S.; Meng, S.; Fang, F.; Yilmaz, E.; Vivanco, I.; Janakiraman, M.; Schultz, N.; Hanrahan, A.J.; Pao, W.; et al. Somatic mutations of the Parkinson’s disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat. Genet. 2010, 42, 77–82. [Google Scholar] [CrossRef]
- Kerr, J.S.; Adriaanse, B.A.; Greig, N.H.; Mattson, M.P.; Cader, M.Z.; Bohr, V.A.; Fang, E.F. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci. 2017, 40, 151–166. [Google Scholar] [CrossRef]
- Lou, G.; Palikaras, K.; Lautrup, S.; Scheibye-Knudsen, M.; Tavernarakis, N.; Fang, E.F. Mitophagy and Neuroprotection. Trends Mol. Med. 2020, 26, 8–20. [Google Scholar] [CrossRef]
- Hoshino, A.; Mita, Y.; Okawa, Y.; Ariyoshi, M.; Iwai-Kanai, E.; Ueyama, T.; Ikeda, K.; Ogata, T.; Matoba, S. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat. Commun. 2013, 4, 2308. [Google Scholar] [CrossRef]
- Garcia-Prat, L.; Martinez-Vicente, M.; Perdiguero, E.; Ortet, L.; Rodriguez-Ubreva, J.; Rebollo, E.; Ruiz-Bonilla, V.; Gutarra, S.; Ballestar, E.; Serrano, A.L.; et al. Autophagy maintains stemness by preventing senescence. Nature 2016, 529, 37–42. [Google Scholar] [CrossRef]
- Wang, S.; Long, H.; Hou, L.; Feng, B.; Ma, Z.; Wu, Y.; Zeng, Y.; Cai, J.; Zhang, D.W.; Zhao, G. The mitophagy pathway and its implications in human diseases. Signal Transduct. Target. Ther. 2023, 8, 304. [Google Scholar] [CrossRef]
- Baker, D.J.; Betik, A.C.; Krause, D.J.; Hepple, R.T. No decline in skeletal muscle oxidative capacity with aging in long-term calorically restricted rats: Effects are independent of mitochondrial DNA integrity. J. Gerontol. A Biol. Sci. Med. Sci. 2006, 61, 675–684. [Google Scholar] [CrossRef]
- Hepple, R.T.; Baker, D.J.; McConkey, M.; Murynka, T.; Norris, R. Caloric restriction protects mitochondrial function with aging in skeletal and cardiac muscles. Rejuven. Res. 2006, 9, 219–222. [Google Scholar] [CrossRef]
- Liu, Y.J.; McIntyre, R.L.; Janssens, G.E.; Houtkooper, R.H. Mitochondrial fission and fusion: A dynamic role in aging and potential target for age-related disease. Mech. Ageing Dev. 2020, 186, 111212. [Google Scholar] [CrossRef] [PubMed]
- Fields, J.A.; Serger, E.; Campos, S.; Divakaruni, A.S.; Kim, C.; Smith, K.; Trejo, M.; Adame, A.; Spencer, B.; Rockenstein, E.; et al. HIV alters neuronal mitochondrial fission/fusion in the brain during HIV-associated neurocognitive disorders. Neurobiol. Dis. 2016, 86, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Shirihai, O.S. The interplay between mitochondrial dynamics and mitophagy. Antioxid. Redox Signal. 2011, 14, 1939–1951. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Lee, H.; Chanda, D.; Thoudam, T.; Kang, H.J.; Harris, R.A.; Lee, I.K. The Role of Pyruvate Metabolism in Mitochondrial Quality Control and Inflammation. Mol. Cells 2023, 46, 259–267. [Google Scholar] [CrossRef]
- Kim, J.S.; Lee, S.; Kim, W.K.; Han, B.S. Mitochondrial transplantation: An overview of a promising therapeutic approach. BMB Rep. 2023, 56, 488–495. [Google Scholar] [CrossRef]
- McCully, J.D.; Cowan, D.B.; Pacak, C.A.; Toumpoulis, I.K.; Dayalan, H.; Levitsky, S. Injection of isolated mitochondria during early reperfusion for cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H94–H105. [Google Scholar] [CrossRef]
- Kaza, A.K.; Wamala, I.; Friehs, I.; Kuebler, J.D.; Rathod, R.H.; Berra, I.; Ericsson, M.; Yao, R.; Thedsanamoorthy, J.K.; Zurakowski, D.; et al. Myocardial rescue with autologous mitochondrial transplantation in a porcine model of ischemia/reperfusion. J. Thorac. Cardiovasc. Surg. 2017, 153, 934–943. [Google Scholar] [CrossRef]
- Guariento, A.; Piekarski, B.L.; Doulamis, I.P.; Blitzer, D.; Ferraro, A.M.; Harrild, D.M.; Zurakowski, D.; Del Nido, P.J.; McCully, J.D.; Emani, S.M. Autologous mitochondrial transplantation for cardiogenic shock in pediatric patients following ischemia-reperfusion injury. J. Thorac. Cardiovasc. Surg. 2021, 162, 992–1001. [Google Scholar] [CrossRef] [PubMed]
- Emani, S.M.; Piekarski, B.L.; Harrild, D.; Del Nido, P.J.; McCully, J.D. Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury. J. Thorac. Cardiovasc. Surg. 2017, 154, 286–289. [Google Scholar] [CrossRef] [PubMed]
- Taner, O.F.; Ulger, O.; Ersahin, S.; Baser, N.T.; Genc, O.; Kubat, G.B. Effects of mitochondrial transplantation on chronic pressure wound healing in a human patient. Cytotherapy 2024, 26, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Baharvand, F.; Habibi Roudkenar, M.; Pourmohammadi-Bejarpasi, Z.; Najafi-Ghalehlou, N.; Feizkhah, A.; Bashiri Aliabadi, S.; Salari, A.; Mohammadi Roushandeh, A. Safety and efficacy of platelet-derived mitochondrial transplantation in ischaemic heart disease. Int. J. Cardiol. 2024, 410, 132227. [Google Scholar] [CrossRef]
- Lin, R.Z.; Im, G.B.; Luo, A.C.; Zhu, Y.; Hong, X.; Neumeyer, J.; Tang, H.W.; Perrimon, N.; Melero-Martin, J.M. Mitochondrial transfer mediates endothelial cell engraftment through mitophagy. Nature 2024, 629, 660–668. [Google Scholar] [CrossRef]
- Baldwin, J.G.; Heuser-Loy, C.; Saha, T.; Schelker, R.C.; Slavkovic-Lukic, D.; Strieder, N.; Hernandez-Lopez, I.; Rana, N.; Barden, M.; Mastrogiovanni, F.; et al. Intercellular nanotube-mediated mitochondrial transfer enhances T cell metabolic fitness and antitumor efficacy. Cell 2024, 187, 6614–6630.e21. [Google Scholar] [CrossRef]
- Liu, Y.; Dissanayaka, W.L.; Yiu, C. Therapeutic implications of mitochondrial transfer on stem cell fate in regenerative medicine. J. Transl. Med. 2025, 23, 568. [Google Scholar] [CrossRef]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA heteroplasmy in disease and targeted nuclease-based therapeutic approaches. EMBO Rep. 2020, 21, e49612. [Google Scholar] [CrossRef]
- Lim, K. Mitochondrial genome editing: Strategies, challenges, and applications. BMB Rep. 2024, 57, 19–29. [Google Scholar] [CrossRef]
- Brestoff, J.R.; Singh, K.K.; Aquilano, K.; Becker, L.B.; Berridge, M.V.; Boilard, E.; Caicedo, A.; Crewe, C.; Enriquez, J.A.; Gao, J.; et al. Recommendations for mitochondria transfer and transplantation nomenclature and characterization. Nat. Metab. 2025, 7, 53–67. [Google Scholar] [CrossRef]
- Borcherding, N.; Jia, W.; Giwa, R.; Field, R.L.; Moley, J.R.; Kopecky, B.J.; Chan, M.M.; Yang, B.Q.; Sabio, J.M.; Walker, E.C.; et al. Dietary lipids inhibit mitochondria transfer to macrophages to divert adipocyte-derived mitochondria into the blood. Cell Metab. 2022, 34, 1499–1513.e8. [Google Scholar] [CrossRef]
- Lin, M.W.; Fang, S.Y.; Hsu, J.C.; Huang, C.Y.; Lee, P.H.; Huang, C.C.; Chen, H.F.; Lam, C.F.; Lee, J.S. Mitochondrial Transplantation Attenuates Neural Damage and Improves Locomotor Function After Traumatic Spinal Cord Injury in Rats. Front. Neurosci. 2022, 16, 800883. [Google Scholar] [CrossRef] [PubMed]
- Huynh, K. Artificially transplanted mitochondria in endothelial cells promote mitophagy. Nat. Rev. Cardiol. 2024, 21, 439. [Google Scholar] [CrossRef] [PubMed]
- Nicolas-Avila, J.A.; Lechuga-Vieco, A.V.; Esteban-Martinez, L.; Sanchez-Diaz, M.; Diaz-Garcia, E.; Santiago, D.J.; Rubio-Ponce, A.; Li, J.L.; Balachander, A.; Quintana, J.A.; et al. A Network of Macrophages Supports Mitochondrial Homeostasis in the Heart. Cell 2020, 183, 94–109.e23. [Google Scholar] [CrossRef]
- Babenko, V.A.; Silachev, D.N.; Popkov, V.A.; Zorova, L.D.; Pevzner, I.B.; Plotnikov, E.Y.; Sukhikh, G.T.; Zorov, D.B. Miro1 Enhances Mitochondria Transfer from Multipotent Mesenchymal Stem Cells (MMSC) to Neural Cells and Improves the Efficacy of Cell Recovery. Molecules 2018, 23, 687. [Google Scholar] [CrossRef]
- Yu, M.; Wang, D.; Chen, X.; Zhong, D.; Luo, J. BMSCs-derived Mitochondria Improve Osteoarthritis by Ameliorating Mitochondrial Dysfunction and Promoting Mitochondrial Biogenesis in Chondrocytes. Stem Cell Rev. Rep. 2022, 18, 3092–3111. [Google Scholar] [CrossRef]
- Kim, Y.S.; Yoo, S.; Jung, Y.J.; Yoon, J.W.; Kwon, Y.S.; Lee, N.; Cheon, C.K.; Kim, J.H. Allogenic mitochondria transfer improves cardiac function in iPS-cell-differentiated cardiomyocytes of a patient with Barth syndrome. Exp. Mol. Med. 2025, 57, 1260–1271. [Google Scholar] [CrossRef]
- Yamada, Y.; Ito, M.; Arai, M.; Hibino, M.; Tsujioka, T.; Harashima, H. Challenges in Promoting Mitochondrial Transplantation Therapy. Int. J. Mol. Sci. 2020, 21, 6365. [Google Scholar] [CrossRef]
- Huang, T.; Zhang, T.; Gao, J. Targeted mitochondrial delivery: A therapeutic new era for disease treatment. J. Control. Release 2022, 343, 89–106. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Margreiter, R. Heterogeneity of mitochondria and mitochondrial function within cells as another level of mitochondrial complexity. Int. J. Mol. Sci. 2009, 10, 1911–1929. [Google Scholar] [CrossRef]
- Iorio, R.; Petricca, S.; Di Emidio, G.; Falone, S.; Tatone, C. Mitochondrial Extracellular Vesicles (mitoEVs): Emerging mediators of cell-to-cell communication in health, aging and age-related diseases. Ageing Res. Rev. 2024, 101, 102522. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Li, J.; Zhang, J.; Li, P.; Bai, H.; Fang, B.; Fang, H.; Huang, K.; Wang, G.; Nowell, C.J.; et al. Mitochondrial segmentation and function prediction in live-cell images with deep learning. Nat. Commun. 2025, 16, 743. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.C.; Bergamini, C.; Fato, R.; Pon, L.A.; Pallotti, F. Isolation of mitochondria from cells and tissues. Methods Cell Biol. 2020, 155, 3–31. [Google Scholar] [CrossRef] [PubMed]
- Bodenstein, D.F.; Powlowski, P.; Zachos, K.A.; El Soufi El Sabbagh, D.; Jeong, H.; Attisano, L.; Edgar, L.; Wallace, D.C.; Andreazza, A.C. Optimization of differential filtration-based mitochondrial isolation for mitochondrial transplant to cerebral organoids. Stem Cell Res. Ther. 2023, 14, 202. [Google Scholar] [CrossRef]
- Zhang, A.; Liu, Y.; Pan, J.; Pontanari, F.; Chia-Hao Chang, A.; Wang, H.; Gao, S.; Wang, C.; Chang, A.C. Delivery of mitochondria confers cardioprotection through mitochondria replenishment and metabolic compliance. Mol. Ther. 2023, 31, 1468–1479. [Google Scholar] [CrossRef]
- Lin, H.C.; Lai, I.R. Isolated mitochondria infusion mitigates ischemia-reperfusion injury of the liver in rats: Reply. Shock 2013, 39, 543. [Google Scholar] [CrossRef]
- Chen, T.; Zhu, Y.; Jia, J.; Meng, H.; Xu, C.; Xian, P.; Li, Z.; Tang, Z.; Wu, Y.; Liu, Y. Mitochondrial Transplantation Promotes Remyelination and Long-Term Locomotion Recovery following Cerebral Ischemia. Mediat. Inflamm. 2022, 2022, 1346343. [Google Scholar] [CrossRef]
- Bamshad, C.; Habibi Roudkenar, M.; Abedinzade, M.; Yousefzadeh Chabok, S.; Pourmohammadi-Bejarpasi, Z.; Najafi-Ghalehlou, N.; Sato, T.; Tomita, K.; Jahanian-Najafabadi, A.; Feizkhah, A.; et al. Human umbilical cord-derived mesenchymal stem cells-harvested mitochondrial transplantation improved motor function in TBI models through rescuing neuronal cells from apoptosis and alleviating astrogliosis and microglia activation. Int. Immunopharmacol. 2023, 118, 110106. [Google Scholar] [CrossRef]
- Hayashida, K.; Takegawa, R.; Endo, Y.; Yin, T.; Choudhary, R.C.; Aoki, T.; Nishikimi, M.; Murao, A.; Nakamura, E.; Shoaib, M.; et al. Exogenous mitochondrial transplantation improves survival and neurological outcomes after resuscitation from cardiac arrest. BMC Med. 2023, 21, 56. [Google Scholar] [CrossRef]
- Chen, T.H.; Chang, C.J.; Hung, P.H. Possible Pathogenesis and Prevention of Long COVID: SARS-CoV-2-Induced Mitochondrial Disorder. Int. J. Mol. Sci. 2023, 24, 8034. [Google Scholar] [CrossRef]
- Chang, X.; Ismail, N.I.; Rahman, A.; Xu, D.; Chan, R.W.Y.; Ong, S.G.; Ong, S.B. Long COVID-19 and the Heart: Is Cardiac Mitochondria the Missing Link? Antioxid. Redox Signal. 2023, 38, 599–618. [Google Scholar] [CrossRef]
- Srinivasan, K.; Pandey, A.K.; Livingston, A.; Venkatesh, S. Roles of host mitochondria in the development of COVID-19 pathology: Could mitochondria be a potential therapeutic target? Mol. Biomed. 2021, 2, 38. [Google Scholar] [CrossRef]
- Ryback, R.; Eirin, A. Mitochondria, a Missing Link in COVID-19 Heart Failure and Arrest? Front. Cardiovasc. Med. 2021, 8, 830024. [Google Scholar] [CrossRef] [PubMed]
- Masters, M.C.; Landay, A.L.; Robbins, P.D.; Tchkonia, T.; Kirkland, J.L.; Kuchel, G.A.; Niedernhofer, L.J.; Palella, F.J. Chronic HIV Infection and Aging: Application of a Geroscience-Guided Approach. J. Acquir. Immune Defic. Syndr. 2022, 89, S34–S46. [Google Scholar] [CrossRef] [PubMed]
- Pinti, M.; Salomoni, P.; Cossarizza, A. Anti-HIV drugs and the mitochondria. Biochim. Biophys. Acta 2006, 1757, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Zhang, W.; Wu, S.; Qi, K.; Zhu, S.; Zhang, X.; Chen, Y.; Chen, X.; Li, Y.; Liu, F.; et al. Mitochondrial transplantation reconstructs the oxidative microenvironment within fibroblasts to reverse photoaging. Biochem. Biophys. Res. Commun. 2025, 745, 151214. [Google Scholar] [CrossRef]
- Hwang, J.W.; Lee, M.J.; Chung, T.N.; Lee, H.A.R.; Lee, J.H.; Choi, S.Y.; Park, Y.J.; Kim, C.H.; Jin, I.; Kim, S.H.; et al. The immune modulatory effects of mitochondrial transplantation on cecal slurry model in rat. Crit. Care 2021, 25, 20. [Google Scholar] [CrossRef]
- Aoki, T.; Endo, Y.; Nakamura, E.; Kuschner, C.E.; Kazmi, J.; Singh, P.; Yin, T.; Becker, L.B.; Hayashida, K. Therapeutic potential of mitochondrial transplantation in modulating immune responses post-cardiac arrest: A narrative review. J. Transl. Med. 2024, 22, 230. [Google Scholar] [CrossRef]
- Eo, H.; Yu, S.H.; Choi, Y.; Kim, Y.; Kang, Y.C.; Lee, H.; Kim, J.H.; Han, K.; Lee, H.K.; Chang, M.Y.; et al. Mitochondrial transplantation exhibits neuroprotective effects and improves behavioral deficits in an animal model of Parkinson’s disease. Neurotherapeutics 2024, 21, e00355. [Google Scholar] [CrossRef]
- Paiardini, M.; Muller-Trutwin, M. HIV-associated chronic immune activation. Immunol. Rev. 2013, 254, 78–101. [Google Scholar] [CrossRef]
- Tandon, P.; Abrams, N.D.; Avula, L.R.; Carrick, D.M.; Chander, P.; Divi, R.L.; Dwyer, J.T.; Gannot, G.; Gordiyenko, N.; Liu, Q.; et al. Unraveling Links between Chronic Inflammation and Long COVID: Workshop Report. J. Immunol. 2024, 212, 505–512. [Google Scholar] [CrossRef]
- The PHOSP-COVID Collaborative Group. Clinical characteristics with inflammation profiling of long COVID and association with 1-year recovery following hospitalisation in the UK: A prospective observational study. Lancet Respir. Med. 2022, 10, 761–775. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Hwang, J.W.; Kim, M.J.; Jung, S.Y.; Kim, K.S.; Ahn, E.H.; Min, K.; Choi, Y.S. Mitochondrial Transplantation Modulates Inflammation and Apoptosis, Alleviating Tendinopathy Both In Vivo and In Vitro. Antioxidants 2021, 10, 696. [Google Scholar] [CrossRef] [PubMed]
- Court, A.C.; Le-Gatt, A.; Luz-Crawford, P.; Parra, E.; Aliaga-Tobar, V.; Batiz, L.F.; Contreras, R.A.; Ortuzar, M.I.; Kurte, M.; Elizondo-Vega, R.; et al. Mitochondrial transfer from MSCs to T cells induces Treg differentiation and restricts inflammatory response. EMBO Rep. 2020, 21, e48052. [Google Scholar] [CrossRef] [PubMed]
- Tolomeo, M.; Cascio, A. The Complex Dysregulations of CD4 T Cell Subtypes in HIV Infection. Int. J. Mol. Sci. 2024, 25, 7512. [Google Scholar] [CrossRef]
- Pokharel, M.D.; Fu, P.; Garcia-Flores, A.; Yegambaram, M.; Lu, Q.; Sun, X.; Unwalla, H.; Aggarwal, S.; Fineman, J.R.; Wang, T.; et al. Inflammatory lung injury is associated with endothelial cell mitochondrial fission and requires the nitration of RhoA and cytoskeletal remodeling. Free Radic. Biol. Med. 2024, 221, 125–135. [Google Scholar] [CrossRef]
- Cloonan, S.M.; Kim, K.; Esteves, P.; Trian, T.; Barnes, P.J. Mitochondrial dysfunction in lung ageing and disease. Eur. Respir. Rev. 2020, 29, 200165. [Google Scholar] [CrossRef]
- Roda, R.H.; Hoke, A. Mitochondrial dysfunction in HIV-induced peripheral neuropathy. Int. Rev. Neurobiol. 2019, 145, 67–82. [Google Scholar] [CrossRef]
- Chapman, J.; Fielder, E.; Passos, J.F. Mitochondrial dysfunction and cell senescence: Deciphering a complex relationship. FEBS Lett. 2019, 593, 1566–1579. [Google Scholar] [CrossRef]
- Kang, E.; Kang, C.; Lee, Y.S.; Lee, S.V. Brief guide to senescence assays using cultured mammalian cells. Mol. Cells 2024, 47, 100102. [Google Scholar] [CrossRef]
- Cohen, J.; Torres, C. HIV-associated cellular senescence: A contributor to accelerated aging. Ageing Res. Rev. 2017, 36, 117–124. [Google Scholar] [CrossRef]
- Lin, L.; Xu, H.; Bishawi, M.; Feng, F.; Samy, K.; Truskey, G.; Barbas, A.S.; Kirk, A.D.; Brennan, T.V. Circulating mitochondria in organ donors promote allograft rejection. Am. J. Transplant. 2019, 19, 1917–1929. [Google Scholar] [CrossRef]
- Pollara, J.; Edwards, R.W.; Lin, L.; Bendersky, V.A.; Brennan, T.V. Circulating mitochondria in deceased organ donors are associated with immune activation and early allograft dysfunction. JCI Insight 2018, 3, e121622. [Google Scholar] [CrossRef]
- Dabravolski, S.A.; Khotina, V.A.; Sukhorukov, V.N.; Kalmykov, V.A.; Mikhaleva, L.M.; Orekhov, A.N. The Role of Mitochondrial DNA Mutations in Cardiovascular Diseases. Int. J. Mol. Sci. 2022, 23, 952. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.F.; Tseng, L.M.; Lee, H.C. Role of mitochondrial alterations in human cancer progression and cancer immunity. J. Biomed. Sci. 2023, 30, 61. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Yang, B.; Huang, Y.; Zhang, Y.; Jiang, Y.; Ma, L.; Shen, Y.Q. Mitochondrial DNA-targeted therapy: A novel approach to combat cancer. Cell Insight 2023, 2, 100113. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.H.; Le, Q.; Barboza, R.; Morin, A.; Singh, S.M.; Castellani, C.A. Mitochondrial transplantation: Triumphs, challenges, and impacts on nuclear genome remodelling. Mitochondrion 2025, 84, 102042. [Google Scholar] [CrossRef]
- Blanco, A.; Coronado, R.A.; Arun, N.; Ma, K.; Dar, R.D.; Kieffer, C. Monocyte to macrophage differentiation and changes in cellular redox homeostasis promote cell type-specific HIV latency reactivation. Proc. Natl. Acad. Sci. USA 2024, 121, e2313823121. [Google Scholar] [CrossRef]
- Masuzawa, A.; Black, K.M.; Pacak, C.A.; Ericsson, M.; Barnett, R.J.; Drumm, C.; Seth, P.; Bloch, D.B.; Levitsky, S.; Cowan, D.B.; et al. Transplantation of autologously derived mitochondria protects the heart from ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H966–H982. [Google Scholar] [CrossRef]
- Doulamis, I.P.; Nomoto, R.S.; Tzani, A.; Hong, X.; Duignan, T.; Celik, A.; Del Nido, P.J.; McCully, J.D. Transcriptomic and proteomic pathways of diabetic and non-diabetic mitochondrial transplantation. Sci. Rep. 2022, 12, 22101. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).