

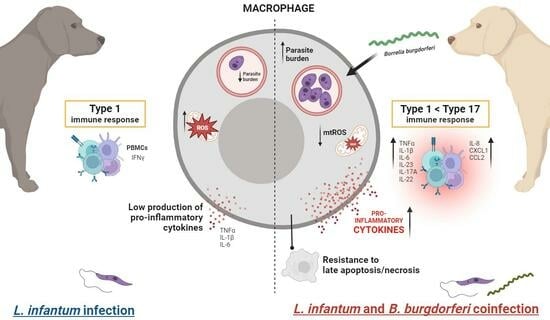
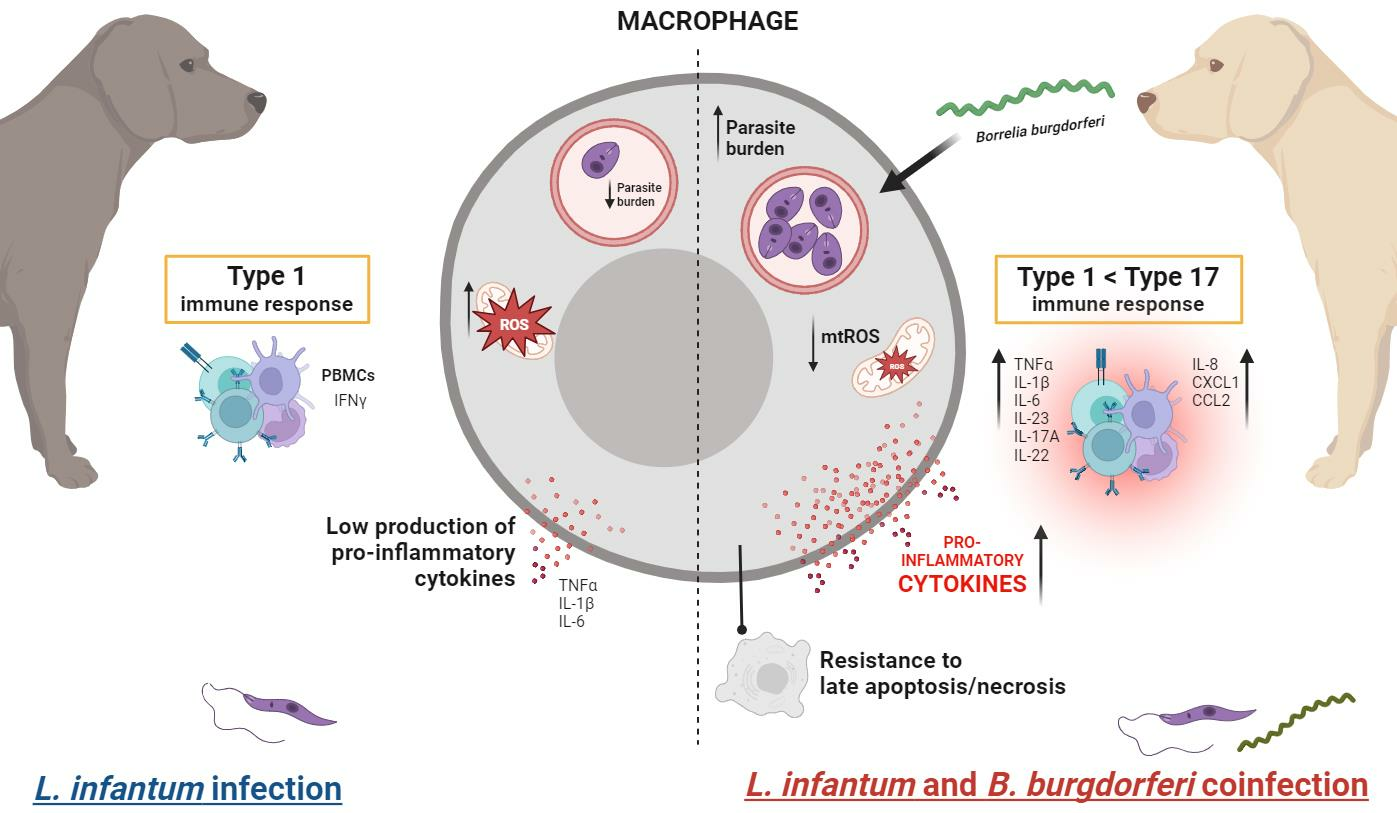
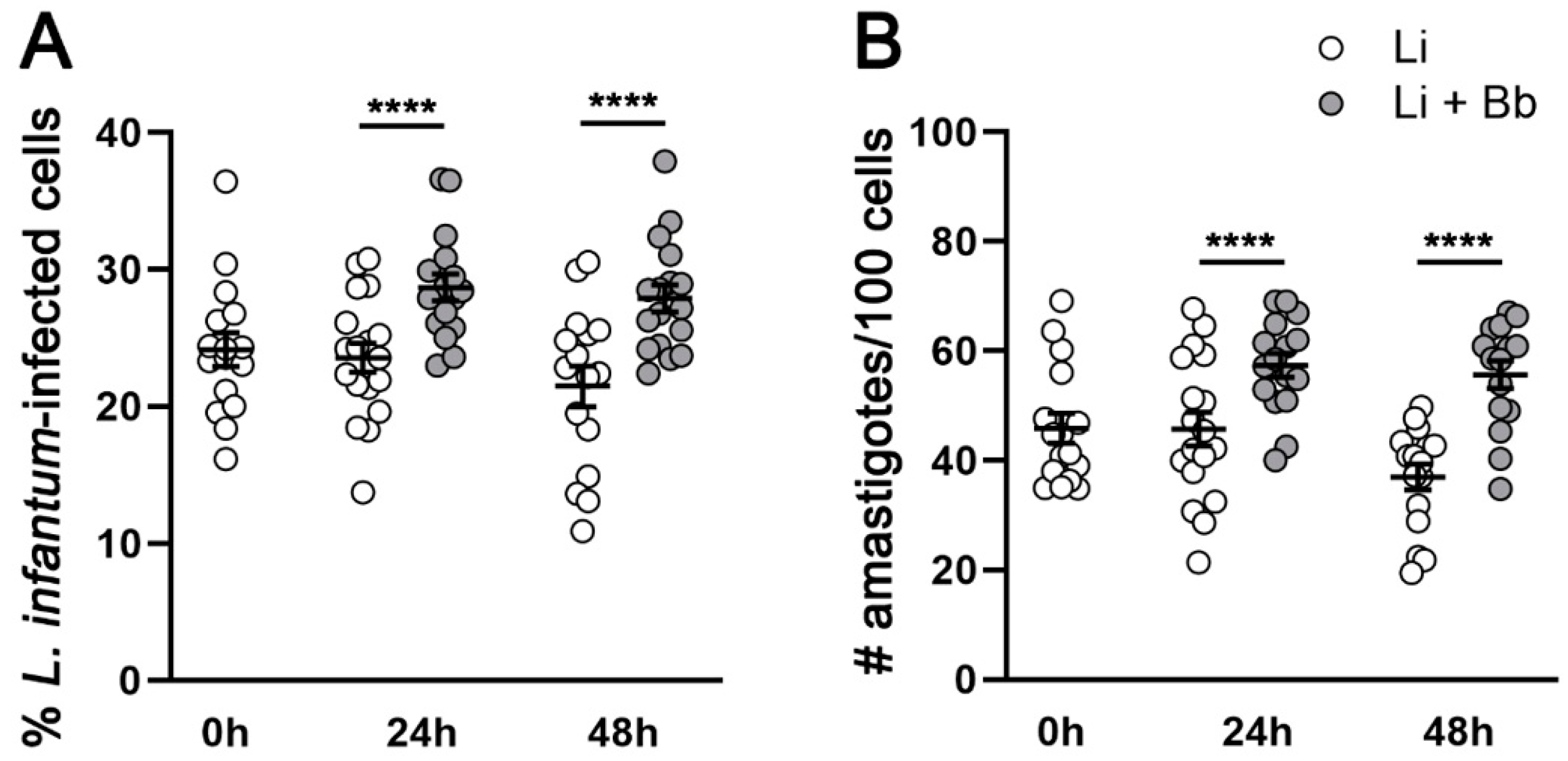
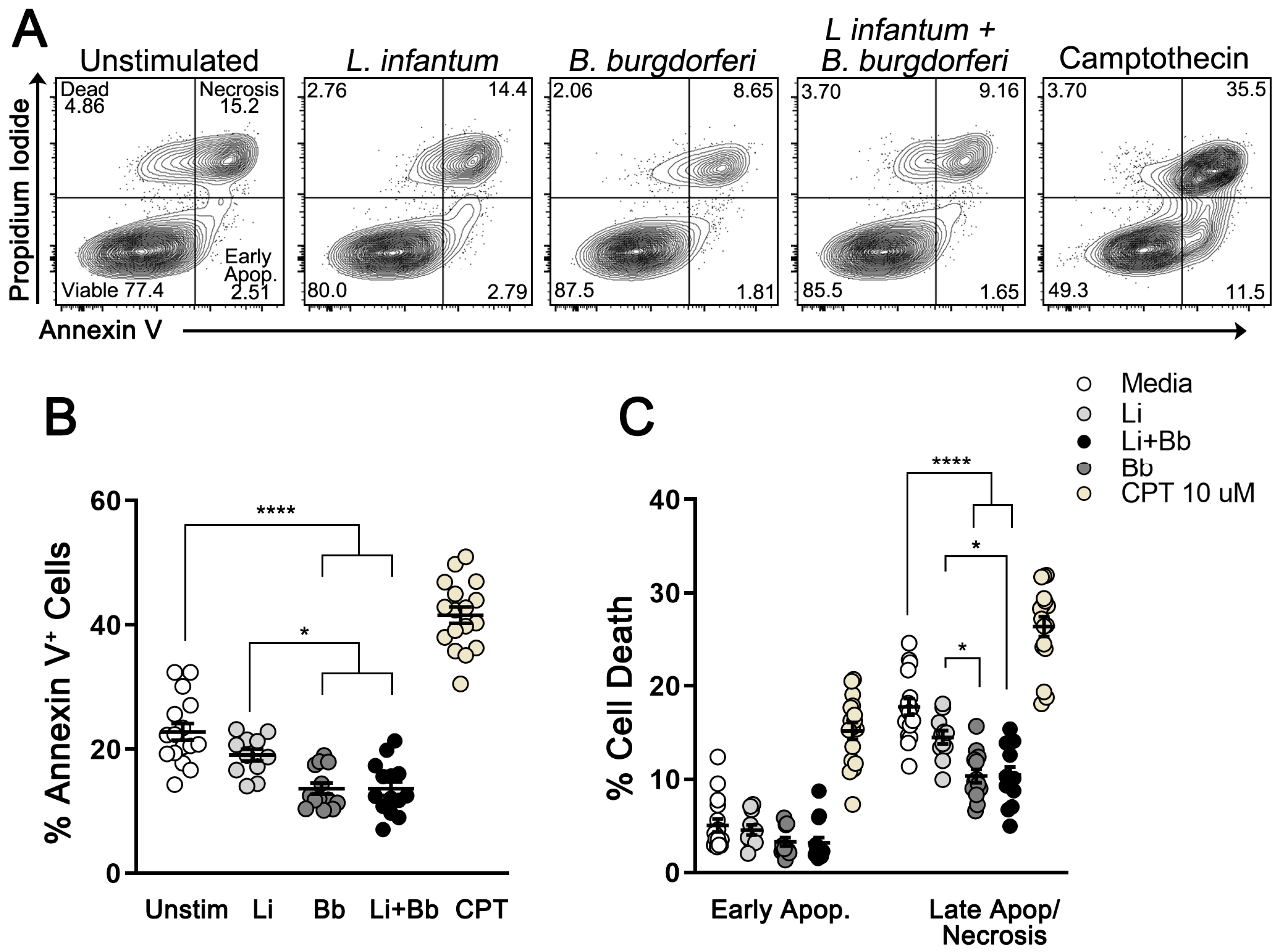
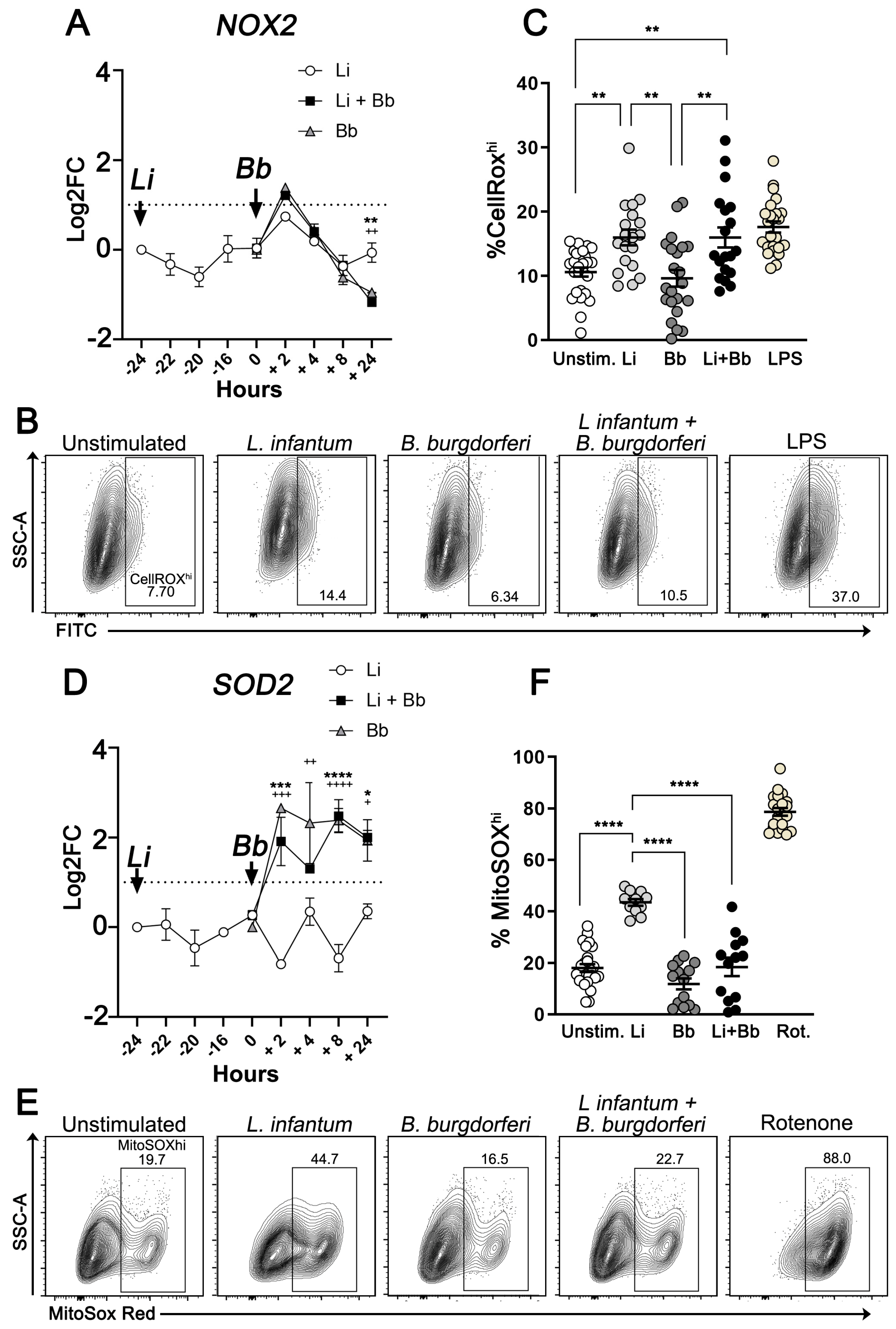
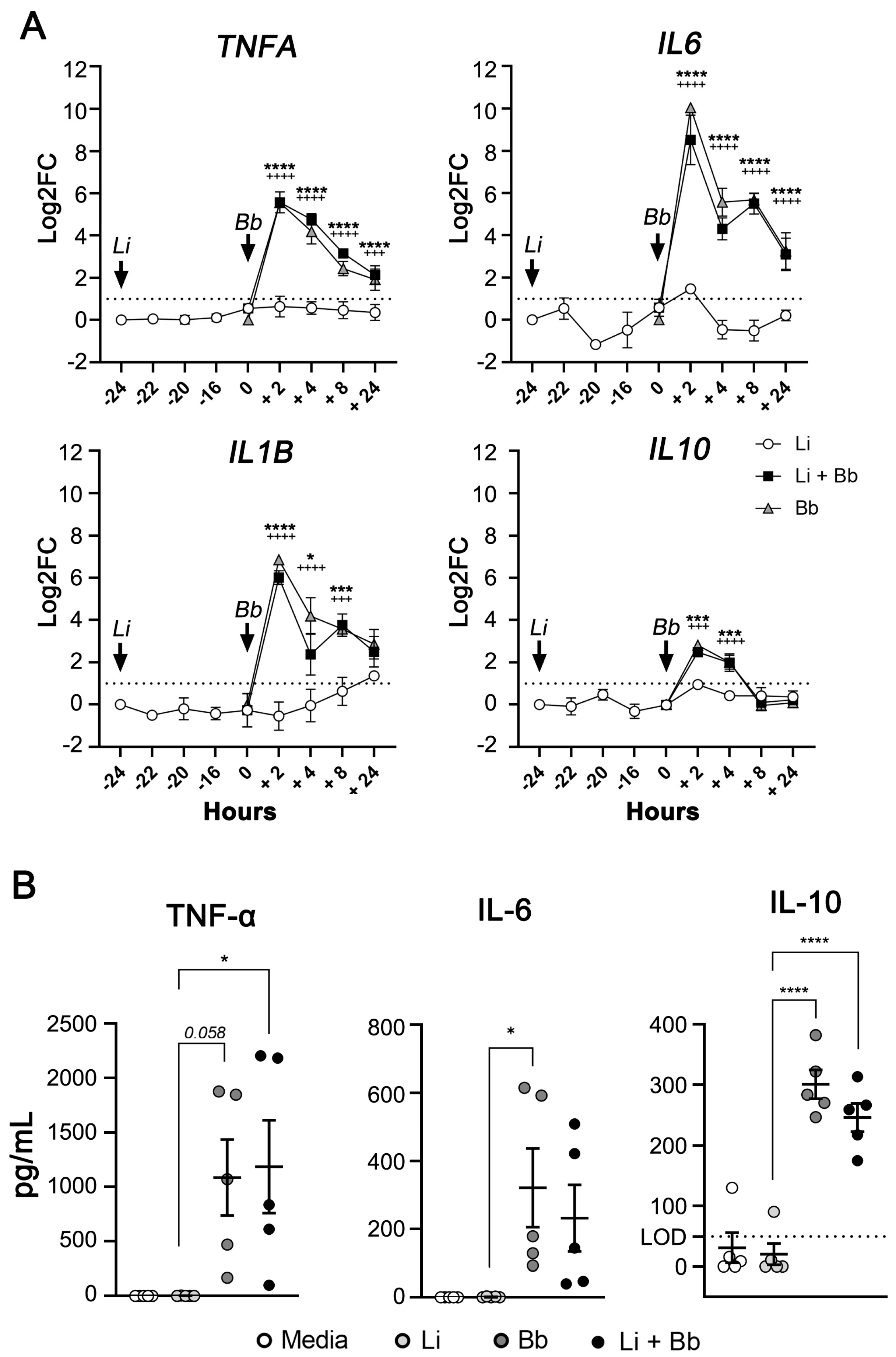
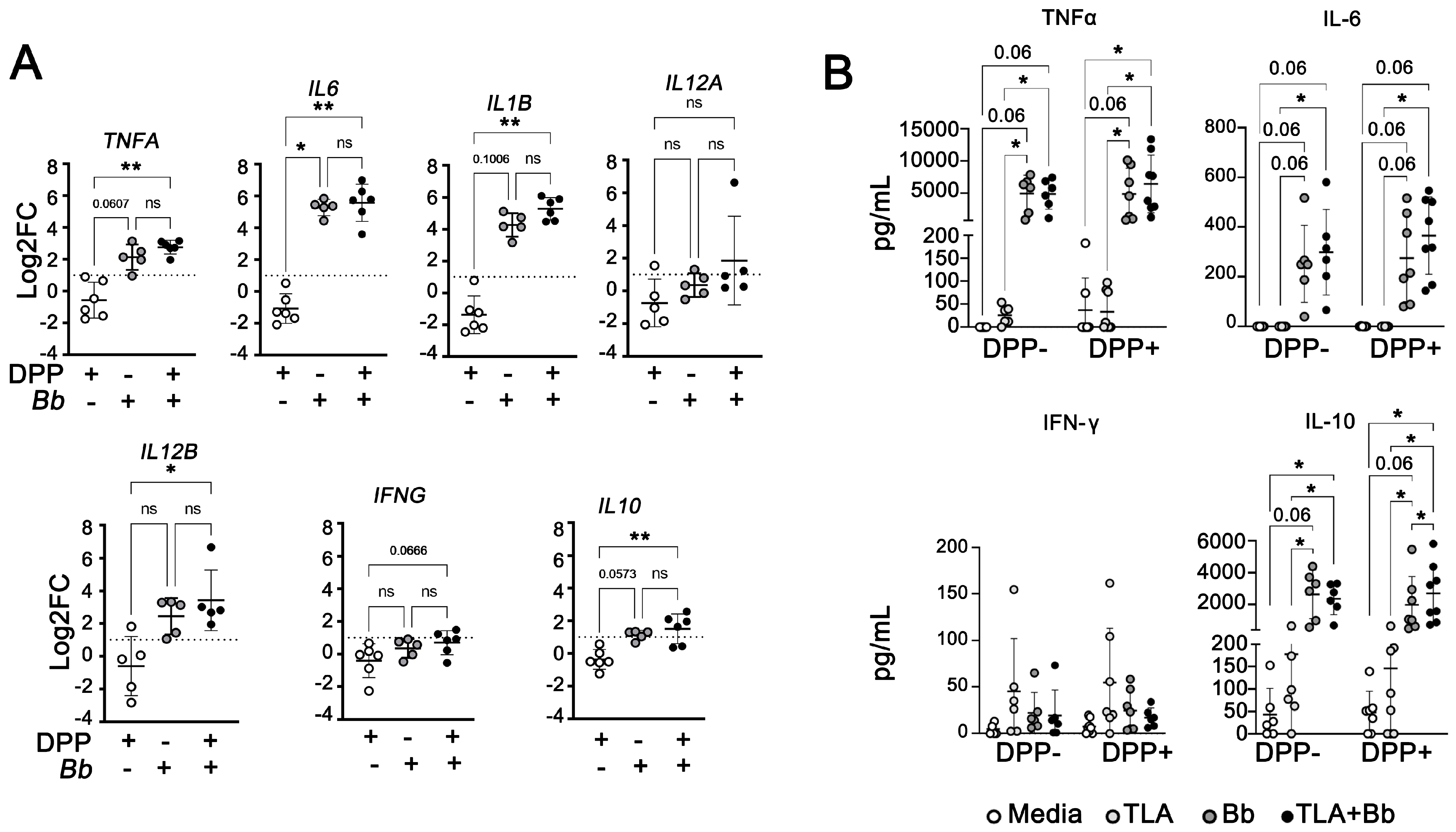
Modulation of Macrophage Redox and Apoptotic Processes to Leishmania infantum during Coinfection with the Tick-Borne Bacteria Borrelia burgdorferi

 ,
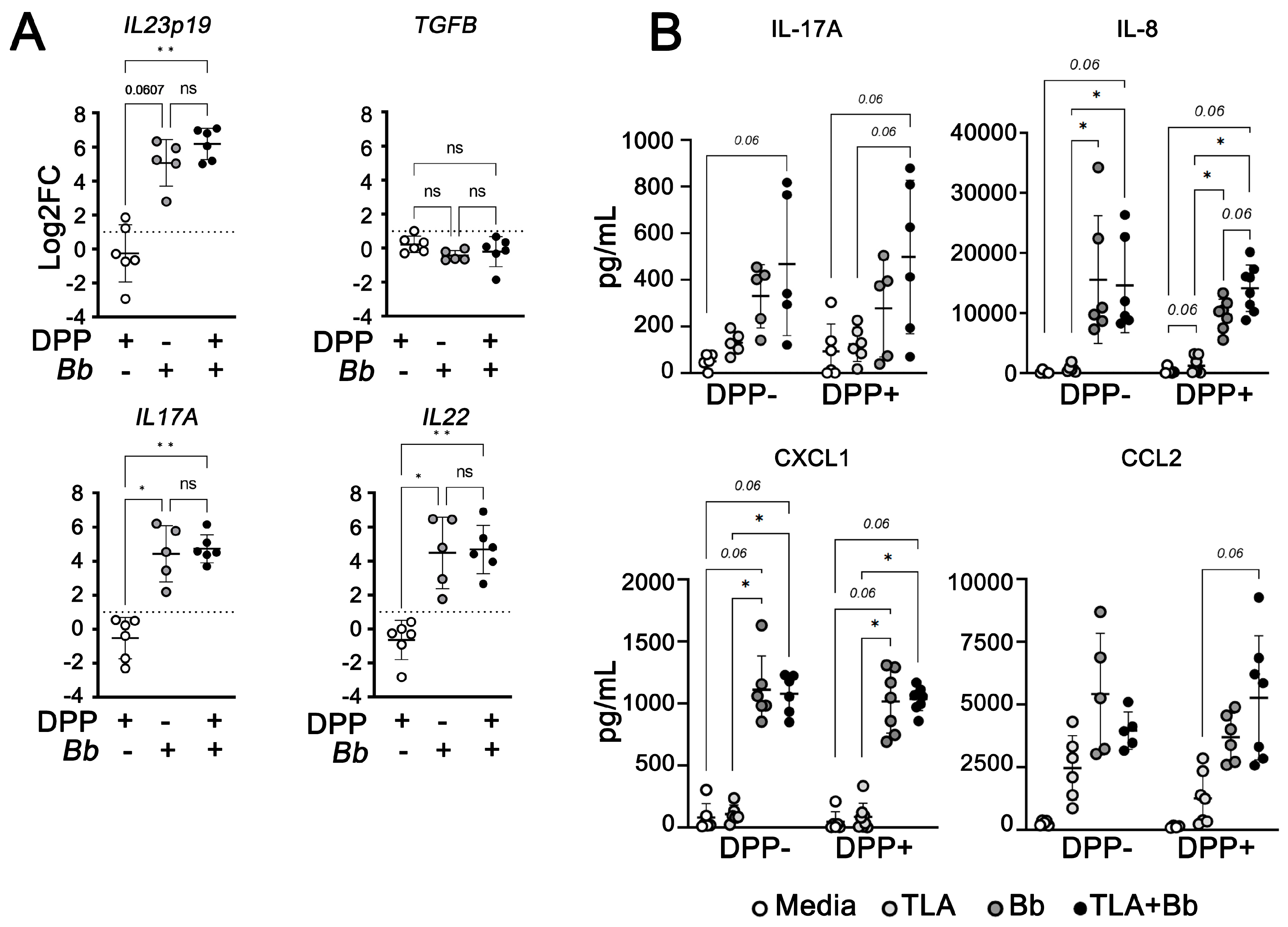
, Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Share and Cite
Pessôa-Pereira, D.; Scorza, B.M.; Cyndari, K.I.; Beasley, E.A.; Petersen, C.A. Modulation of Macrophage Redox and Apoptotic Processes to Leishmania infantum during Coinfection with the Tick-Borne Bacteria Borrelia burgdorferi. Pathogens 2023, 12, 1128. https://doi.org/10.3390/pathogens12091128
Pessôa-Pereira D, Scorza BM, Cyndari KI, Beasley EA, Petersen CA. Modulation of Macrophage Redox and Apoptotic Processes to Leishmania infantum during Coinfection with the Tick-Borne Bacteria Borrelia burgdorferi. Pathogens. 2023; 12(9):1128. https://doi.org/10.3390/pathogens12091128
Chicago/Turabian StylePessôa-Pereira, Danielle, Breanna M. Scorza, Karen I. Cyndari, Erin A. Beasley, and Christine A. Petersen. 2023. "Modulation of Macrophage Redox and Apoptotic Processes to Leishmania infantum during Coinfection with the Tick-Borne Bacteria Borrelia burgdorferi" Pathogens 12, no. 9: 1128. https://doi.org/10.3390/pathogens12091128
APA StylePessôa-Pereira, D., Scorza, B. M., Cyndari, K. I., Beasley, E. A., & Petersen, C. A. (2023). Modulation of Macrophage Redox and Apoptotic Processes to Leishmania infantum during Coinfection with the Tick-Borne Bacteria Borrelia burgdorferi. Pathogens, 12(9), 1128. https://doi.org/10.3390/pathogens12091128