


The Impact of Pseudomonas aeruginosa Infection in Adult Cystic Fibrosis Patients—A Single Polish Centre Study
 ,
,  , , ,
, , ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Clinical Material
2.3. Microbiological Methods of the Pseudomonas aeruginosa Isolates Characteristics
2.3.1. Culture Conditions
2.3.2. Antimicrobial Susceptibility Testing
2.3.3. Proteolytic Activity
2.3.4. Mucoidy Ability
2.3.5. Biofilm Assays
2.3.6. Motility Tests
2.3.7. Genetic Analysis
2.4. Genetic Differences of the Clinical Isolates
2.5. Statistical Analysis and Visualization
3. Results
3.1. Clinical Parameters of the CF Patient Cohort
3.2. Variants Related to CF
3.3. Microbiological Properties of the P. aeruginosa Isolates
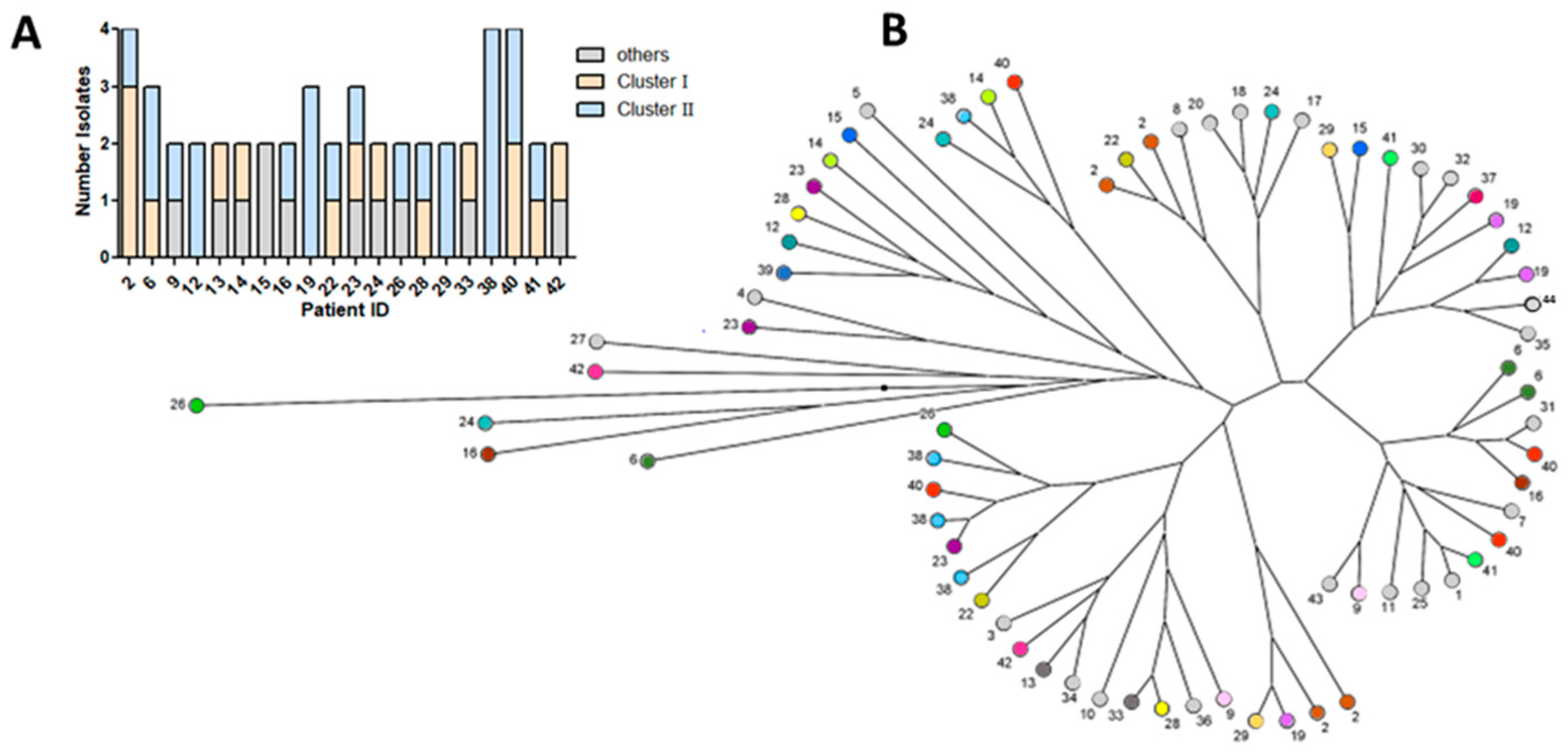
3.4. Genotypic Differences of Clinical P. aeruginosa Isolates
3.5. Correlation of Patients and Pathogen Characteristics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Davies, J.C.; Ebdon, A.M.; Orchard, C. Recent advances in the management of cystic fibrosis. Arch. Dis. Child. 2014, 99, 1033–1036. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, B.P.; Freedman, S.D. Cystic fibrosis. Lancet 2009, 373, 1891–1904. [Google Scholar] [CrossRef]
- Cystic Fibrosis Foundation Patient Registry 2020 Annual Data Report. Available online: https://www.cff.org/sites/default/files/2021-11/Patient-Registry-Annual-Data-Report.pdf (accessed on 14 April 2022).
- Voynow, J.A.; Fischer, B.M.; Zheng, S. Proteases and cystic fibrosis. Int. J. Biochem. Cell Biol. 2008, 40, 1238–1245. [Google Scholar] [CrossRef] [PubMed]
- Kidd, T.J.; Ramsay, K.A.; Vidmar, S.; Carlin, J.B.; Bell, S.C.; Wainwright, C.E.; Grimwood, K.; Francis, P.W.; Dakin, C.; Cheney, J.; et al. Pseudomonas aeruginosa genotypes acquired by children with cystic fibrosis by age 5-years. J. Cyst. Fibros. 2015, 14, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Orenti, A.; Zolin, A.; Jung, A.; van Rens, J.; Fox, A.; Krasnyk, M.; Daneau, G.; Hatziagorou, E.; Mei-Zahav, M.; Naehrlich, L.; et al. ECFSPR Annual Report 2020, published June 2022. Version 1.0; Available online: https://www.ecfs.eu/sites/default/files/ECFSPR_Report_2020_v1.0%20%2807Jun2022%29_website.pdf (accessed on 20 October 2023).
- Durda-Masny, M.; Gozdzik-Spychalska, J.; John, A.; Czainski, W.; Strozewska, W.; Pawlowska, N.; Wlizlo, J.; Batura-Gabryel, H.; Szwed, A. The determinants of survival among adults with cystic fibrosis-a cohort study. J. Physiol. Anthropol. 2021, 40, 19. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, B.C.; McColley, S.A.; Kissner, D.G.; Rolfe, M.W.; Rosen, J.M.; McKevitt, M.; Moorehead, L.; Montgomery, A.B.; Geller, D.E. Fosfomycin/tobramycin for inhalation in patients with cystic fibrosis with pseudomonas airway infection. Am. J. Respir. Crit. Care Med. 2012, 185, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Castellani, C.; Duff, A.J.A.; Bell, S.C.; Heijerman, H.G.M.; Munck, A.; Ratjen, F.; Sermet-Gaudelus, I.; Southern, K.W.; Barben, J.; Flume, P.A.; et al. ECFS best practice guidelines: The 2018 revision. J. Cyst. Fibros. 2018, 17, 153–178. [Google Scholar] [CrossRef]
- Kapnadak, S.G.; Dimango, E.; Hadjiliadis, D.; Hempstead, S.E.; Tallarico, E.; Pilewski, J.M.; Faro, A.; Albright, J.; Benden, C.; Blair, S.; et al. Cystic Fibrosis Foundation consensus guidelines for the care of individuals with advanced cystic fibrosis lung disease. J. Cyst. Fibros. 2020, 19, 344–354. [Google Scholar] [CrossRef]
- Cos, P.; Tote, K.; Horemans, T.; Maes, L. Biofilms: An extra hurdle for effective antimicrobial therapy. Curr. Pharm. Des. 2010, 16, 2279–2295. [Google Scholar] [CrossRef]
- Jennings, L.K.; Dreifus, J.E.; Reichhardt, C.; Storek, K.M.; Secor, P.R.; Wozniak, D.J.; Hisert, K.B.; Parsek, M.R. Pseudomonas aeruginosa aggregates in cystic fibrosis sputum produce exopolysaccharides that likely impede current therapies. Cell Rep. 2021, 34, 108782. [Google Scholar] [CrossRef]
- Harshey, R.M. Bacterial motility on a surface: Many ways to a common goal. Annu. Rev. Microbiol. 2003, 57, 249–273. [Google Scholar] [CrossRef] [PubMed]
- Koch, B.; Jensen, L.E.; Nybroe, O. A panel of Tn7-based vectors for insertion of the gfp marker gene or for delivery of cloned DNA into Gram-negative bacteria at a neutral chromosomal site. J. Microbiol. Methods 2001, 45, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Sauer, K.; Camper, A.K.; Ehrlich, G.D.; Costerton, J.W.; Davies, D.G. Pseudomonas aeruginosa displays multiple phenotypes during development as a biofilm. J. Bacteriol. 2002, 184, 1140–1154. [Google Scholar] [CrossRef] [PubMed]
- Deligianni, E.; Pattison, S.; Berrar, D.; Ternan, N.G.; Haylock, R.W.; Moore, J.E.; Elborn, S.J.; Dooley, J.S. Pseudomonas aeruginosa cystic fibrosis isolates of similar RAPD genotype exhibit diversity in biofilm forming ability in vitro. BMC Microbiol. 2010, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- EUCAST. The European Committee on Antimicrobial Susceptibility Testing, Clinical Breakpoints—Breakpoints and Guidance. Available online: https://www.eucast.org/clinical_breakpoints (accessed on 1 March 2023).
- Kessler, E.; Safrin, M. Elastinolytic and proteolytic enzymes. Methods Mol. Biol. 2014, 1149, 135–169. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Lu, M.; Qiu, H.; Li, Y.; Huang, L.; Dai, W. Spontaneous quorum-sensing hierarchy reprogramming in Pseudomonas aeruginosa laboratory strain PAO1. AMB Express 2022, 12, 6. [Google Scholar] [CrossRef]
- Filloux, A.; Ramos, J.L. Preface. In Pseudomonas Methods and Protocols; Methods in Molecular Biology; Humana: New York, NY, USA, 2014; Volume 1149, p. v. [Google Scholar] [CrossRef]
- Cullen, L.; Weiser, R.; Olszak, T.; Maldonado, R.F.; Moreira, A.S.; Slachmuylders, L.; Brackman, G.; Paunova-Krasteva, T.S.; Zarnowiec, P.; Czerwonka, G.; et al. Phenotypic characterization of an international Pseudomonas aeruginosa reference panel: Strains of cystic fibrosis (CF) origin show less in vivo virulence than non-CF strains. Microbiology 2015, 161, 1961–1977. [Google Scholar] [CrossRef]
- Murray, T.S.; Ledizet, M.; Kazmierczak, B.I. Swarming motility, secretion of type 3 effectors and biofilm formation phenotypes exhibited within a large cohort of Pseudomonas aeruginosa clinical isolates. J. Med. Microbiol. 2010, 59, 511–520. [Google Scholar] [CrossRef]
- Lee, B.; Haagensen, J.A.; Ciofu, O.; Andersen, J.B.; Hoiby, N.; Molin, S. Heterogeneity of biofilms formed by nonmucoid Pseudomonas aeruginosa isolates from patients with cystic fibrosis. J. Clin. Microbiol. 2005, 43, 5247–5255. [Google Scholar] [CrossRef]
- Matar, G.M.; Chaar, M.H.; Araj, G.F.; Srour, Z.; Jamaleddine, G.; Hadi, U. Detection of a highly prevalent and potentially virulent strain of Pseudomonas aeruginosa from nosocomial infections in a medical center. BMC Microbiol. 2005, 5, 29. [Google Scholar] [CrossRef]
- Yang, X.; Lai, Y.; Li, C.; Yang, J.; Jia, M.; Sheng, J. Molecular epidemiology of Pseudomonas aeruginosa isolated from lower respiratory tract of ICU patients. Braz. J. Biol. 2021, 81, 351–360. [Google Scholar] [CrossRef] [PubMed]
- Auda, I.G.; Al-Kadmy, I.M.; Kareem, S.M.; Lafta, A.K.; A’Affus, M.H.; Khit, I.A.; Al Kheraif, A.A.; Divakar, D.D.; Ramakrishnaiah, R. RAPD- and ERIC-Based Typing of Clinical and Environmental Pseudomonas aeruginosa Isolates. J. AOAC Int. 2017, 100, 532–536. [Google Scholar] [CrossRef] [PubMed]
- Hematzadeh, A.; Haghkhah, M. Biotyping of isolates of Pseudomonas aeruginosa isolated from human infections by RAPD and ERIC-PCR. Heliyon 2021, 7, e07967. [Google Scholar] [CrossRef] [PubMed]
- [VCV000802360.1], C. [VCV000802360.1], C. NM_000492.4(CFTR):c.3134C>T (p.Ser1045Phe). Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000802360.1 (accessed on 14 April 2022).
- Bonyadi, P.; Saleh, N.T.; Dehghani, M.; Yamini, M.; Amini, K. Prevalence of antibiotic resistance of Pseudomonas aeruginosa in cystic fibrosis infection: A systematic review and meta-analysis. Microb. Pathog. 2022, 165, 105461. [Google Scholar] [CrossRef] [PubMed]
- Sobczyńska-Tomaszewska, A.; Ołtarzewski, M.; Czerska, K.; Wertheim-Tysarowska, K.; Sands, D.; Walkowiak, J.; Bal, J.; Mazurczak, T.; NBS CF working group. Newborn screening for cystic fibrosis: Polish 4 years’ experience with CFTR sequencing strategy. Eur. J. Hum. Genet. 2013, 21, 391–396. [Google Scholar] [CrossRef] [PubMed]
- De Boeck, K.; Vermeulen, F.; Dupont, L. The diagnosis of cystic fibrosis. Presse Médicale 2017, 46, e97–e108. [Google Scholar] [CrossRef] [PubMed]
- Padoan, R.; Quattrucci, S.; Amato, A.; Carnovale, V.; Salvatore, D.; Salvatore, M.; Campagna, G. The Diagnosis of Cystic Fibrosis in Adult Age. Data from the Italian Registry. Diagnostics 2021, 11, 321. [Google Scholar] [CrossRef] [PubMed]
- Rachel, M.; Topolewicz, S.; Sliwczynski, A.; Galiniak, S. Managing Cystic Fibrosis in Polish Healthcare. Int. J. Environ. Res. Public Health 2020, 17, 7630. [Google Scholar] [CrossRef]
- Duguépéroux, I.; De Braekeleer, M. The CFTR 3849+10kbC->T and 2789+5G->A alleles are associated with a mild CF phenotype. Eur. Respir. J. 2005, 25, 468–473. [Google Scholar] [CrossRef]
- Dörk, T.; Macek, M., Jr.; Mekus, F.; Tümmler, B.; Tzountzouris, J.; Casals, T.; Krebsová, A.; Koudová, M.; Sakmaryová, I.; Macek, M., Sr.; et al. Characterization of a novel 21-kb deletion, CFTRdele2,3(21 kb), in the CFTR gene: A cystic fibrosis mutation of Slavic origin common in Central and East Europe. Hum. Genet. 2000, 106, 259–268. [Google Scholar] [CrossRef]
- Feldmann, D.; Couderc, R.; Audrezet, M.P.; Ferec, C.; Bienvenu, T.; Desgeorges, M.; Claustres, M.; Mittre, H.; Blayau, M.; Bozon, D.; et al. CFTR genotypes in patients with normal or borderline sweat chloride levels. Hum. Mutat. 2003, 22, 340. [Google Scholar] [CrossRef] [PubMed]
- Ozdemir, T.R.; Kanik, A. A Case of Cystic Fibrosis With a Rare Mutation (3849+10 kbC>T) and Normal Sweat Chloride Levels. Iran. J. Pediatr. 2015, 25, e369. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.C.; Shyur, S.D.; Chu, S.H.; Huang, L.H.; Kao, Y.H.; Lei, W.T.; Cheng, C.H.; Lo, C.Y.; Chen, C.K.; Fang, L.C. Cystic fibrosis: Experience in one institution. J. Microbiol. Immunol. Infect. 2014, 47, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Ziętkiewicz, E.; Rutkiewicz, E.; Pogorzelski, A.; Klimek, B.; Voelkel, K.; Witt, M. CFTR Mutations Spectrum and the Efficiency of Molecular Diagnostics in Polish Cystic Fibrosis Patients. PLoS ONE 2014, 9, e89094. [Google Scholar] [CrossRef] [PubMed]
- Elborn, J.S.P. Cystic fibrosis. Lancet 2016, 388, 2519–2531. [Google Scholar] [CrossRef] [PubMed]
- Hodson, M.E.; Gallagher, C.G.; Govan, J.R.W. A randomised clinical trial of nebulised tobramycin or colistin in cystic fibrosis. Eur. Respir. J. 2002, 20, 658–664. [Google Scholar] [CrossRef] [PubMed]
- Bilton, D.; Pressler, T.; Fajac, I.; Clancy, J.P.; Sands, D.; Minic, P.; Cipolli, M.; Galeva, I.; Solé, A.; Quittner, A.L.; et al. Amikacin liposome inhalation suspension for chronic Pseudomonas aeruginosa infection in cystic fibrosis. J. Cyst. Fibros. 2020, 19, 284–291. [Google Scholar] [CrossRef]
- ECDC. Surveillance Atlas of Infectious Diseases. Available online: https://atlas.ecdc.europa.eu/public/index.aspx (accessed on 14 April 2023).
- Lim, J.; Cui, Y.; Oh, Y.J.; Park, J.R.; Jo, W.; Cho, Y.H.; Park, S. Studying the effect of alginate overproduction on Pseudomonas aeruginosa biofilm by atomic force microscopy. J. Nanosci. Nanotechnol. 2011, 11, 5676–5681. [Google Scholar] [CrossRef]
- Mann, E.E.; Wozniak, D.J. Pseudomonas biofilm matrix composition and niche biology. FEMS Microbiol. Rev. 2012, 36, 893–916. [Google Scholar] [CrossRef]
- Jones, C.J.; Wozniak, D.J. Psl Produced by Mucoid Pseudomonas aeruginosa Contributes to the Establishment of Biofilms and Immune Evasion. mBio 2017, 8, e00864-17. [Google Scholar] [CrossRef]
- Malhotra, S.; Hayes, D., Jr.; Wozniak, D.J. Mucoid Pseudomonas aeruginosa and regional inflammation in the cystic fibrosis lung. J. Cyst. Fibros. 2019, 18, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Atkin, S.D.; Abid, S.; Foster, M.; Bose, M.; Keller, A.; Hollaway, R.; Sader, H.S.; Greenberg, D.E.; Finklea, J.D.; Castanheira, M.; et al. Multidrug-resistant Pseudomonas aeruginosa from sputum of patients with cystic fibrosis demonstrates a high rate of susceptibility to ceftazidime-avibactam. Infect. Drug Resist. 2018, 11, 1499–1510. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Barat, L.; Motos, A.; Ranzani, O.T.; Bassi, G.L.; Aguilera Xiol, E.; Senussi, T.; Travierso, C.; Chiurazzi, C.; Idone, F.; Muñoz, L.; et al. Diagnostic Value of Endotracheal Aspirates Sonication on Ventilator-Associated Pneumonia Microbiologic Diagnosis. Microorganisms 2017, 5, 62. [Google Scholar] [CrossRef] [PubMed]
- Buyck, J.M.; Tulkens, P.M.; Van Bambeke, F. Activities of antibiotic combinations against resistant strains of Pseudomonas aeruginosa in a model of infected THP-1 monocytes. Antimicrob. Agents Chemother. 2015, 59, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Kashyap, S.; Kaur, S.; Sharma, P.; Capalash, N. Combination of colistin and tobramycin inhibits persistence of Acinetobacter baumannii by membrane hyperpolarization and down-regulation of efflux pumps. Microbes Infect. 2021, 23, 104795. [Google Scholar] [CrossRef]
- Warren, A.E.; Boulianne-Larsen, C.M.; Chandler, C.B.; Chiotti, K.; Kroll, E.; Miller, S.R.; Taddei, F.; Sermet-Gaudelus, I.; Ferroni, A.; McInnerney, K.; et al. Genotypic and phenotypic variation in Pseudomonas aeruginosa reveals signatures of secondary infection and mutator activity in certain cystic fibrosis patients with chronic lung infections. Infect. Immun. 2011, 79, 4802–4818. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Ashcroft, P.; Ackermann, M.; Bonhoeffer, S. Stochastic Gene Expression Influences the Selection of Antibiotic Resistance Mutations. Mol. Biol. Evol. 2020, 37, 58–70. [Google Scholar] [CrossRef] [PubMed]
- Rojas, L.J.; Yasmin, M.; Benjamino, J.; Marshall, S.M.; DeRonde, K.J.; Krishnan, N.P.; Perez, F.; Colin, A.A.; Cardenas, M.; Martinez, O.; et al. Genomic heterogeneity underlies multidrug resistance in Pseudomonas aeruginosa: A population-level analysis beyond susceptibility testing. PLoS ONE 2022, 17, e0265129. [Google Scholar] [CrossRef]
- Rivas Caldas, R.; Le Gall, F.; Revert, K.; Rault, G.; Virmaux, M.; Gouriou, S.; Héry-Arnaud, G.; Barbier, G.; Boisramé, S. Pseudomonas aeruginosa and Periodontal Pathogens in the Oral Cavity and Lungs of Cystic Fibrosis Patients: A Case-Control Study. J. Clin. Microbiol. 2015, 53, 1898–1907. [Google Scholar] [CrossRef]
- Dosanjh, A.; Lakhani, S.; Elashoff, D.; Chin, C.; Hsu, V.; Hilman, B. A comparison of microbiologic flora of the sinuses and airway among cystic fibrosis patients with maxillary antrostomies. Pediatr. Transplant. 2000, 4, 182–185. [Google Scholar] [CrossRef]
- Aanæs, K. Bacterial sinusitis can be a focus for initial lung colonisation and chronic lung infection in patients with cystic fibrosis. J. Cyst. Fibros. 2013, 12, S1–S20. [Google Scholar] [CrossRef]
- Emaneini, M.; Kalantar-Neyestanaki, D.; Jabalameli, L.; Hashemi, M.; Beigverdi, R.; Jabalameli, F. Molecular analysis and antimicrobial resistance pattern of distinct strains of Pseudomonas aeruginosa isolated from cystic fibrosis patients in Iran. Iran. J. Microbiol. 2019, 11, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Rueda-Nieto, S.; Mondejar-Lopez, P.; Mira-Escolano, M.P.; Cutillas-Tolin, A.; Maceda-Roldan, L.A.; Arense-Gonzalo, J.J.; Palomar-Rodriguez, J.A. Analysis of the genotypic profile and its relationship with the clinical manifestations in people with cystic fibrosis: Study from a rare disease registry. Orphanet J. Rare Dis. 2022, 17, 222. [Google Scholar] [CrossRef] [PubMed]
- Winstanley, C.; O’Brien, S.; Brockhurst, M.A. Pseudomonas aeruginosa Evolutionary Adaptation and Diversification in Cystic Fibrosis Chronic Lung Infections. Trends Microbiol. 2016, 24, 327–337. [Google Scholar] [CrossRef] [PubMed]
- Spencer, D.H.; Kas, A.; Smith, E.E.; Raymond, C.K.; Sims, E.H.; Hastings, M.; Burns, J.L.; Kaul, R.; Olson, M.V. Whole-genome sequence variation among multiple isolates of Pseudomonas aeruginosa. J. Bacteriol. 2003, 185, 1316–1325. [Google Scholar] [CrossRef]
- Jarych, D.; Augustynowicz-Kopec, E.; Iwanska, A.; Parniewski, P.; Majchrzak, M. Molecular analysis of Pseudomonas aeruginosa strains isolated from cystic fibrosis patients. Sci. Rep. 2021, 11, 15460. [Google Scholar] [CrossRef]
- Vaněrková, M.; Mališová, B.; Kotásková, I.; Holá, V.; Růžička, F.; Freiberger, T. Biofilm formation, antibiotic susceptibility and RAPD genotypes in Pseudomonas aeruginosa clinical strains isolated from single centre intensive care unit patients. Folia Microbiol. 2017, 62, 531–538. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Total CF Population, n | 44 |
|---|---|
| Female, n (%) | 24 (54.5) |
| Male, n (%) | 20 (45.5) |
| Pancreatic insufficiency, n (%) | 32 (72.7) |
| Diabetes, n (%) | 15 (34.1) |
| Qualification for lung transplantation, n (%) | 7 (15.9) |
| Genetic characterization of the CFTR (%) | 38 (86.4) |
| average ± SD (median, min–max) | |
| Age in the study period average | 28.3 ± 7.1 (27, 18–53) |
| Age at CF diagnosis | 7.8 ± 11 (2, 0.01–46) |
| Years under therapy | 9.1 ± 4.6 (8.5, 1–20) |
| Hospitalization frequency [n/a] | 1.1 ± 1.2 (0.9, 0–4.8) |
| Visits frequency at CF-OOC [n/a] | 12.7 ± 14.2 (8.6, 3.7–73) |
| BMI [kg/m2] | 19.8 ± 3.0 (19.9, 13.5–28) |
| FEV1 [L] | 52.1 ± 22.2 (48.2, 14.7–99.8) |
| FVC average [L] | 71.6 ± 19.2 (69.2, 28.5–116.9) |
| FEV1/FVC [%] | 70.3 ± 15.9 (72.2, 37.9–100) |
| Phenotype | Isolates with Respective Phenotype, % (n) |
|---|---|
| Mucoidal phenotype | 33.8 (25) |
| Protease activity | 60.8 (45) |
| Motility (total) | 78.4 (58) |
| Swimming | 67.6 (50) |
| Swarming | 62.2 (46) |
| Biofilm formation (in total) | 41.9 (31) |
| Weak biofilm producers | 16.2 (12) |
| Moderate biofilm producers | 8.1 (6) |
| Strong biofilm producers | 17.6 (13) |
| Number of resistances per isolate against the tested compounds: | |
| 0 | 39.2 (29) |
| 1 | 21.6 (16) |
| 2 | 6.8 (5) |
| 3 | 5.4 (4) |
| 4 | 4.1 (3) |
| 5 | 12.2 (9) |
| 6 | 9.5 (7) |
| 7 | 1.4 (1) |
| Resistances Against | In the Study Cohort, % (n) | Weighted Pooled Resistance Prevalence 2011–2021 [29], % (95% CI) | Non-Mucoid, % (n) | Mucoid, % (n) |
|---|---|---|---|---|
| Piperazillin/tazobactam | 29.7 (22) | 19 (15–24) | 24.3 (18) | 5.4 (4) |
| Ceftazidime | 32.4 (24) | 36 (29–44) | 25.7 (19) | 6.8 (5) |
| Meropenem * | 25.7 (19) | 19 (15–24) | 21.6 (16) | 4.1 (3) |
| Ciprofloxacin ** | 36.5 (27) | 28 (33–36) | 27 (20) | 9.5 (7) |
| Colistin # | 5.6 (4) | 5 (2–8) | 5.4 (4) | 0 |
| Tobramycin | 27.3 (20) | 22 (17–28) | 24.3 (18) | 2.7 (2) |
| Amikacin *** | 37.8 (28) | 38 (24–58) | 32.4 (24) | 5.4 (4) |
| Biofilm Ability | Mucoid | Proteolysis | Swimming | Swarming | No. Resistances | 4MRGN | PIP/TAZ | CAZ | MEM | CIP | TB | AK | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age | 0.03 | 0.10 | −0.12 | −0.12 | −0.14 | −0.43 | −0.37 | −0.03 | −0.03 | −0.18 | −0.29 | −0.16 | −0.06 |
| Sex | 0.10 | 0.08 | −0.22 | −0.13 | −0.08 | 0.08 | −0.20 | −0.22 | −0.02 | −0.04 | −0.27 | −0.07 | −0.11 |
| BMI | −0.03 | 0.02 | −0.11 | 0.04 | 0.13 | −0.03 | −0.26 | −0.18 | −0.15 | −0.50 | −0.20 | −0.18 | −0.33 |
| AD | −0.12 | 0.28 | −0.12 | 0.14 | −0.04 | −0.25 | −0.16 | −0.10 | −0.06 | −0.08 | 0.03 | −0.10 | −0.12 |
| YT | 0.11 | 0.02 | −0.11 | −0.23 | −0.28 | −0.31 | −0.25 | 0.01 | 0.04 | −0.04 | −0.06 | −0.01 | 0.14 |
| HF | 0.03 | 0.03 | −0.08 | −0.40 | −0.24 | 0.08 | 0.35 | 0.38 | 0.54 | 0.53 | 0.32 | 0.46 | 0.35 |
| FEV1 | 0.06 | 0.06 | −0.17 | 0.20 | 0.19 | 0.07 | −0.13 | −0.41 | −0.35 | −0.34 | −0.06 | −0.04 | −0.12 |
| FVC | 0.02 | −0.05 | −0.10 | 0.17 | 0.04 | −0.04 | −0.07 | −0.25 | −0.28 | −0.28 | 0.02 | 0.00 | −0.01 |
| FEV1/ FVC | 0.08 | 0.16 | −0.25 | 0.19 | 0.22 | 0.14 | −0.11 | −0.41 | −0.32 | −0.30 | −0.10 | −0.04 | −0.15 |
| LT | 0.08 | 0.19 | 0.09 | −0.21 | −0.19 | −0.01 | 0.12 | 0.13 | 0.21 | 0.30 | 0.09 | 0.01 | 0.17 |
| Diabetes | −0.12 | −0.14 | −0.12 | 0.11 | 0.05 | −0.27 | 0.03 | 0.16 | 0.09 | 0.07 | −0.05 | −0.01 | 0.12 |
| PI | −0.17 | 0.04 | −0.17 | −0.12 | 0.07 | 0.14 | 0.16 | 0.06 | 0.23 | 0.26 | −0.04 | 0.26 | 0.17 |
| VF | −0.11 | −0.02 | 0.11 | 0.23 | 0.28 | 0.31 | 0.25 | −0.01 | −0.04 | 0.04 | 0.06 | 0.01 | −0.14 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jarzynka, S.; Makarewicz, O.; Weiss, D.; Minkiewicz-Zochniak, A.; Iwańska, A.; Skorupa, W.; Padzik, M.; Augustynowicz-Kopeć, E.; Olędzka, G. The Impact of Pseudomonas aeruginosa Infection in Adult Cystic Fibrosis Patients—A Single Polish Centre Study. Pathogens 2023, 12, 1440. https://doi.org/10.3390/pathogens12121440
Jarzynka S, Makarewicz O, Weiss D, Minkiewicz-Zochniak A, Iwańska A, Skorupa W, Padzik M, Augustynowicz-Kopeć E, Olędzka G. The Impact of Pseudomonas aeruginosa Infection in Adult Cystic Fibrosis Patients—A Single Polish Centre Study. Pathogens. 2023; 12(12):1440. https://doi.org/10.3390/pathogens12121440
Chicago/Turabian StyleJarzynka, Sylwia, Oliwia Makarewicz, Daniel Weiss, Anna Minkiewicz-Zochniak, Agnieszka Iwańska, Wojciech Skorupa, Marcin Padzik, Ewa Augustynowicz-Kopeć, and Gabriela Olędzka. 2023. "The Impact of Pseudomonas aeruginosa Infection in Adult Cystic Fibrosis Patients—A Single Polish Centre Study" Pathogens 12, no. 12: 1440. https://doi.org/10.3390/pathogens12121440
APA StyleJarzynka, S., Makarewicz, O., Weiss, D., Minkiewicz-Zochniak, A., Iwańska, A., Skorupa, W., Padzik, M., Augustynowicz-Kopeć, E., & Olędzka, G. (2023). The Impact of Pseudomonas aeruginosa Infection in Adult Cystic Fibrosis Patients—A Single Polish Centre Study. Pathogens, 12(12), 1440. https://doi.org/10.3390/pathogens12121440