
Assessing the Potential Role of Cats (Felis catus) as Generators of Relevant SARS-CoV-2 Lineages during the Pandemic


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Viral Sequences and Metadata Information
2.2. Hierarchical Cluster Analysis
2.3. Correlation between Genetic Groups Affecting Humans and Cat Populations during the Pandemic
2.4. Phylogenetic Analysis
2.5. Evolutionary Analysis
3. Results
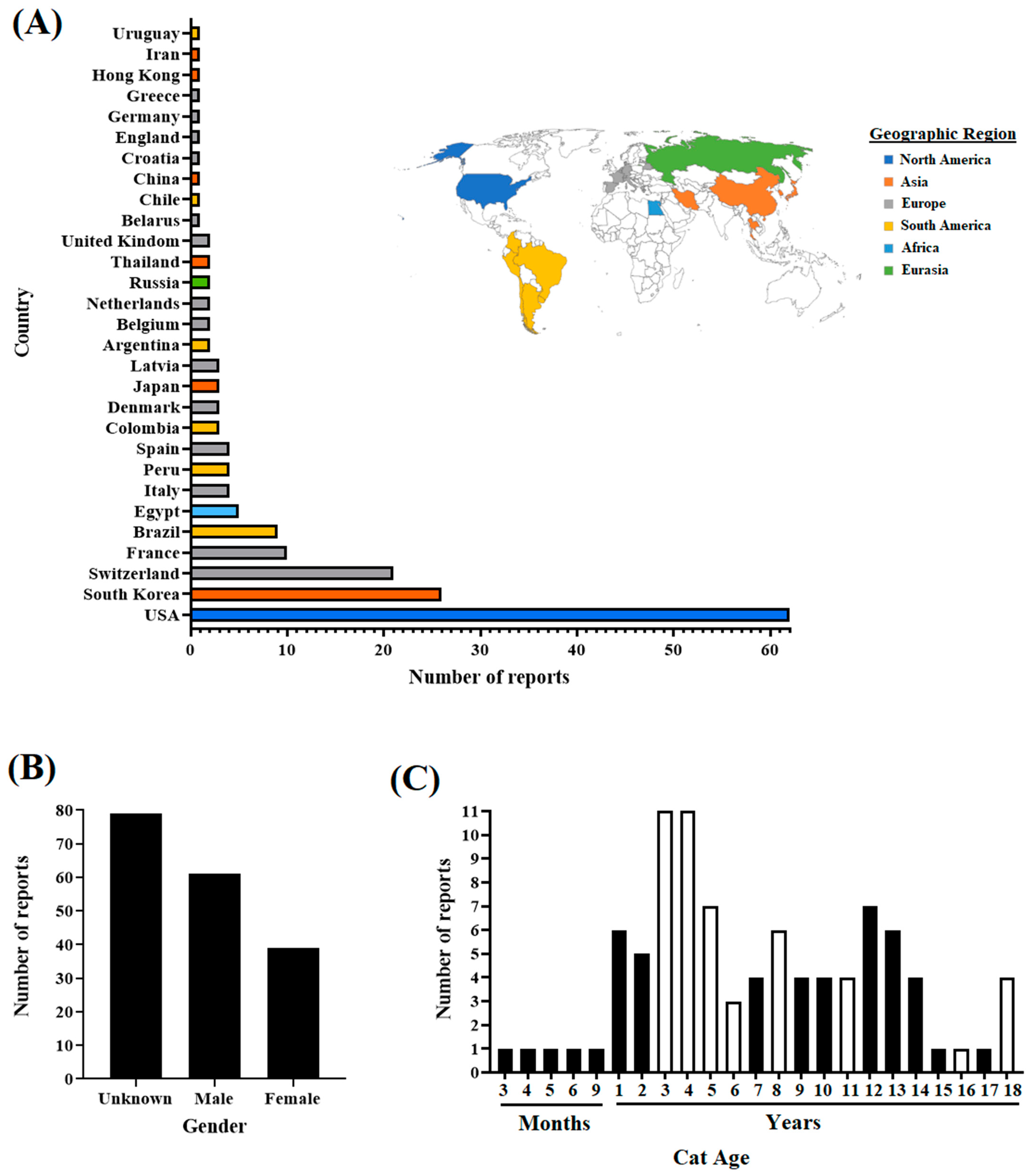
3.1. Overview of the SARS-CoV-2 Lineages Isolated from Cat Infections around the World
3.2. Assessing the Prevalence of SARS-CoV-2 Lineages in Cats and Other Animal Species during the Pandemic
3.3. Evolutionary Dynamics of Viral Lineages Affecting Cat Populations
3.4. Evaluating the Hypothesis about Independent Evolution of SARS-CoV-2 in Cat Populations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Velazquez-Salinas, L.; Zarate, S.; Eberl, S.; Gladue, D.P.; Novella, I.; Borca, M.V. Positive Selection of ORF1ab, ORF3a, and ORF8 Genes Drives the Early Evolutionary Trends of SARS-CoV-2 During the 2020 COVID-19 Pandemic. Front. Microbiol. 2020, 11, 550674. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Weaver, S.; Tegally, H.; San, J.E.; Shank, S.D.; Wilkinson, E.; Lucaci, A.G.; Giandhari, J.; Naidoo, S.; Pillay, Y.; et al. The emergence and ongoing convergent evolution of the SARS-CoV-2 N501Y lineages. Cell 2021, 184, 5189–5200.e5187. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, A.; Pybus, O.G.; Abram, M.E.; Kelly, E.J.; Rambaut, A. Pango lineage designation and assignment using SARS-CoV-2 spike gene nucleotide sequences. BMC Genom. 2022, 23, 121. [Google Scholar] [CrossRef]
- Velazquez-Salinas, L. The complex evolutionary dynamics of SARS-CoV-2, a big challenge to control the pandemic of COVID-19. J. Med. Virol. 2022, 94, 5082–5085. [Google Scholar] [CrossRef] [PubMed]
- Lenharo, M. WHO declares end to COVID-19′s emergency phase. Nature, 2023; Advance online publication. [Google Scholar] [CrossRef]
- Mahdy, M.A.A.; Younis, W.; Ewaida, Z. An Overview of SARS-CoV-2 and Animal Infection. Front. Veter Sci. 2020, 7, 596391. [Google Scholar] [CrossRef]
- Bashor, L.; Gagne, R.B.; Bosco-Lauth, A.; Stenglein, M.; VandeWoude, S. Rapid evolution of SARS-CoV-2 in domestic cats. Virus Evol. 2022, 8, veac092. [Google Scholar] [CrossRef]
- Bashor, L.; Gagne, R.B.; Bosco-Lauth, A.M.; Bowen, R.A.; Stenglein, M.; VandeWoude, S. SARS-CoV-2 evolution in animals suggests mechanisms for rapid variant selection. Proc. Natl. Acad. Sci. USA 2021, 118, e2105253118. [Google Scholar] [CrossRef]
- McBride, D.S.; Garushyants, S.K.; Franks, J.; Magee, A.F.; Overend, S.H.; Huey, D.; Williams, A.M.; Faith, S.A.; Kandeil, A.; Trifkovic, S.; et al. Accelerated evolution of SARS-CoV-2 in free-ranging white-tailed deer. Nat. Commun. 2023, 14, 5105. [Google Scholar] [CrossRef]
- Prince, T.; Smith, S.L.; Radford, A.D.; Solomon, T.; Hughes, G.L.; Patterson, E.I. SARS-CoV-2 Infections in Animals: Reservoirs for Reverse Zoonosis and Models for Study. Viruses 2021, 13, 494. [Google Scholar] [CrossRef]
- Navarro-Lopez, R.; Solis-Hernandez, M.; Rocha-Martinez, M.K.; Eberl, S.; Gomez-Romero, N.; Velazquez-Salinas, L.; Estrada-Franco, J.G. Near-Full-Length Genome Sequences Representing an Event of Zooanthroponotic Transmission of SARS-CoV-2 Lineage B.1.189 in Mexico during 2020. Genome Announc. 2022, 11, e0049722. [Google Scholar] [CrossRef]
- Koeppel, K.N.; Mendes, A.; Strydom, A.; Rotherham, L.; Mulumba, M.; Venter, M. SARS-CoV-2 Reverse Zoonoses to Pumas and Lions, South Africa. Viruses 2022, 14, 120. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, L.; Wernike, K.; Hoffmann, D.; Mettenleiter, T.C.; Beer, M. Experimental Infection of Cattle with SARS-CoV-2. Emerg. Infect. Dis. 2020, 26, 2979–2981. [Google Scholar] [CrossRef]
- EFSA Panel on Animal Health and Welfare (AHAW); Nielsen, S.S.; Alvarez, J.; Bicout, D.J.; Calistri, P.; Canali, E.; Drewe, J.A.; Garin-Bastuji, B.; Rojas, J.L.G.; Gortázar, C.; et al. SARS-CoV-2 in animals: Susceptibility of animal species, risk for animal and public health, monitoring, prevention and control. EFSA J. 2023, 21, e07822. [Google Scholar] [CrossRef] [PubMed]
- Frazzini, S.; Amadori, M.; Turin, L.; Riva, F. SARS-CoV-2 infections in animals, two years into the pandemic. Arch. Virol. 2022, 167, 2503–2517. [Google Scholar] [CrossRef] [PubMed]
- Cool, K.; Gaudreault, N.N.; Morozov, I.; Trujillo, J.D.; Meekins, D.A.; McDowell, C.; Carossino, M.; Bold, D.; Mitzel, D.; Kwon, T.; et al. Infection and transmission of ancestral SARS-CoV-2 and its alpha variant in pregnant white-tailed deer. Emerg. Microbes Infect. 2021, 11, 95–112. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Wen, Z.; Zhong, G.; Yang, H.; Wang, C.; Huang, B.; Liu, R.; He, X.; Shuai, L.; Sun, Z.; et al. Susceptibility of ferrets, cats, dogs, and other domesticated animals to SARS-coronavirus 2. Science 2020, 368, 1016–1020. [Google Scholar] [CrossRef]
- Bosco-Lauth, A.M.; Hartwig, A.E.; Porter, S.M.; Gordy, P.W.; Nehring, M.; Byas, A.D.; VandeWoude, S.; Ragan, I.K.; Maison, R.M.; Bowen, R.A. Experimental infection of domestic dogs and cats with SARS-CoV-2: Pathogenesis, transmission, and response to reexposure in cats. Proc. Natl. Acad. Sci. USA 2020, 117, 26382–26388. [Google Scholar] [CrossRef]
- Neira, V.; Brito, B.; Agüero, B.; Berrios, F.; Valdés, V.; Gutierrez, A.; Ariyama, N.; Espinoza, P.; Retamal, P.; Holmes, E.C.; et al. A household case evidences shorter shedding of SARS-CoV-2 in naturally infected cats compared to their human owners. Emerg. Microbes Infect. 2021, 10, 376–383. [Google Scholar] [CrossRef]
- Garigliany, M.; Van Laere, A.S.; Clercx, C.; Giet, D.; Escriou, N.; Huon, C.; van der Werf, S.; Eloit, M.; Desmecht, D. SARS-CoV-2 Natural Transmission from Human to Cat, Belgium, March 2020. Emerg. Infect. Dis. 2020, 26, 3069–3071. [Google Scholar] [CrossRef]
- Newman, A.; Smith, D.; Ghai, R.R.; Wallace, R.M.; Torchetti, M.K.; LoIacono, C.; Murrell, L.S.; Carpenter, A.; Moroff, S.; Rooney, J.A.; et al. First Reported Cases of SARS-CoV-2 Infection in Companion Animals—New York, March–April 2020. Morb. Mortal. Wkly. Rep. MMWR 2020, 69, 710–713. [Google Scholar] [CrossRef]
- Sailleau, C.; Dumarest, M.; Vanhomwegen, J.; Delaplace, M.; Caro, V.; Kwasiborski, A.; Hourdel, V.; Chevaillier, P.; Barbarino, A.; Comtet, L.; et al. First detection and genome sequencing of SARS-CoV-2 in an infected cat in France. Transbound. Emerg. Dis. 2020, 67, 2324–2328. [Google Scholar] [CrossRef] [PubMed]
- Musso, N.; Costantino, A.; La Spina, S.; Finocchiaro, A.; Andronico, F.; Stracquadanio, S.; Liotta, L.; Visalli, R.; Emmanuele, G. New SARS-CoV-2 Infection Detected in an Italian Pet Cat by RT-qPCR from Deep Pharyngeal Swab. Pathogens 2020, 9, 746. [Google Scholar] [CrossRef] [PubMed]
- Segalés, J.; Puig, M.; Rodon, J.; Avila-Nieto, C.; Carrillo, J.; Cantero, G.; Terrón, M.T.; Cruz, S.; Parera, M.; Noguera-Julián, M.; et al. Detection of SARS-CoV-2 in a cat owned by a COVID-19−affected patient in Spain. Proc. Natl. Acad. Sci. USA 2020, 117, 24790–24793. [Google Scholar] [CrossRef] [PubMed]
- Botero, Y.; Ramírez, J.D.; Serrano-Coll, H.; Aleman, A.; Ballesteros, N.; Martinez, C.; Muñoz, M.; Calderon, A.; Patiño, L.H.; Guzman, C.; et al. First report and genome sequencing of SARS-CoV-2 in a cat (Felis catus) in Colombia. Mem. Do Inst. Oswaldo Cruz 2022, 117, e210375. [Google Scholar] [CrossRef] [PubMed]
- Carlos, R.S.A.; Mariano, A.P.M.; Maciel, B.M.; Gadelha, S.R.; Silva, M.d.M.; Belitardo, E.M.M.A.; Gil Rocha, D.J.P.; Almeida, J.P.P.; Pacheco, L.G.C.; Aguiar, E.R.G.R.; et al. First genome sequencing of SARS-CoV-2 recovered from an infected cat and its owner in Latin America. Transbound. Emerg. Dis. 2021, 68, 3070–3074. [Google Scholar] [CrossRef]
- Ruiz-Arrondo, I.; Portillo, A.; Palomar, A.M.; Santibáñez, S.; Santibáñez, P.; Cervera, C.; Oteo, J.A. Detection of SARS-CoV-2 in pets living with COVID-19 owners diagnosed during the COVID-19 lockdown in Spain: A case of an asymptomatic cat with SARS-CoV-2 in Europe. Transbound. Emerg. Dis. 2020, 68, 973–976. [Google Scholar] [CrossRef]
- Carvallo, F.R.; Martins, M.; Joshi, L.R.; Caserta, L.C.; Mitchell, P.K.; Cecere, T.; Hancock, S.; Goodrich, E.L.; Murphy, J.; Diel, D.G. Severe SARS-CoV-2 Infection in a Cat with Hypertrophic Cardiomyopathy. Viruses 2021, 13, 1510. [Google Scholar] [CrossRef]
- Tewari, D.; Boger, L.; Brady, S.; Livengood, J.; Killian, M.L.; Nair, M.S.; Thirumalapura, N.; Kuchipudi, S.V.; Zellers, C.; Schroder, B.; et al. Transmission of SARS-CoV-2 from humans to a 16-year-old domestic cat with comorbidities in Pennsylvania, USA. Veter Med. Sci. 2021, 8, 899–906. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, H.; Gao, J.; Huang, K.; Yang, Y.; Hui, X.; He, X.; Li, C.; Gong, W.; Zhang, Y.; et al. A serological survey of SARS-CoV-2 in cat in Wuhan. Emerg. Microbes Infect. 2020, 9, 2013–2019. [Google Scholar] [CrossRef]
- Patterson, E.I.; Elia, G.; Grassi, A.; Giordano, A.; Desario, C.; Medardo, M.; Smith, S.L.; Anderson, E.R.; Prince, T.; Patterson, G.T.; et al. Evidence of exposure to SARS-CoV-2 in cats and dogs from households in Italy. Nat. Commun. 2020, 11, 6231. [Google Scholar] [CrossRef]
- Fritz, M.; Rosolen, B.; Krafft, E.; Becquart, P.; Elguero, E.; Vratskikh, O.; Denolly, S.; Boson, B.; Vanhomwegen, J.; Gouilh, M.A.; et al. High prevalence of SARS-CoV-2 antibodies in pets from COVID-19+ households. One Health 2020, 11, 100192. [Google Scholar] [CrossRef] [PubMed]
- Hamer, S.A.; Pauvolid-Corrêa, A.; Zecca, I.B.; Davila, E.; Auckland, L.D.; Roundy, C.M.; Tang, W.; Torchetti, M.K.; Killian, M.L.; Jenkins-Moore, M.; et al. SARS-CoV-2 Infections and Viral Isolations among Serially Tested Cats and Dogs in Households with Infected Owners in Texas, USA. Viruses 2021, 13, 938. [Google Scholar] [CrossRef] [PubMed]
- Villanueva-Saz, S.; Giner, J.; Tobajas, A.P.; Pérez, M.D.; González-Ramírez, A.M.; Macías-León, J.; González, A.; Verde, M.; Yzuel, A.; Hurtado-Guerrero, R.; et al. Serological evidence of SARS-CoV-2 and co-infections in stray cats in Spain. Transbound. Emerg. Dis. 2021, 69, 1056–1064. [Google Scholar] [CrossRef]
- Michelitsch, A.; Hoffmann, D.; Wernike, K.; Beer, M. Occurrence of Antibodies against SARS-CoV-2 in the Domestic Cat Population of Germany. Vaccines 2020, 8, 772. [Google Scholar] [CrossRef]
- Michelitsch, A.; Schön, J.; Hoffmann, D.; Beer, M.; Wernike, K. The Second Wave of SARS-CoV-2 Circulation-Antibody Detection in the Domestic Cat Population in Germany. Viruses 2021, 13, 1009. [Google Scholar] [CrossRef] [PubMed]
- Martins, M.; do Nascimento, G.M.; Nooruzzaman, M.; Yuan, F.; Chen, C.; Caserta, L.C.; Miller, A.D.; Whittaker, G.R.; Fang, Y.; Diel, D.G. The Omicron Variant BA.1.1 Presents a Lower Pathogenicity than B.1 D614G and Delta Variants in a Feline Model of SARS-CoV-2 Infection. J. Virol. 2022, 96, e0096122. [Google Scholar] [CrossRef] [PubMed]
- Zoccola, R.; Beltramo, C.; Magris, G.; Peletto, S.; Acutis, P.; Bozzetta, E.; Radovic, S.; Zappulla, F.; Porzio, A.M.; Gennero, M.S.; et al. First detection of an Italian human-to-cat outbreak of SARS-CoV-2 Alpha variant—Lineage B.1.1.7. One Health. 2021, 13, 100295. [Google Scholar] [CrossRef]
- Schiaffino, F.; Ferradas, C.; Jara, L.M.; Salvatierra, G.; Dávila-Barclay, A.; Sanchez-Carrion, C.; Ulloa, A.; Mascaro, L.; Pajuelo, M.J.; Sarmiento, L.G.; et al. First Detection and Genome Sequencing of SARS-CoV-2 Lambda (C.37) Variant in Symptomatic Domestic Cats in Lima, Peru. Front. Veter Sci. 2021, 8, 737350. [Google Scholar] [CrossRef]
- Kuhlmeier, E.; Chan, T.; Agüí, C.V.; Willi, B.; Wolfensberger, A.; Beisel, C.; Topolsky, I.; Beerenwinkel, N.; Stadler, T.; Swiss SARS-CoV-2 Sequencing Consortium; et al. Detection and Molecular Characterization of the SARS-CoV-2 Delta Variant and the Specific Immune Response in Companion Animals in Switzerland. Viruses 2023, 15, 245. [Google Scholar] [CrossRef]
- Ferasin, L.; Fritz, M.; Ferasin, H.; Becquart, P.; Corbet, S.; Gouilh, M.A.; Legros, V.; Leroy, E.M. Infection with SARS-CoV-2 variant B.1.1.7 detected in a group of dogs and cats with suspected myocarditis. Veter Rec. 2021, 189, e944. [Google Scholar] [CrossRef]
- Gaudreault, N.N.; Trujillo, J.D.; Carossino, M.; Meekins, D.A.; Morozov, I.; Madden, D.W.; Indran, S.V.; Bold, D.; Balaraman, V.; Kwon, T.; et al. SARS-CoV-2 infection, disease and transmission in domestic cats. Emerg. Microbes Infect. 2020, 9, 2322–2332. [Google Scholar] [CrossRef]
- Khare, S.; Gurry, C.; Freitas, L.; Schultz, M.B.; Bach, G.; Diallo, A.; Akite, N.; Ho, J.; Lee, R.T.; Yeo, W.; et al. GISAID’s Role in Pandemic Response. China CDC Wkly 2021, 3, 1049–1051. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Kosakovsky Pond, S.L.; Frost, S.D. Not so different after all: A comparison of methods for detecting amino acid sites under selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky Pond, S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef]
- Smith, M.D.; Wertheim, J.O.; Weaver, S.; Murrell, B.; Scheffler, K.; Kosakovsky Pond, S.L. Less is more: An adaptive branch-site random effects model for efficient detection of episodic diversifying selection. Mol. Biol. Evol. 2015, 32, 1342–1353. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Weaver, S.; Smith, M.D.; Wertheim, J.O.; Murrell, S.; Aylward, A.; Eren, K.; Pollner, T.; Martin, D.P.; Smith, D.M.; et al. Gene-Wide Identification of Episodic Selection. Mol. Biol. Evol. 2015, 32, 1365–1371. [Google Scholar] [CrossRef]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A Modern Web Application for Characterizing Selective and Other Evolutionary Processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef]
- Velazquez-Salinas, L. Deciphering the evolutionary mechanisms of SARS-CoV-2: Absence of ORF8 protein and its potential advantage in the emergence of viral lineages. J. Med. Virol. 2023, 95, e29002. [Google Scholar] [CrossRef]
- Hale, V.L.; Dennis, P.M.; McBride, D.S.; Nolting, J.M.; Madden, C.; Huey, D.; Ehrlich, M.; Grieser, J.; Winston, J.; Lombardi, D.; et al. SARS-CoV-2 infection in free-ranging white-tailed deer. Nature 2022, 602, 481–486. [Google Scholar] [CrossRef]
- Gonzales, J.L.; de Jong, M.C.M.; Gerhards, N.M.; Van der Poel, W.H.M. The SARS-CoV-2 Reproduction Number R0 in Cats. Viruses 2021, 13, 2480. [Google Scholar] [CrossRef] [PubMed]
- Gerhards, N.M.; Gonzales, J.L.; Vreman, S.; Ravesloot, L.; Brand, J.M.A.v.D.; Doekes, H.P.; Egberink, H.F.; Stegeman, A.; Oreshkova, N.; van der Poel, W.H.M.; et al. Efficient Direct and Limited Environmental Transmission of SARS-CoV-2 Lineage B.1.22 in Domestic Cats. Microbiol. Spectr. 2023, 11, e0255322. [Google Scholar] [CrossRef] [PubMed]
- Michelitsch, A.; Allendorf, V.; Conraths, F.J.; Gethmann, J.; Schulz, J.; Wernike, K.; Denzin, N. SARS-CoV-2 Infection and Clinical Signs in Cats and Dogs from Confirmed Positive Households in Germany. Viruses 2023, 15, 837. [Google Scholar] [CrossRef] [PubMed]
- Lott, N.; Gebhard, C.E.; Bengs, S.; Haider, A.; Kuster, G.M.; Regitz-Zagrosek, V.; Gebhard, C. Sex hormones in SARS-CoV-2 susceptibility: Key players or confounders? Nat. Rev. Endocrinol. 2022, 19, 217–231. [Google Scholar] [CrossRef]
- Duong, D. Alpha, Beta, Delta, Gamma: What’s important to know about SARS-CoV-2 variants of concern? Can. Med. Assoc. J. 2021, 193, E1059–E1060. [Google Scholar] [CrossRef]
- Han, A.X.; Parker, E.; Scholer, F.; Maurer-Stroh, S.; Russell, C.A. Phylogenetic Clustering by Linear Integer Programming (PhyCLIP). Mol. Biol. Evol. 2019, 36, 1580–1595. [Google Scholar] [CrossRef]
- Braun, K.M.; Moreno, G.K.; Halfmann, P.J.; Hodcroft, E.B.; Baker, D.A.; Boehm, E.C.; Weiler, A.M.; Haj, A.K.; Hatta, M.; Chiba, S.; et al. Transmission of SARS-CoV-2 in domestic cats imposes a narrow bottleneck. PLoS Pathog. 2021, 17, e1009373. [Google Scholar] [CrossRef]
- Lin, X.; Sha, Z.; Trimpert, J.; Kunec, D.; Jiang, C.; Xiong, Y.; Xu, B.; Zhu, Z.; Xue, W.; Wu, H. The NSP4 T492I mutation increases SARS-CoV-2 infectivity by altering non-structural protein cleavage. Cell Host Microbe 2023, 31, 1170–1184.e7. [Google Scholar] [CrossRef]
- Abduljaleel, Z.; Melebari, S.; Athar, M.; Dehlawi, S.; Udhaya Kumar, S.; Aziz, S.A.; Dannoun, A.I.; Malik, S.M.; Thasleem, J.; George Priya Doss, C. SARS-CoV-2 vaccine breakthrough infections (VBI) by Omicron variant (B.1.1.529) and consequences in structural and functional impact. Cell. Signal. 2023, 109, 110798. [Google Scholar] [CrossRef]
- Greaney, A.J.; Starr, T.N.; Gilchuk, P.; Zost, S.J.; Binshtein, E.; Loes, A.N.; Hilton, S.K.; Huddleston, J.; Eguia, R.; Crawford, K.H.; et al. Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition. Cell Host Microbe 2021, 29, 44–57.e9. [Google Scholar] [CrossRef]
- Starr, T.N.; Greaney, A.J.; Hilton, S.K.; Ellis, D.; Crawford, K.H.D.; Dingens, A.S.; Navarro, M.J.; Bowen, J.E.; Tortorici, M.A.; Walls, A.C.; et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell 2020, 182, 1295–1310.e20. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Fan, Q.; Song, S.; Shen, S.; Zhou, B.; Wang, H.; Cheng, L.; Ge, X.; Ju, B.; Zhang, Z. Increased resistance of SARS-CoV-2 Lambda variant to antibody neutralization. J. Clin. Virol. 2022, 150–151, 105162. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Zhang, L.; Zhang, Y.; Wang, H.; Ding, R.; Nie, J.; Li, Q.; Liu, S.; Yu, Y.; Yang, X.; et al. The Antigenicity of Epidemic SARS-CoV-2 Variants in the United Kingdom. Front. Immunol. 2021, 12, 687869. [Google Scholar] [CrossRef]
- Wang, H.-X.; Zhang, L.; Liang, Z.-T.; Nie, J.-H.; Wu, J.-J.; Li, Q.-Q.; Ding, R.-X.; Zhang, Y.; Chen, G.-Q.; Wang, Y.-C.; et al. Infectivity and antigenicity of pseudoviruses with high-frequency mutations of SARS-CoV-2 identified in Portugal. Arch. Virol. 2022, 167, 459–470. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, J.; Plante, K.S.; Plante, J.A.; Xie, X.; Zhang, X.; Ku, Z.; An, Z.; Scharton, D.; Schindewolf, C.; et al. The N501Y spike substitution enhances SARS-CoV-2 infection and transmission. Nature 2021, 602, 294–299. [Google Scholar] [CrossRef] [PubMed]
- de Silva, T.I.; Liu, G.; Lindsey, B.B.; Dong, D.; Moore, S.C.; Hsu, N.S.; Shah, D.; Wellington, D.; Mentzer, A.J.; Angyal, A.; et al. The impact of viral mutations on recognition by SARS-CoV-2 specific T cells. iScience 2021, 24, 103353. [Google Scholar] [CrossRef]
- Yalcinkaya, M.; Liu, W.; Islam, M.N.; Kotini, A.G.; Gusarova, G.A.; Fidler, T.P.; Papapetrou, E.P.; Bhattacharya, J.; Wang, N.; Tall, A.R. Modulation of the NLRP3 inflammasome by SARS-CoV-2 Envelope protein. Sci. Rep. 2021, 11, 24432. [Google Scholar] [CrossRef]
- Li, X.; Hou, P.; Ma, W.; Wang, X.; Wang, H.; Yu, Z.; Chang, H.; Wang, T.; Jin, S.; Wang, X.; et al. SARS-CoV-2 ORF10 suppresses the antiviral innate immune response by degrading MAVS through mitophagy. Cell. Mol. Immunol. 2021, 19, 67–78. [Google Scholar] [CrossRef]
- Zhou, S.; Lv, P.; Li, M.; Chen, Z.; Xin, H.; Reilly, S.; Zhang, X. SARS-CoV-2 E protein: Pathogenesis and potential therapeutic development. Biomed. Pharmacother. 2023, 159, 114242. [Google Scholar] [CrossRef]
- Colson, P.; Gautret, P.; Delerce, J.; Chaudet, H.; Pontarotti, P.; Forterre, P.; Tola, R.; Bedotto, M.; Delorme, L.; Bader, W.; et al. The emergence, spread and vanishing of a French SARS-CoV-2 variant exemplifies the fate of RNA virus epidemics and obeys the Mistigri rule. J. Med. Virol. 2022, 95, e28102. [Google Scholar] [CrossRef]
- Pereira, F. SARS-CoV-2 variants lacking ORF8 occurred in farmed mink and pangolin. Gene 2021, 784, 145596. [Google Scholar] [CrossRef] [PubMed]
- Young, B.E.; Fong, S.-W.; Chan, Y.-H.; Mak, T.-M.; Ang, L.W.; Anderson, D.E.; Lee, C.Y.-P.; Amrun, S.N.; Lee, B.; Goh, Y.S.; et al. Effects of a major deletion in the SARS-CoV-2 genome on the severity of infection and the inflammatory response: An observational cohort study. Lancet 2020, 396, 603–611. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomez-Romero, N.; Basurto-Alcantara, F.J.; Velazquez-Salinas, L. Assessing the Potential Role of Cats (Felis catus) as Generators of Relevant SARS-CoV-2 Lineages during the Pandemic. Pathogens 2023, 12, 1361. https://doi.org/10.3390/pathogens12111361
Gomez-Romero N, Basurto-Alcantara FJ, Velazquez-Salinas L. Assessing the Potential Role of Cats (Felis catus) as Generators of Relevant SARS-CoV-2 Lineages during the Pandemic. Pathogens. 2023; 12(11):1361. https://doi.org/10.3390/pathogens12111361
Chicago/Turabian StyleGomez-Romero, Ninnet, Francisco Javier Basurto-Alcantara, and Lauro Velazquez-Salinas. 2023. "Assessing the Potential Role of Cats (Felis catus) as Generators of Relevant SARS-CoV-2 Lineages during the Pandemic" Pathogens 12, no. 11: 1361. https://doi.org/10.3390/pathogens12111361
APA StyleGomez-Romero, N., Basurto-Alcantara, F. J., & Velazquez-Salinas, L. (2023). Assessing the Potential Role of Cats (Felis catus) as Generators of Relevant SARS-CoV-2 Lineages during the Pandemic. Pathogens, 12(11), 1361. https://doi.org/10.3390/pathogens12111361