


The Cultured Microbiome of Pollinated Maize Silks Shifts after Infection with Fusarium graminearum and Varies by Distance from the Site of Pathogen Inoculation
Abstract
1. Introduction
2. Materials and Methods
2.1. Overview of Methods
2.2. Comparison to V4-MiSeq Results
2.3. Fusarium graminearum Field Treatments
2.4. Source and Preparation of F. graminearum Treatment

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inbred/Hybrid Line | Year Released | Derivation | Heterotic Group | Heterotic Group Description | Grain Type | Days to Silking | * Gibberella Ear Rot/(Silk) | * Gibberella Ear Rot/(Kernel) |
|---|---|---|---|---|---|---|---|---|
| CO462 | 2016 | CO388 × W153R | BSSS/Minnesota 13 | US Hybrid era stiff stalk/Minnesota US Pre-Hybrid era | Dent | 75 | S | S |
| CO452 | 2014 | (CO388 × CO328) × CO388(4) | BSSS | US Hybrid era stiff stalk | Dent | 80 | I | I |
| CO444 | 2007 | S1381 × CO382 | European flint | European flint, Pre-Hybrid era | Flint | 79 | I | I |
| CO448 | 2012 | CO273 × CO431 | P3990/Iodent | Pioneer Hybrid/US Pre-Hybrid era Corn Belt Dent | Dent | 70 | I | I |
| CO325 | 1991 | (CO256 × CO264) × CO264 (2) | Early Butler | New York, Pennsylvania US Pre-Hybrid era | Dent | 76 | I | I |
| CO449 | 2012 | CO432 × CO433 | Minnesota 13 | Minnesota US Pre-Hybrid era | Dent | 75 | MR-R | MR-R |
| CO441 | 2002 | Jacques 7700 × CO298 | Lancaster | Pennsylvania US Pre-Hybrid Corn Belt Dent | Dent | 72 | R | R |
| CO431 | 1999 | Fusarium Resistant Synthetic | Iodent | US Pre-Hybrid Corn Belt Dent | Dent | 71 | R | I |
| CO433 | 2000 | Pride K127 | Minnesota 13 | Minnesota US Pre-Hybrid era | Dent | 77 | R | R |
| CO430 | 1999 | Fusarium Resistant Synthetic | P3990 | Pioneer Hybrid | Dent | 69 | HR | HR |
| CO432 | 2000 | Fusarium Resistant Synthetic C1 | Minnesota 13 | Minnesota US Pre-Hybrid era | Dent | 74 | HR | I |
| P35837◊ | NA | NA | NA | Pioneer Hybrid | Dent | NA | NA | NA |
| P38157◊ | NA | NA | NA | Pioneer Hybrid | Dent | NA | NA | NA |
| P9855HR◊ | NA | NA | NA | Pioneer Hybrid | Dent | NA | NA | NA |
3. Results
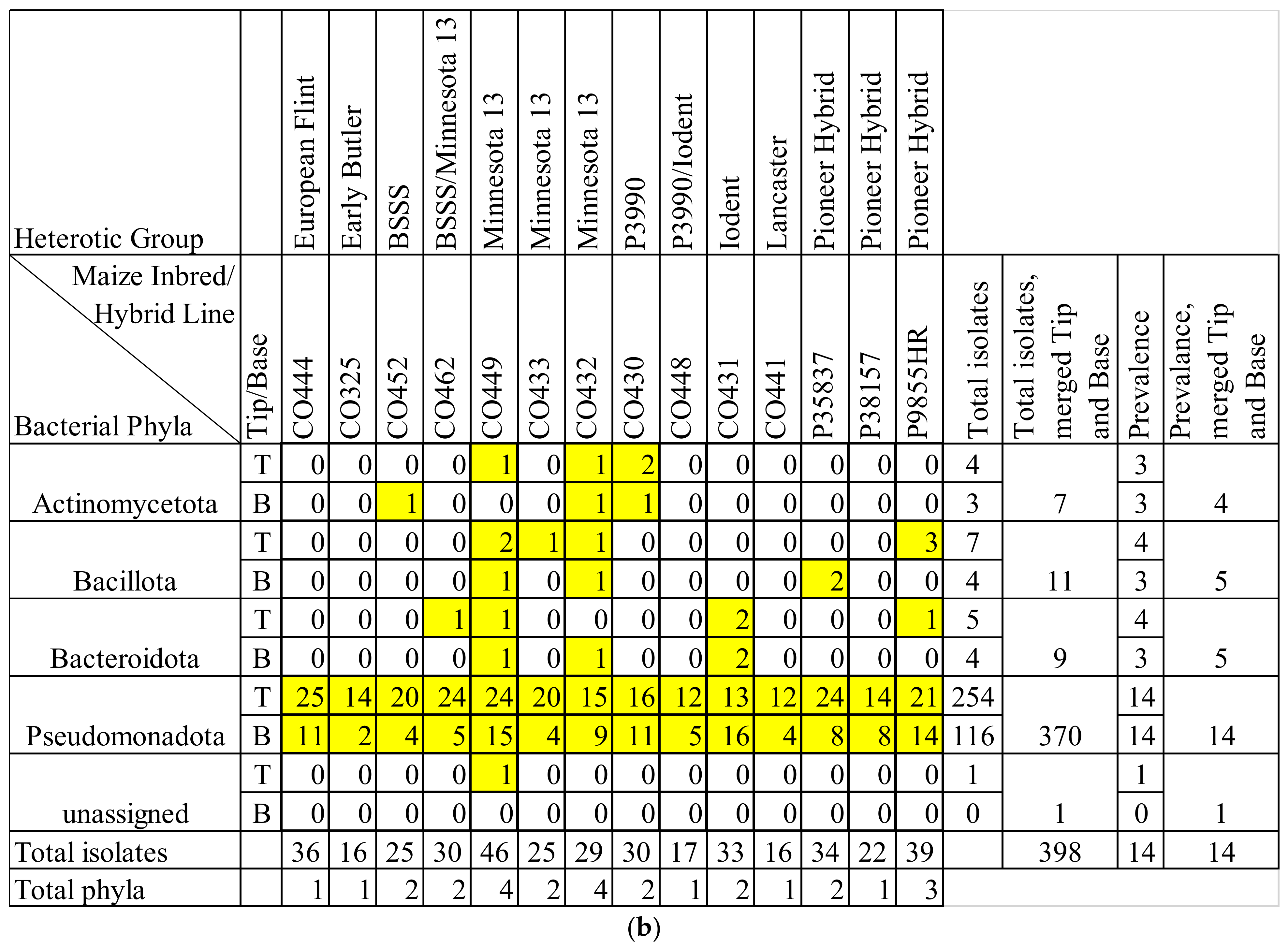
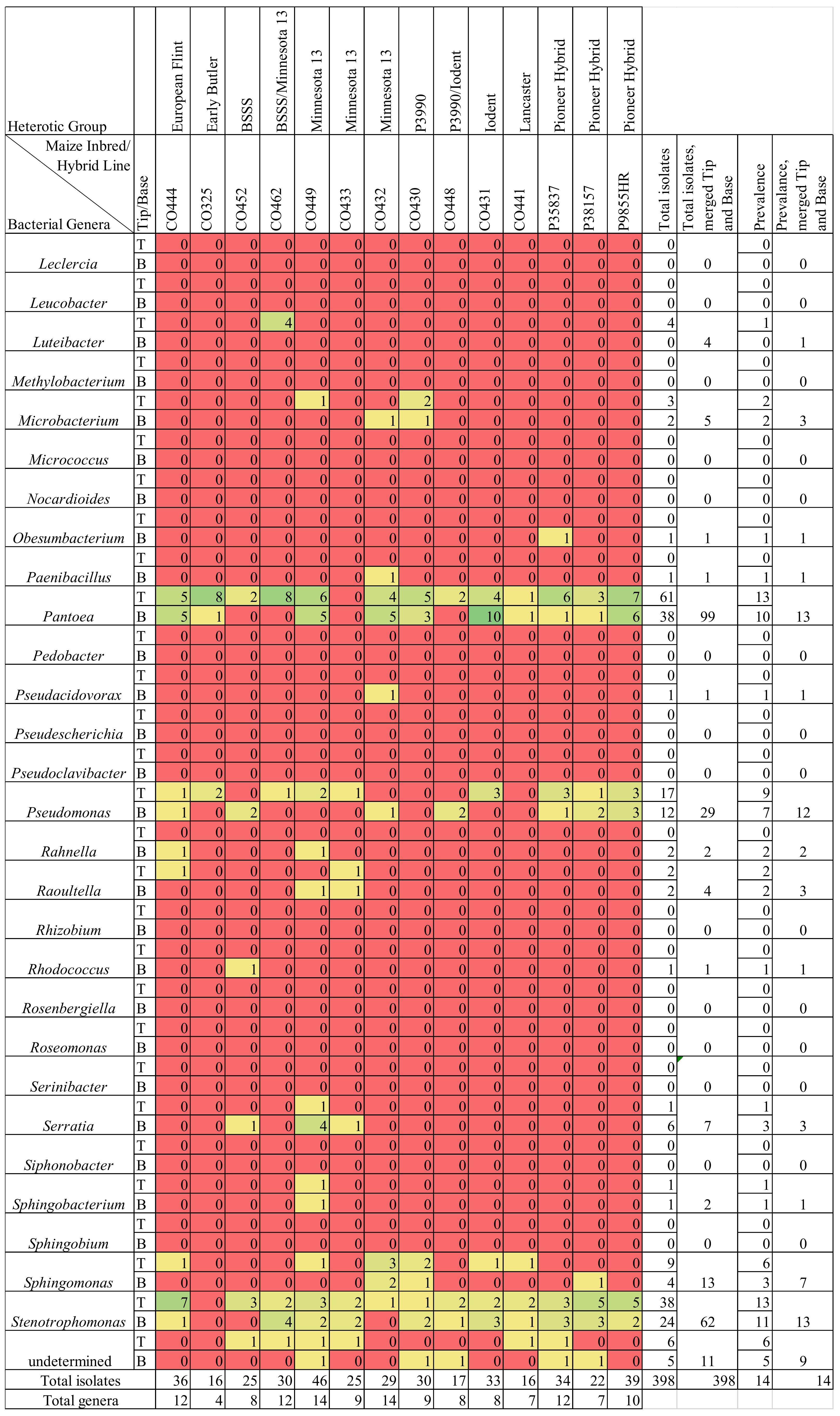
3.1. Overview of Culturing, Sequencing, and Taxonomy
3.2. Silk Tip versus Base Comparisons
3.3. Maize Genotype Comparisons
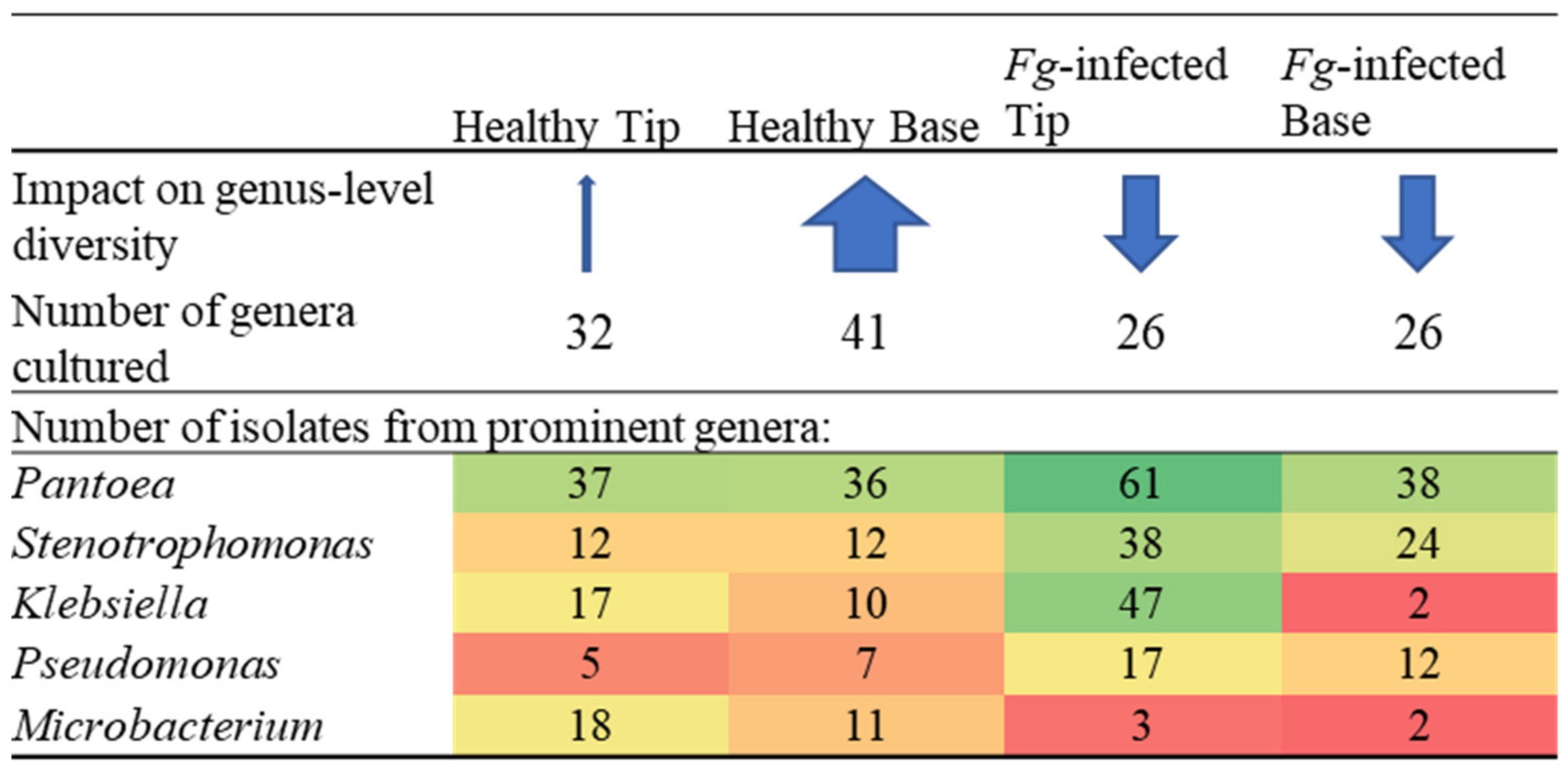
3.4. Comparing Taxonomy of Healthy and F. graminearum-Infected Silks




3.5. F. graminearum-Treated Transmitting Silk Taxonomy at the Cultured OTU Level
3.6. Comparison of Prevalent OTUs in Healthy versus F. graminearum-Treated Silks



3.7. Comparison of V4-MiSeq Predicted Taxa and Isolates Cultured from the Transmitting Silk Microbiome
3.7.1. Overview
3.7.2. Improved Species Level Taxonomic Resolution within the Previously Defined V4-MiSeq Transmitting Silk Microbiome for Core Taxa and F. graminearum-Induced Taxa
3.7.3. Diversity amongst Cultured OTUs That Matched Previously Defined V4-MiSeq Core and F. graminearum-Induced Taxa in the Transmitting Silk Microbiome
3.7.4. Comparing F. graminearum-Treated and Healthy Silk Isolates Matching V4-MiSeq Core and V4-MiSeq F. graminearum-Predictors
3.7.5. Prevalent Individual Cultured OTUs That Matched Previously Defined V4-MiSeq Core and F. graminearum-Induced Taxa in the Transmitting Silk Microbiome
- Multiple prevalent cultured OTUs matched core V4-MiSeq OTU 25 (Pantoea), the most prevalent and abundant silk OTU identified by Khalaf et al. [42]: cultured OTU 291 was found in 11 maize genotypes; cultured OTU 289 was found in nine maize genotypes; cultured OTU 290 was found in eight maize genotypes, and cultured OTU 292 was found in eight maize genotypes and also appeared in two genotypes in healthy silks. Cultured OTU 271 (predicted as Pantoea ananatis) matched V4-MiSeq OTU 25 and was isolated from three maize genotypes in the healthy silks and five host genotypes in the F. graminearum-treated silks.
- Cultured OTU 375 (identified as Stenotrophomonas pavanii) matched core V4-MiSeq OTU 34, and was cultured from six maize genotypes in the healthy silks; in the F. graminearum-treated silks, cultured OTU 375 appeared in 12 genotypes.
- Cultured OTU 117 matched V4-MiSeq OTU 3 and was isolated from three maize genotypes in the healthy silks. Cultured OTU 117 was also cultured from three maize genotypes in the F. graminearum-treated silks, although only one of the genotypes (CO452, of BSSS heterotic group) overlapped with those in the healthy silks.
- There were five isolates from the F. graminearum-treated silks that matched V4-MiSeq OTU 28 (V4-MiSeq core taxa), and of these, four matched a single cultured OTU (OTU 317) (Figure S10). By contrast, in the healthy silks, V4-MiSeq OTU 28 was only cultured from one sample, which did not match cultured OTU 317.
- Similarly, of the isolates matching V4-MiSeq OTU 31 (V4-MiSeq core taxa), three of the four isolates from the F. graminearum-treated silks corresponded to a single cultured OTU (OTU 7). In the healthy silks, one of the two isolates matching V4-MiSeq OTU 31 also matched cultured OTU 7.
- Cultured OTU 246 (a taxon of Pantoea agglomerans) matched core V4-MiSeq OTU 4, and was cultured from four maize genotypes in healthy silks. However, cultured OTU 246 was only identified once in F. graminearum-treated silks.
4. Discussion
4.1. Overview
4.2. Effects of F. graminearum Treatment on the TSM in Comparison to Healthy Silks
Species Level Differences
4.3. Tip versus Base Location Effect on the Cultured TSM
4.4. Maize Genotype Effect on the Cultured TSM
4.5. Cultured Core and V4-MiSeq Taxa Evaluations
4.5.1. Defining the Cultured Core Transmitting Silk Microbiome at the OTU Level
4.5.2. Evaluating Diversity within V4-MiSeq Taxa at the Cultured OTU Level
4.5.3. Conserved Cultures at the OTU Level within V4-MiSeq Taxa
4.5.4. Overview of Cultured Bacteria Matching Non-Core V4-MiSeq F. graminearum Indicators
4.5.5. V4-MiSeq Taxa in the Context of Silk Tip and Base Locations
4.6. Study Limitations and Future Experiments
4.7. Lessons and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ngugi, H.K.; Scherm, H. Biology of Flower-Infecting Fungi. Annu. Rev. Phytopathol. 2006, 44, 261–282. [Google Scholar] [CrossRef] [PubMed]
- Aleklett, K.; Hart, M.; Shade, A. The Microbial Ecology of Flowers: An Emerging Frontier in Phyllosphere Research. Botany 2014, 92, 253–266. [Google Scholar] [CrossRef]
- Thompson, M.E.H.; Raizada, M.N. Fungal Pathogens of Maize Gaining Free Passage along the Silk Road. Pathogens 2018, 7, 81. [Google Scholar] [CrossRef] [PubMed]
- Ali, M.L.; Taylor, J.H.; Jie, L.; Sun, G.; William, M.; Kasha, K.J.; Reid, L.M.; Pauls, K.P. Molecular Mapping of QTLs for Resistance to Gibberella Ear Rot, in Corn, Caused by Fusarium graminearum. Genome 2005, 48, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.J.; Dobson, A.D.W. Mycotoxin Production by Aspergillus, Fusarium and Penicillium Species. Int. J. Food Microbiol. 1998, 43, 141–158. [Google Scholar] [CrossRef] [PubMed]
- Vesonder, R.F.; Ellis, J.J.; Rohwedder, W.K. Elaboration of Vomitoxin and Zearalenone by Fusarium Isolates and the Biological Activity of Fusarium-Produced Toxins. Appl. Environ. Microbiol. 1981, 42, 1132–1134. [Google Scholar] [CrossRef]
- Abbas, H.K.; Shier, W.T.; Plasencia, J.; Weaver, M.A.; Bellaloui, N.; Kotowicz, J.K.; Butler, A.M.; Accinelli, C.; de la Torre-Hernandez, M.E.; Zablotowicz, R.M. Mycotoxin Contamination in Corn Smut (Ustilago maydis) Galls in the Field and in the Commercial Food Products. Food Control 2017, 71, 57–63. [Google Scholar] [CrossRef]
- Miller, S.S.; Reid, L.M.; Harris, L.J. Colonization of Maize Silks by Fusarium graminearum, the Causative Organism of Gibberella Ear Rot. Can. J. Bot. 2007, 85, 369–376. [Google Scholar] [CrossRef]
- Reid, L.M.; Bolton, A.T.; Hamilton, R.I.; Woldemariam, T.; Mather, D.E. Effect of Silk Age on Resistance of Maize to Fusarium graminearum. Can. J. Plant Pathol. 1992, 14, 293–298. [Google Scholar] [CrossRef]
- Reid, L.M.; Sinha, R.C. Maize Maturity and the Development of Gibberella Ear Rot Symptoms and Deoxynivalenol after Inoculation. Eur. J. Plant Pathol. 1998, 104, 147–154. [Google Scholar] [CrossRef]
- Schaafsma, A.W.; Nicol, R.W.; Reid, L.M. Evaluating Commercial Maize Hybrids for Resistance to Gibberella Ear Rot. Eur. J. Plant Pathol. 1997, 103, 737–746. [Google Scholar] [CrossRef]
- Mueller, D.; Wise, K.A.; Sisson, A.J.; Allen, T.W.; Bergstrom, G.C.; Bissonnette, K.M.; Bradley, C.A.; Byamukama, E.; Chilvers, M.I.; Collins, A.A.; et al. Corn Yield Loss Estimates Due to Diseases in the United States and Ontario, Canada, from 2016 to 2019. Plant Health Prog. 2020, 21, 238–247. [Google Scholar] [CrossRef]
- Mueller, D.; Wise, K.; Sisson, A. Corn Disease Loss Estimates from the United States and Ontario, Canada—2021. Crop Prot. Netw. 2022, CPN-2007-21. [Google Scholar] [CrossRef]
- Mueller, D.; Wise, K.; Sisson, A. Corn Disease Loss Estimates from the United States and Ontario, Canada—2020. Crop Prot. Netw. 2021, CPN-2007-20. [Google Scholar] [CrossRef]
- Mueller, D.; Wise, K.; Sisson, A. Corn Disease Loss Estimates from the United States and Ontario, Canada—2018. Crop Prot. Netw. 2018, CPN-2007-18. [Google Scholar] [CrossRef]
- De Ruyck, K.; De Boevre, M.; Huybrechts, I.; De Saeger, S. Dietary Mycotoxins, Co-Exposure, and Carcinogenesis in Humans: Short Review. Mutat. Res. Rev. Mutat. Res. 2015, 766, 32–41. [Google Scholar] [CrossRef]
- Powlson, D.S.; Stirling, C.M.; Jat, M.L.; Gerard, B.G.; Palm, C.A.; Sanchez, P.A.; Cassman, K.G. Limited Potential of No-till Agriculture for Climate Change Mitigation. Nat. Clim. Change 2014, 4, 678–683. [Google Scholar] [CrossRef]
- Windels, C.E. Economic and Social Impacts of Fusarium Head Blight: Changing Farms and Rural Communities in the Northern Great Plains. Phytopathology 2000, 90, 17–21. [Google Scholar] [CrossRef]
- Ni, X.; Krakowsky, M.D.; Buntin, G.D.; Rector, B.G.; Guo, B.; Snook, M.E. Identification of Multiple Ear-Colonizing Insect and Disease Resistance in CIMMYT Maize Inbred Lines with Varying Levels of Silk Maysin. J. Econ. Entomol. 2008, 101, 1455–1465. [Google Scholar] [CrossRef]
- Waiss, A.C., Jr.; Chan, B.G.; Elliger, C.A.; Wiseman, B.R.; McMillian, W.W.; Widstrom, N.W.; Zuber, M.S.; Keaster, A.J. Maysin, a Flavone Glycoside from Corn Silks with Antibiotic Activity toward Corn Earworm. J. Econ. Entomol. 1979, 72, 256–258. [Google Scholar] [CrossRef]
- Ni, X.; Xu, W.; Blanco, M.H.; Wilson, J.P. Evaluation of Corn Germplasm Lines for Multiple Ear-Colonizing Insect and Disease Resistance. J. Econ. Entomol. 2012, 105, 1457–1464. [Google Scholar] [CrossRef] [PubMed]
- Reid, L.M.; Mather, D.E.; Arnason, J.T.; Hamilton, R.I.; Bolton, A.T. Changes in Phenolic Constituents of Maize Silk Infected with Fusarium graminearum. Can. J. Bot. 1992, 70, 1697–1702. [Google Scholar] [CrossRef]
- Reid, L.M.; Zhu, X.; Parker, A.; Yan, W. Increased Resistance to Ustilago zeae and Fusarium verticilliodes in Maize Inbred Lines Bred for Fusarium graminearum Resistance. Euphytica 2009, 165, 567–578. [Google Scholar] [CrossRef]
- Dawlal, P.; Barros, E.; Marais, G.J. Evaluation of Maize Cultivars for Their Susceptibility towards Mycotoxigenic Fungi under Storage Conditions. J. Stored Prod. Res. 2012, 48, 114–119. [Google Scholar] [CrossRef]
- Akohoue, F.; Miedaner, T. Meta-Analysis and Co-Expression Analysis Revealed Stable QTL and Candidate Genes Conferring Resistances to Fusarium and Gibberella Ear Rots While Reducing Mycotoxin Contamination in Maize. Front. Plant Sci. 2022, 13, 4379. [Google Scholar] [CrossRef]
- Reid, L.M.; Hamilton, R.I.; Mather, D.E. Effect of Macroconidial Suspension Volume and Concentration on Expression of Resistance to Fusarium graminearum in Maize. Plant Dis. 1995, 79, 366–461. [Google Scholar] [CrossRef]
- Reid, L.M.; Woldemariam, T.; Zhu, X.; Stewart, D.W.; Schaafsma, A.W. Effect of Inoculation Time and Point of Entry on Disease Severity in Fusarium graminearum, Fusarium verticillioides, or Fusarium subglutinans Inoculated Maize Ears. Can. J. Plant Pathol. 2002, 24, 162–167. [Google Scholar] [CrossRef]
- Munkvold, G.P. Cultural and Genetic Approaches to Managing Mycotoxins in Maize. Annu. Rev. Phytopathol. 2003, 41, 99–116. [Google Scholar] [CrossRef]
- Mesterhazy, A.; Szabó, B.; Szél, S.; Nagy, Z.; Berényi, A.; Tóth, B. Novel Insights into the Inheritance of Gibberella Ear Rot (GER), Deoxynivalenol (DON) Accumulation, and DON Production. Toxins 2022, 14, 583. [Google Scholar] [CrossRef]
- Hou, Y.-P.; Qu, X.-P.; Mao, X.-W.; Kuang, J.; Duan, Y.-B.; Song, X.; Wang, J.-X.; Chen, C.-J.; Zhou, M.-G. Resistance Mechanism of Fusarium fujikuroi to Phenamacril in the Field. Pest. Manag. Sci. 2018, 74, 607–616. [Google Scholar] [CrossRef]
- Spolti, P.; Del Ponte, E.M.; Dong, Y.; Cummings, J.A.; Bergstrom, G.C. Triazole Sensitivity in a Contemporary Population of Fusarium graminearum from New York Wheat and Competitiveness of a Tebuconazole-Resistant Isolate. Plant Dis. 2014, 98, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Eli, K.; Schaafsma, A.W.; Limay-Rios, V.; Hooker, D.C. Effect of Pydiflumetofen on Gibberella Ear Rot and Fusarium Mycotoxin Accumulation in Maize Grain. World Mycotoxin J. 2021, 14, 495–512. [Google Scholar] [CrossRef]
- Marin, S.; Magan, N.; Ramos, A.J.; Sanchis, V. Fumonisin-Producing Strains of Fusarium: A Review of Their Ecophysiology. J. Food Prot. 2004, 67, 1792–1805. [Google Scholar] [CrossRef] [PubMed]
- Mitter, B.; Pfaffenbichler, N.; Flavell, R.; Compant, S.; Antonielli, L.; Petric, A.; Berninger, T.; Naveed, M.; Sheibani-Tezerji, R.; von Maltzahn, G.; et al. A New Approach to Modify Plant Microbiomes and Traits by Introducing Beneficial Bacteria at Flowering into Progeny Seeds. Front. Microbiol. 2017, 8, 11. [Google Scholar] [CrossRef]
- Turner, T.R.; James, E.K.; Poole, P.S. The Plant Microbiome. Genome Biol. 2013, 14, 209. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.K.; Singh, M.C.; Singh, S.; Singh, A.K.; Kumar, M.; Pathak, M.; Shakywar, R.C.; Pandey, A.K. Inside the Plants: Endophytic Bacteria and Their Functional Attributes for Plant Growth Promotion. Int. J. Curr. Microbiol. Appl. Sci. 2017, 6, 11–21. [Google Scholar] [CrossRef]
- Hardoim, P.R.; van Overbeek, L.S.; Berg, G.; Pirttilä, A.M.; Compant, S.; Campisano, A.; Döring, M.; Sessitsch, A. The Hidden World within Plants: Ecological and Evolutionary Considerations for Defining Functioning of Microbial Endophytes. Microbiol. Mol. Biol. Rev. 2015, 79, 293–320. [Google Scholar] [CrossRef]
- Rebolleda-Gomez, M.; Forrester, N.; Russell, A.; Wei, N.; Fetters, A.M.; Gómez, R.; Forrester, N.J.; Russell, A.L.; Stephens, J.D.; Ashman, T.-L. Gazing into the Anthosphere: Considering How Microbes Influence Floral Evolution Floral Evolution. New Phytol. 2019, 3, 1012–1020. [Google Scholar] [CrossRef]
- Wei, N.; Ashman, T.L. The Effects of Host Species and Sexual Dimorphism Differ among Root, Leaf and Flower Microbiomes of Wild Strawberries in Situ. Sci. Rep. 2018, 8, 5195. [Google Scholar] [CrossRef]
- Marre, M.; Ushio, M.; Sakai, S. Contrasting Microbial Communities on Male and Female Flowers of a Dioecious Plant, Mallotus japonicus (Euphorbiaceae). Environ. DNA 2022, 4, 565–579. [Google Scholar] [CrossRef]
- Cui, Z.; Huntley, R.B.; Zeng, Q.; Steven, B. Temporal and Spatial Dynamics in the Apple Flower Microbiome in the Presence of the Phytopathogen Erwinia amylovora. ISME J. 2020, 15, 318–329. [Google Scholar] [CrossRef] [PubMed]
- Khalaf, E.M.; Shrestha, A.; Rinne, J.; Lynch, M.D.J.; Shearer, C.R.; Limay-Rios, V.; Reid, L.M.; Raizada, M.N. Transmitting Silks of Maize Have a Complex and Dynamic Microbiome. Sci. Rep. 2021, 11, 13215. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, I.; Persson, P.; Friberg, H. Fusarium Head Blight from a Microbiome Perspective. Front. Microbiol. 2021, 12, 371. [Google Scholar] [CrossRef]
- Pieterse, C.M.J.; Zamioudis, C.; Berendsen, R.L.; Weller, D.M.; Van Wees, S.C.M.; Bakker, P.A.H.M. Induced Systemic Resistance by Beneficial Microbes. Annu. Rev. Phytopathol. 2014, 52, 347–375. [Google Scholar] [CrossRef] [PubMed]
- Mousa, W.K.; Shearer, C.; Limay-Rios, V.; Ettinger, C.L.; Eisen, J.A.; Raizada, M.N. Root-Hair Endophyte Stacking in Finger Millet Creates a Physicochemical Barrier to Trap the Fungal Pathogen Fusarium graminearum. Nat. Microbiol. 2016, 1, 16167. [Google Scholar] [CrossRef]
- De Diniz, G.F.D.; Figueiredo, J.E.F.; Lana, U.G.P.; Marins, M.S.; Silva, D.D.; Cota, L.V.; Marriel, I.E.; Oliveira-Paiva, C.A. Microorganisms from Corn Stigma with Biocontrol Potential of Fusarium verticillioides. Braz. J. Biol. 2022, 82, e262567. [Google Scholar] [CrossRef] [PubMed]
- De Diniz, G.F.D.; Cota, L.V.; Figueiredo, J.E.F.; Aguiar, F.M.; da Silva, D.D.; de Paula Lana, U.G.; dos Santos, V.L.; Marriel, I.E.; de Oliveira-Paiva, C.A. Antifungal Activity of Bacterial Strains from Maize Silks against Fusarium verticillioides. Arch. Microbiol. 2022, 204, 89. [Google Scholar] [CrossRef]
- Frank, A.C.; Guzmán, J.P.S.; Shay, J.E. Transmission of Bacterial Endophytes. Microorganisms 2017, 5, 70. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, S.; de Cates, C.; Hodgson, J.; Morley, N.J.; Sutton, B.C.; Gange, A.C. Vertical Transmission of Fungal Endophytes Is Widespread in Forbs. Ecol. Evol. 2014, 4, 1199–1208. [Google Scholar] [CrossRef]
- Zampino, D.; Duro, A.; Sciandrello, S.; Parafati, L.; Restuccia, C. Pollen Viability and Endophytic Yeast Species of Cistus creticus and C. monspeliensis. Plant Biosyst. 2020, 155, 384–393. [Google Scholar] [CrossRef]
- Khalaf, E.M.; Shrestha, A.; Reid, M.; McFadyen, B.J.; Raizada, M.N. Conservation and Diversity of the Pollen Microbiome of Pan-American Maize Using PacBio and MiSeq. Department of Plant Agriculture, University of Guelph, Guelph, ON N1G 2W1, Canada. 2023; in preparation. [Google Scholar]
- Yoshida, S.; Ohba, A.; Liang, Y.M.; Koitabashi, M.; Tsushima, S. Specificity of Pseudomonas Isolates on Healthy and Fusarium Head Blight-Infected Spikelets of Wheat Heads. Microb. Ecol. 2012, 64, 214–225. [Google Scholar] [CrossRef]
- Deroo, W.; De Troyer, L.; Dumoulin, F.; De Saeger, S.; De Boevre, M.; Vandenabeele, S.; De Gelder, L.; Audenaert, K. A Novel in Planta Enrichment Method Employing Fusarium graminearum-Infected Wheat Spikes to Select for Competitive Biocontrol Bacteria. Toxins 2022, 14, 222. [Google Scholar] [CrossRef] [PubMed]
- Mousa, W.K.; Shearer, C.R.; Limay-Rios, V.; Zhou, T.; Raizada, M.N. Bacterial Endophytes from Wild Maize Suppress Fusarium graminearum in Modern Maize and Inhibit Mycotoxin Accumulation. Front. Plant Sci. 2015, 6, 805. [Google Scholar] [CrossRef] [PubMed]
- Mousa, W.K.; Schwan, A.; Raizada, M. Characterization of Antifungal Natural Products Isolated from Endophytic Fungi of Finger Millet (Eleusine coracana). Molecules 2016, 21, 1171. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Mishra, S.; Dixit, V.; Kumar, M.; Agarwal, L.; Chauhan, P.S.; Nautiyal, C.S. Synergistic Effect of Pseudomonas putida and Bacillus amyloliquefaciens Ameliorates Drought Stress in Chickpea (Cicer arietinum L.). Plant Signal Behav. 2016, 11, e1071004. [Google Scholar] [CrossRef]
- Woo, S.L.; Pepe, O. Microbial Consortia: Promising Probiotics as Plant Biostimulants for Sustainable Agriculture. Front. Plant Sci. 2018, 9, 1801. [Google Scholar] [CrossRef]
- Ankati, S.; Srinivas, V.; Pratyusha, S.; Gopalakrishnan, S. Streptomyces Consortia-Mediated Plant Defense against Fusarium Wilt and Plant Growth-Promotion in Chickpea. Microb. Pathog. 2021, 157, 104961. [Google Scholar] [CrossRef]
- Wong, C.K.F.; Saidi, N.B.; Vadamalai, G.; Teh, C.Y.; Zulperi, D. Effect of Bioformulations on the Biocontrol Efficacy, Microbial Viability and Storage Stability of a Consortium of Biocontrol Agents against Fusarium Wilt of Banana. J. Appl. Microbiol. 2019, 127, 544–555. [Google Scholar] [CrossRef]
- Minchev, Z.; Kostenko, O.; Soler, R.; Pozo, M.J. Microbial Consortia for Effective Biocontrol of Root and Foliar Diseases in Tomato. Front. Plant Sci. 2021, 12, 2428. [Google Scholar] [CrossRef]
- Adeniji, A.A.; Babalola, O.O. Evaluation of Pseudomonas fulva PS9.1 and Bacillus velezensis NWUMFkBS10.5 as Candidate Plant Growth Promoters during Maize-Fusarium Interaction. Plants 2022, 11, 324. [Google Scholar] [CrossRef]
- Schlatter, D.; Kinkel, L.; Thomashow, L.; Weller, D.; Paulitz, T. Disease Suppressive Soils: New Insights from the Soil Microbiome. Phytopathology 2017, 107, 1284–1297. [Google Scholar] [CrossRef] [PubMed]
- Gopal, M.; Gupta, A. Microbiome Selection Could Spur Next-Generation Plant Breeding Strategies. Front. Microbiol. 2016, 7, 1971. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.; Jousset, A. Plant Breeding Goes Microbial. Trends Plant Sci. 2017, 22, 555–558. [Google Scholar] [CrossRef]
- Kinnunen-Grubb, M.; Sapkota, R.; Vignola, M.; Nunes, I.M.; Nicolaisen, M. Breeding Selection Imposed a Differential Selective Pressure on the Wheat Root-Associated Microbiome. FEMS Microbiol. Ecol. 2020, 96, 196. [Google Scholar] [CrossRef]
- Alegria Terrazas, R.; Balbirnie-Cumming, K.; Morris, J.; Hedley, P.E.; Russell, J.; Paterson, E.; Baggs, E.M.; Fridman, E.; Bulgarelli, D. A Footprint of Plant Eco-Geographic Adaptation on the Composition of the Barley Rhizosphere Bacterial Microbiota. Sci. Rep. 2020, 10, 12916. [Google Scholar] [CrossRef]
- Schlechter, R.O.; Miebach, M.; Remus-Emsermann, M.N.P. Driving Factors of Epiphytic Bacterial Communities: A Review. J. Adv. Res. 2019, 19, 57–65. [Google Scholar] [CrossRef]
- Simmons, T.; Styer, A.B.; Pierroz, G.; Gonçalves, A.P.; Pasricha, R.; Hazra, A.B.; Bubner, P.; Coleman-Derr, D. Drought Drives Spatial Variation in the Millet Root Microbiome. Front. Plant Sci. 2020, 11, 599. [Google Scholar] [CrossRef]
- Ramírez-Puebla, S.T.; Weigel, B.L.; Jack, L.; Schlundt, C.; Pfister, C.A.; Mark Welch, J.L. Spatial Organization of the Kelp Microbiome at Micron Scales. Microbiome 2022, 10, 52. [Google Scholar] [CrossRef]
- Thompson, M.E.H. Discovery and Testing of Pollinated Maize Silk-Associated Microbes including Microbiome Assisted Selection of Biocontrol Agents against Fusarium graminearum. Ph.D. Thesis, University of Guelph, Guelph, ON, Canada, 2023. [Google Scholar]
- Priest, F.G.; Barker, M. Gram-Negative Bacteria Associated with Brewery Yeasts: Reclassification of Obesumbacterium proteus Biogroup 2 as Shimwellia pseudoproteus Gen. Nov., Sp. Nov., and Transfer of Escherichia blattae to Shimwellia blattae Comb. Nov. Int. J. Syst. Evol. Microbiol. 2010, 60, 828–833. [Google Scholar] [CrossRef]
- Kuhnem, P.R.; Del Ponte, E.M.; Dong, Y.; Bergstrom, G.C. Fusarium graminearum Isolates from Wheat and Maize in New York Show Similar Range of Aggressiveness and Toxigenicity in Cross-Species Pathogenicity Tests. Phytopathology 2015, 105, 441–448. [Google Scholar] [CrossRef]
- Troyer, A.F.; Hendrickson, L.G. Background and Importance of ‘Minnesota 13′ Corn. Crop Sci. 2007, 47, 905–914. [Google Scholar] [CrossRef]
- Beckett, T.J.; Morales, A.J.; Koehler, K.L.; Rocheford, T.R. Genetic Relatedness of Previously Plant-Variety-Protected Commercial Maize Inbreds. PLoS ONE 2017, 12, e0189277. [Google Scholar] [CrossRef] [PubMed]
- White, M.R.; Mikel, M.A.; de Leon, N.; Kaeppler, S.M. Diversity and Heterotic Patterns in North American Proprietary Dent Maize Germplasm. Crop Sci. 2020, 60, 100–114. [Google Scholar] [CrossRef]
- Borrás, L.; Vitantonio-Mazzini, L.N. Maize Reproductive Development and Kernel Set under Limited Plant Growth Environments. J. Exp. Bot. 2018, 69, 3235–3243. [Google Scholar] [CrossRef]
- Pasley, H.R.; Camberato, J.J.; Cairns, J.E.; Zaman-Allah, M.; Das, B.; Vyn, T.J. Nitrogen Rate Impacts on Tropical Maize Nitrogen Use Efficiency and Soil Nitrogen Depletion in Eastern and Southern Africa. Nutr. Cycl. Agroecosyst. 2020, 116, 397. [Google Scholar] [CrossRef]
- Debruin, J.L.; Hemphill, B.; Schussler, J.R. Silk Development and Kernel Set in Maize as Related to Nitrogen Stress. Crop Sci. 2018, 58, 2581–2592. [Google Scholar] [CrossRef]
- Edmeades, G.O.; Bolanos, J.; Hernandez, M.; Bello, S. Causes for Silk Delay in a Lowland Tropical Maize Population. Crop Sci. 1993, 33, 1029–1035. [Google Scholar] [CrossRef]
- Lemcoff, J.H.; Loomis, R.S. Nitrogen and Density Influences on Silk Emergence, Endosperm Development, and Grain Yield in Maize (Zea mays L.). Field Crops Res. 1994, 38, 63–72. [Google Scholar] [CrossRef]
- Bakker, M.G.; McCormick, S.P. Microbial Correlates of Fusarium Load and Deoxynivalenol Content in Individual Wheat Kernels. Phytopathology 2019, 109, 993–1002. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, J.; Liu, F.; Liang, J.; Zhao, P.; Tsui, C.K.M.; Cai, L. Cross-Kingdom Synthetic Microbiota Supports Tomato Suppression of Fusarium Wilt Disease. Nat. Commun. 2022, 13, 7890. [Google Scholar] [CrossRef]
- Johnston-Monje, D.; Raizada, M.N. Conservation and Diversity of Seed Associated Endophytes in Zea across Boundaries of Evolution, Ethnography and Ecology. PLoS ONE 2011, 6, e20396. [Google Scholar] [CrossRef] [PubMed]
- Engelhard, M.; Hurek, T.; Reinhold-Hurek, B. Preferential Occurrence of Diazotrophic Endophytes, Azoarcus Spp., in Wild Rice Species and Land Races of Oryza sativa in Comparison with Modern Races. Environ. Microbiol. 2000, 2, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Sun, L.; Dong, X.; Cai, Z.; Sun, X.; Yang, H.; Wang, Y.; Song, W. Characterization of a Novel Plant Growth-Promoting Bacteria Strain Delftia tsuruhatensis HR4 Both as a Diazotroph and a Potential Biocontrol Agent against Various Plant Pathogens. Syst. Appl. Microbiol. 2005, 28, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, R.; Nazzi, F.; Locci, R.; Firrao, G. Degradation of Fumonisin B1 by a Bacterial Strain Isolated from Soil. Biodegradation 2006, 17, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Da Silveira, A.P.D.; de Iório, R.P.F.; Marcos, F.C.C.; Fernandes, A.O.; de Souza, S.A.C.D.; Kuramae, E.E.; Cipriano, M.A.P. Exploitation of New Endophytic Bacteria and Their Ability to Promote Sugarcane Growth and Nitrogen Nutrition. Antonie Van. Leeuwenhoek 2019, 112, 283–295. [Google Scholar] [CrossRef]
- Aldwinckle, H.S.; Beer, S.B.; Billing, E.; Bogdanove, A.J.; Bonn, W.G.; Dellagi, A.; Eden-Green, S.; Expert, D.; Geider, K.; Johnson, K.B.; et al. Fire Blight: The Disease and Its Causative Agent, Erwinia Amylovora; Vanneste, J.L., Ed.; CABI Publishing: New York, NY, USA, 2000. [Google Scholar]
- Sheibani-Tezerji, R.; Naveed, M.; Jehl, M.A.; Sessitsch, A.; Rattei, T.; Mitter, B. The Genomes of Closely Related Pantoea ananatis Maize Seed Endophytes Having Different Effects on the Host Plant Differ in Secretion System Genes and Mobile Genetic Elements. Front. Microbiol. 2015, 6, 440. [Google Scholar] [CrossRef]
- Egamberdieva, D.; Shurigin, V.; Alaylar, B.; Ma, H.; Müller, M.E.H.; Wirth, S.; Reckling, M.; Bellingrath-Kimura, S.D. The Effect of Biochars and Endophytic Bacteria on Growth and Root Rot Disease Incidence of Fusarium Infested Narrow-Leafed Lupin (Lupinus angustifolius L.). Microorganisms 2020, 8, 496. [Google Scholar] [CrossRef]
- Higdon, S.M.; Pozzo, T.; Kong, N.; Huang, B.C.; Yang, M.L.; Jeannotte, R.; Brown, C.T.; Bennett, A.B.; Weimer, B.C. Genomic Characterization of a Diazotrophic Microbiota Associated with Maize Aerial Root Mucilage. PLoS ONE 2020, 15, e0239677. [Google Scholar] [CrossRef]
- Higdon, S.M.; Huang, B.C.; Bennett, A.B.; Weimer, B.C. Identification of Nitrogen Fixation Genes in Lactococcus Isolated from Maize Using Population Genomics and Machine Learning. Microorganisms 2020, 8, 2043. [Google Scholar] [CrossRef]
- Ali, H.; Pei, M.; Li, H.; Fang, W.; Khan, H.A.; Nadeem, T.; Olsson, S. The Wheat Head Blight Pathogen Fusarium graminearum Recruits Facultative Endohyphal Bacteria from the Soil, Making the Fungal-Bacterial Holobiont Nitrogen-Fixing and Increasing the Fungal Pathogenicity. bioRxiv 2022. [Google Scholar] [CrossRef]
- Agostini, R.B.; Postigo, A.; Rius, S.P.; Rech, G.E.; Campos-Bermudez, V.A.; Vargas, W.A. Long-Lasting Primed State in Maize Plants: Salicylic Acid and Steroid Signaling Pathways as Key Players in the Early Activation of Immune Responses in Silks. Mol. Plant-Microbe Interact. 2019, 32, 90–106. [Google Scholar] [CrossRef]
- Ramos, P.L.; van Trappen, S.; Thompson, F.L.; Rocha, R.C.S.; Barbosa, H.R.; de Vos, P.; Moreira-Filho, C.A. Screening for Endophytic Nitrogen-Fixing Bacteria in Brazilian Sugar Cane Varieties Used in Organic Farming and Description of Stenotrophomonas pavanii sp. nov. Int. J. Syst. Evol. Microbiol. 2011, 61, 926–931. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, N.; Alikhani, H.A.; Pourbabaee, A.A.; Besharati, H. Study the Effects of Siderophore-Producing Bacteria on Zinc and Phosphorous Nutrition of Canola and Maize Plants. Commun. Soil. Sci. Plant Anal. 2016, 47, 1517–1527. [Google Scholar] [CrossRef]
- Dal Bello, G.M.; Mónaco, C.I.; Simón, M.R. Biological Control of Seedling Blight of Wheat Caused by Fusarium graminearum with Beneficial Rhizosphere Microorganisms. World J. Microbiol. Biotechnol. 2002, 18, 627–636. [Google Scholar] [CrossRef]
- Liu, H.; Li, J.; Carvalhais, L.C.; Percy, C.D.; Prakash Verma, J.; Schenk, P.M.; Singh, B.K. Evidence for the Plant Recruitment of Beneficial Microbes to Suppress Soil-Borne Pathogens. New Phytol. 2021, 229, 2873–2885. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, J.D.; O’Keeffe, T.L.; Abbas, H.K. Isolation of Maize Soil and Rhizosphere Bacteria with Antagonistic Activity against Aspergillus flavus and Fusarium verticillioides. J. Food Prot. 2007, 70, 1615–1621. [Google Scholar] [CrossRef]
- Pérez-Pérez, R.; Oudot, M.; Hernández, I.; Nápoles, M.C.; Sosa-Del Castillo, D. Isolation and Characterization of Stenotrophomonas Associated to Maize (Zea mays L.) Rhizosphere. Cultiv. Trop. 2020, 41, 3. [Google Scholar]
- Brady, C.; Venter, S.N.; Cleenwerck, I.; Engelbeen, K.; Vancanneyt, M.; Swings, J.; Coutinho, T.A. Pantoea vagans sp. nov., Pantoea eucalypti. nov., Pantoea deleyi sp. nov. and Pantoea sp. nov. Int. J. Syst. Evol. Microbiol. 2009, 59, 2339–2345. [Google Scholar] [CrossRef]
- Araujo, S.H.; Júnior, O.F.; Viteri, L.O.; Aguiar, R.W.; Oliveira, E.E.; Santos, G.R. Efficiency of Inoculation Methods for Genotypes Selection in Corn Ear Rot Disease Studies. Rev. Cienc. Agrícolas 2022, 39, 160–173. [Google Scholar] [CrossRef]
- Feng, C.; Liu, H.; Tang, Z. Fusarium graminearum Inoculation on Wheat Head. Bio Protoc. 2018, 8, e2964. [Google Scholar] [CrossRef]
- Sutton, J.C.; Baliko, W.; Funnell, H.S. Relation of Weather Variables to Incidence of Zearalenone in Corn in Southern Ontario. Can. J. Plant Sci. 1980, 60, 149–155. [Google Scholar] [CrossRef]
- Paul, P. Gibberella Ear Rots Showing up in Corn: How to Tell It Apart from Other Ear Rots. Agron. Crops Netw. Ohio State Univ. Ext. 2020, 34, 2. [Google Scholar]
- Shrestha, A. Discovery and Testing of Bacteria from Pollen and Unpollinated Silks of Pan-American Maize to Combat Fusarium graminearum. Ph.D. Thesis, University of Guelph, Guelph, ON, Canada, 2023. [Google Scholar]
- Raaijmakers, J.M.; Kiers, E.T. Rewilding Plant Microbiomes. Science 2022, 378, 599–600. [Google Scholar] [CrossRef] [PubMed]
- Pusey, P.L. Crab Apple Blossoms as a Model for Research on Biological Control of Fire Blight. Phytopathology 1997, 87, 1096–1102. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Pérez-Cobas, A.E.; Gomez-Valero, L.; Buchrieser, C. Metagenomic Approaches in Microbial Ecology: An Update on Whole-Genome and Marker Gene Sequencing Analyses. Microb. Genom. 2020, 6, mgen000409. [Google Scholar] [CrossRef]
- Corbin, K.R.; Bolt, B.; Rodríguez López, C.M. Breeding for Beneficial Microbial Communities Using Epigenomics. Front. Microbiol. 2020, 11, 937. [Google Scholar] [CrossRef] [PubMed]
- Kroll, S.; Agler, M.T.; Kemen, E. Genomic Dissection of Host–Microbe and Microbe–Microbe Interactions for Advanced Plant Breeding. Curr. Opin. Plant Biol. 2017, 36, 71–78. [Google Scholar] [CrossRef]
- Contreras-Liza, S.E. Plant Breeding and Microbiome. Plant Breed. Curr. Future Views 2021. [Google Scholar] [CrossRef]
- Khalaf, E.M.; Raizada, M.N. Taxonomic and Functional Diversity of Cultured Seed Associated Microbes of the Cucurbit Family. BMC Microbiol. 2016, 16, 131. [Google Scholar] [CrossRef]
- Shehata, H.R.; Lyons, E.M.; Jordan, K.S.; Raizada, M.N. Bacterial Endophytes from Wild and Ancient Maize Are Able to Suppress the Fungal Pathogen Sclerotinia Homoeocarpa. J. Appl. Microbiol. 2016, 120, 756–769. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit Sequence Alignment Editor: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA 11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar]
- Letunic, I.; Bork, P. Interactive Tree of Life (ITOL) v5: An Online Tool for Phylogenetic Tree Display and Annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Gdanetz, K.; Trail, F. The Wheat Microbiome under Four Management Strategies, and Potential for Endophytes in Disease Protection. Phytobiomes J. 2017, 1, 158–168. [Google Scholar] [CrossRef]
- Yamamoto, M.; Takai, K. Sulfur Metabolisms in Epsilon-and Gamma-Proteobacteria in Deep-Sea Hydrothermal Fields. Front. Microbiol. 2011, 2, 192. [Google Scholar] [CrossRef]
- Jorquera, M.A.; Maruyama, F.; Ogram, A.V.; Navarrete, O.U.; Lagos, L.M.; Inostroza, N.G.; Acuña, J.J.; Rilling, J.I.; de La Luz Mora, M. Rhizobacterial Community Structures Associated with Native Plants Grown in Chilean Extreme Environments. Microb. Ecol. 2016, 72, 633–646. [Google Scholar] [CrossRef]
- Haack, F.S.; Poehlein, A.; Kröger, C.; Voigt, C.A.; Piepenbring, M.; Bode, H.B.; Daniel, R.; Schäfer, W.; Streit, W.R. Molecular Keys to the Janthinobacterium and Duganella Spp. Interaction with the Plant Pathogen Fusarium graminearum. Front. Microbiol. 2016, 7, 1668. [Google Scholar] [CrossRef]
- Zhang, Y.-L.; Guo, X.-J.; Huang, X.; Guo, R.-J.; Lu, X.-H.; Li, S.-D.; Zhang, H. The Co-Association of Enterobacteriaceae and Pseudomonas with Specific Resistant Cucumber against Fusarium Wilt Disease. Biology 2023, 12, 143. [Google Scholar] [CrossRef]
- Huo, Y.; Kang, J.P.; Park, J.K.; Li, J.; Chen, L.; Yang, D.C. Rhodanobacter Ginsengiterrae Sp. Nov., an Antagonistic Bacterium against Root Rot Fungal Pathogen Fusarium solani, Isolated from Ginseng Rhizospheric Soil. Arch. Microbiol. 2018, 200, 1457–1463. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, M.T.; Arnold, A.E. Diverse Bacteria Inhabit Living Hyphae of Phylogenetically Diverse Fungal Endophytes. Appl. Environ. Microbiol. 2010, 76, 4063. [Google Scholar] [CrossRef]
- Ofek, M.; Hadar, Y.; Minz, D. Ecology of Root Colonizing Massilia (Oxalobacteraceae). PLoS ONE 2012, 7, e40117. [Google Scholar] [CrossRef]
- Yu, P.; He, X.; Baer, M.; Beirinckx, S.; Tian, T.; Moya, Y.A.T.; Zhang, X.; Deichmann, M.; Frey, F.P.; Bresgen, V.; et al. Plant Flavones Enrich Rhizosphere Oxalobacteraceae to Improve Maize Performance under Nitrogen Deprivation. Nat. Plants 2021, 7, 481–499. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhu, A.; Tan, H.; Cao, L.; Zhang, R. Engineering Banana Endosphere Microbiome to Improve Fusarium Wilt Resistance in Banana. Microbiome 2019, 7, 74. [Google Scholar] [CrossRef]
- Yuan, J.; Wen, T.; Zhang, H.; Zhao, M.; Ryan Penton, C.; Thomashow, L.S.; Shen, Q. Predicting Disease Occurrence with High Accuracy Based on Soil Macroecological Patterns of Fusarium Wilt. ISME J. 2020, 14, 2936–2950. [Google Scholar] [CrossRef]
- Wu, X.; Shan, Y.; Li, Y.; Li, Q.; Wu, C. The Soil Nutrient Environment Determines the Strategy by Which Bacillus velezensis HN03 Suppresses Fusarium Wilt in Banana Plants. Front. Plant Sci. 2020, 11, 599904. [Google Scholar] [CrossRef]
- Sun, X.; Zhang, C.; Bei, S.; Wang, G.; Geisen, S.; Bedoussac, L.; Christie, P.; Zhang, J. High Bacterial Diversity and Siderophore-Producing Bacteria Collectively Suppress Fusarium oxysporum in Maize/Faba Bean Intercropping. Front. Microbiol. 2022, 13, 2930. [Google Scholar] [CrossRef]
- Cobo-Díaz, J.F.; Baroncelli, R.; Le Floch, G.; Picot, A. Combined Metabarcoding and Co-Occurrence Network Analysis to Profile the Bacterial, Fungal and Fusarium Communities and Their Interactions in Maize Stalks. Front. Microbiol. 2019, 10, 261. [Google Scholar] [CrossRef]
- Wu, D.; Wang, L.; Zhang, Y.; Bai, L.; Yu, F. Emerging Roles of Pathogen-Secreted Host Mimics in Plant Disease Development. Trends Parasitol. 2021, 37, 1082–1095. [Google Scholar] [CrossRef]
- Wen, T.; Yuan, J.; He, X.; Lin, Y.; Huang, Q.; Shen, Q. Enrichment of Beneficial Cucumber Rhizosphere Microbes Mediated by Organic Acid Secretion. Hortic. Res. 2020, 7, 154. [Google Scholar] [CrossRef]
- Durán, P.; Thiergart, T.; Garrido-Oter, R.; Agler, M.; Kemen, E.; Schulze-Lefert, P.; Hacquard, S. Microbial Interkingdom Interactions in Roots Promote Arabidopsis Survival. Cell 2018, 175, 973–983. [Google Scholar] [CrossRef]
- Lemanceau, P.; Alabouvette, C. Biological Control of Fusarium Diseases by Fluorescent Pseudomonas and Non-Pathogenic Fusarium. Crop Prot. 1991, 10, 279–286. [Google Scholar] [CrossRef]
- Fatima, S.; Anjum, T. Identification of a Potential ISR Determinant from Pseudomonas aeruginosa PM12 against Fusarium Wilt in Tomato. Front. Plant Sci. 2017, 8, 848. [Google Scholar] [CrossRef] [PubMed]
- De Boer, M.; Van Der Sluis, I.; Van Loon, L.C.; Bakker, P.A.H.M. Combining Fluorescent Pseudomonas Spp. Strains to Enhance Suppression of Fusarium Wilt of Radish. Eur. J. Plant Pathol. 1999, 105, 201–210. [Google Scholar] [CrossRef]
- Tao, C.; Li, R.; Xiong, W.; Shen, Z.; Liu, S.; Wang, B.; Ruan, Y.; Geisen, S.; Shen, Q.; Kowalchuk, G.A. Bio-Organic Fertilizers Stimulate Indigenous Soil Pseudomonas Populations to Enhance Plant Disease Suppression. Microbiome 2020, 8, 137. [Google Scholar] [CrossRef]
- Hu, W.; Gao, Q.; Sobhy Hamada, M.; Hosni Dawood, D.; Zheng, J.; Chen, Y.; Ma, Z. Potential of Pseudomonas chlororaphis Subsp. Aurantiaca Strain Pcho10 as a Biocontrol Agent against Fusarium graminearum. Phytopathology 2014, 104, 1289–1297. [Google Scholar] [CrossRef]
- Huang, M.; He, P.; Munir, S.; Wu, Y.; Li, X.; He, P.; He, Y. Ecology and Etiology of Bacterial Top Rot in Maize Caused by Klebsiella pneumoniae KpC4. Microb. Pathog. 2020, 139, 103906. [Google Scholar] [CrossRef]
- Huang, M.; Li, L.; Yi-Xin, W.U.; Ho, H.; Peng-Fei, H.E.; Li, G.-Z.; Peng-Bo, H.E.; Xiong, G.-R.; Yuan, Y.; Yue-Qiu, H.E. Pathogenicity of Klebsiella pneumonia (KpC4) Infecting Maize and Mice. J. Integr. Agric. 2016, 15, 1510–1520. [Google Scholar] [CrossRef]
- Liu, D.; Chen, L.; Zhu, X.; Wang, Y.; Xuan, Y.; Liu, X.; Chen, L.; Duan, Y. Klebsiella pneumoniae SnebYK Mediates Resistance against Heterodera glycines and Promotes Soybean Growth. Front. Microbiol. 2018, 9, 1134. [Google Scholar] [CrossRef]
- Ji, S.H.; Gururani, M.A.; Chun, S.C. Isolation and Characterization of Plant Growth Promoting Endophytic Diazotrophic Bacteria from Korean Rice Cultivars. Microbiol. Res. 2014, 169, 83–98. [Google Scholar] [CrossRef]
- Figueroa-López, A.M.; Cordero-Ramírez, J.D.; Martínez-Álvarez, J.C.; López-Meyer, M.; Lizárraga-Sánchez, G.J.; Félix-Gastélum, R.; Castro-Martínez, C.; Maldonado-Mendoza, I.E. Rhizospheric Bacteria of Maize with Potential for Biocontrol of Fusarium verticillioides. Springerplus 2016, 5, 330. [Google Scholar] [CrossRef]
- Rosenblueth, M.; Martínez, L.; Silva, J.; Martínez-Romero, E. Klebsiella variicola, a Novel Species with Clinical and Plant-Associated Isolates. Syst. Appl. Microbiol. 2004, 27, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Yang, K. Biological Function of Klebsiella variicola and Its Effect on the Rhizosphere Soil of Maize Seedlings. PeerJ 2020, 8, e9894. [Google Scholar] [CrossRef] [PubMed]
- Johnston-Monje, D.; Gutiérrez, J.P.; Lopez-Lavalle, L.A.B. Seed-Transmitted Bacteria and Fungi Dominate Juvenile Plant Microbiomes. Front. Microbiol. 2021, 12, 2945. [Google Scholar] [CrossRef]
- Dumigan, C.R.; Muileboom, J.; Gregory, J.; Shrestha, A.; Hewedy, O.A.; Raizada, M.N. Ancient Relatives of Modern Maize from the Center of Maize Domestication and Diversification Host Endophytic Bacteria That Confer Tolerance to Nitrogen Starvation. Front. Plant Sci. 2021, 12, 1908. [Google Scholar] [CrossRef]
- Krawczyk, K.; Foryś, J.; Nakonieczny, M.; Tarnawska, M.; Bereś, P.K. Transmission of Pantoea ananatis, the Causal Agent of Leaf Spot Disease of Maize (Zea mays), by Western Corn Rootworm (Diabrotica virgifera virgifera LeConte). Crop Prot. 2021, 141, 105431. [Google Scholar] [CrossRef]
- Paccola-Meirelles, L.D.; Ferreira, A.S.; Meirelles, W.F.; Marriel, I.E.; Casela, C.R. Detection of a Bacterium Associated with a Leaf Spot Disease of Maize in Brazil. J. Phytopathol. 2001, 149, 275–279. [Google Scholar] [CrossRef]
- Lana, U.G.D.P.; Gomes, E.A.; Silva, D.D.; Costa, R.V.; Cota, L.V.; Parreira, D.F.; Souza, I.R.P.; Teixeira Guimaraes, C. Detection and Molecular Diversity of Pantoea ananatis Associated with White Spot Disease in Maize, Sorghum and Crabgrass in Brazil. J. Phytopathol. 2012, 160, 441–448. [Google Scholar] [CrossRef]
- Pomini, A.M.; Paccola-Meirelles, L.D.; Marsaioli, A.J. Acyl-Homoserine Lactones Produced by Pantoea sp. Isolated from the “Maize White Spot” Foliar Disease. J. Agric. Food Chem. 2007, 55, 1200–1204. [Google Scholar] [CrossRef]
- Coutinho, T.A.; Venter, S.N. Pantoea ananatis: An Unconventional Plant Pathogen. Mol. Plant Pathol. 2009, 10, 325–335. [Google Scholar] [CrossRef]
- Goszczynska, T.; Botha, W.J.; Venter, S.N.; Coutinho, T.A. Isolation and Identification of the Causal Agent of Brown Stalk Rot, a New Disease of Maize in South Africa. Plant Dis. 2007, 91, 711–718. [Google Scholar] [CrossRef]
- Dutkiewicz, J.; Mackiewicz, B.; Lemieszek, M.K.; Golec, M.; Milanowski, J. Pantoea agglomerans: A Mysterious Bacterium of Evil and Good. Part IV. Beneficial Effects. Ann. Agric. Environ. Med. 2016, 23, 206–222. [Google Scholar] [CrossRef] [PubMed]
- Coplin, D.L.; Majerczak, D.R.; Zhang, Y.; Kim, W.S.; Jock, S.; Geider, K. Identification of Pantoea stewartii subsp. stewartii by PCR and Strain Differentiation by PFGE. Plant Dis. 2007, 86, 304–311. [Google Scholar] [CrossRef]
- Ahmad, M.; Majerczak, D.R.; Pike, S.; Hoyos, M.E.; Novacky, A.; Coplin, D.L. Biological Activity of Harpin Produced by Pantoea stewartii subsp. stewartii. Mol. Plant-Microbe Interact. 2001, 14, 1223–1234. [Google Scholar] [PubMed]
- Mishra, A.; Chauhan, P.S.; Chaudhry, V.; Tripathi, M.; Nautiyal, C.S. Rhizosphere Competent Pantoea agglomerans Enhances Maize (Zea mays) and Chickpea (Cicer arietinum L.) Growth, without Altering the Rhizosphere Functional Diversity. Antonie Van Leeuwenhoek 2011, 100, 405–413. [Google Scholar] [CrossRef]
- Gond, S.K.; Torres, M.S.; Bergen, M.S.; Helsel, Z.; White, J.F. Induction of Salt Tolerance and Up-Regulation of Aquaporin Genes in Tropical Corn by Rhizobacterium Pantoea agglomerans. Lett. Appl. Microbiol. 2015, 60, 392–399. [Google Scholar] [CrossRef] [PubMed]
- Smaoui, S.; Agriopoulou, S.; D’Amore, T.; Tavares, L.; Mousavi Khaneghah, A. The Control of Fusarium Growth and Decontamination of Produced Mycotoxins by Lactic Acid Bacteria. Crit. Rev. Food Sci. Nutr. 2022, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Gallo, A.; Bernardes, T.F.; Copani, G.; Fortunati, P.; Giuberti, G.; Bruschi, S.; Bryan, K.A.; Nielsen, N.G.; Witt, K.L.; Masoero, F. Effect of Inoculation with Lactobacillus buchneri LB1819 and Lactococcus lactis O224 on Fermentation and Mycotoxin Production in Maize Silage Compacted at Different Densities. Anim. Feed. Sci. Technol. 2018, 246, 36–45. [Google Scholar] [CrossRef]
- Zebboudj, N.; Yezli, W.; Hamini-Kadar, N.; Kihal, M.; Henni, J.E. Antifungal Activity of Lactic Acid Bacteria against Fusarium oxysporum f. sp. Albedinis Isolated from Diseased Date Palm in South Algeria. Int. J. Biosci. 2014, 5, 99–106. [Google Scholar] [CrossRef]
- Marag, P.S.; Suman, A. Growth Stage and Tissue Specific Colonization of Endophytic Bacteria Having Plant Growth Promoting Traits in Hybrid and Composite Maize (Zea mays L.). Microbiol. Res. 2018, 214, 101–113. [Google Scholar] [CrossRef]
- Zinniel, D.K.; Lambrecht, P.; Harris, N.B.; Feng, Z.; Kuczmarski, D.; Higley, P.; Ishimaru, C.A.; Arunakumari, A.; Barletta, R.G.; Vidaver, A.K. Isolation and Characterization of Endophytic Colonizing Bacteria from Agronomic Crops and Prairie Plants. Appl. Environ. Microbiol. 2002, 68, 2198–2208. [Google Scholar] [CrossRef]
- Lin, S.-Y.; Hameed, A.; Liu, Y.-C.; Hsu, Y.-H.; Hsieh, Y.-T.; Lai, W.-A.; Young, C.-C. Chryseobacterium endophyticum sp. nov. Isolated from a Maize Leaf. Int. J. Syst. Evol. Microbiol. 2017, 67, 570–575. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.K.; Gohel, K.; Patel, H.; Solanki, T. Wheat Growth Dependent Succession of Culturable Endophytic Bacteria and Their Plant Growth Promoting Traits. Curr. Microbiol. 2021, 78, 4103–4114. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thompson, M.E.H.; Shrestha, A.; Rinne, J.; Limay-Rios, V.; Reid, L.; Raizada, M.N. The Cultured Microbiome of Pollinated Maize Silks Shifts after Infection with Fusarium graminearum and Varies by Distance from the Site of Pathogen Inoculation. Pathogens 2023, 12, 1322. https://doi.org/10.3390/pathogens12111322
Thompson MEH, Shrestha A, Rinne J, Limay-Rios V, Reid L, Raizada MN. The Cultured Microbiome of Pollinated Maize Silks Shifts after Infection with Fusarium graminearum and Varies by Distance from the Site of Pathogen Inoculation. Pathogens. 2023; 12(11):1322. https://doi.org/10.3390/pathogens12111322
Chicago/Turabian StyleThompson, Michelle E. H., Anuja Shrestha, Jeffrey Rinne, Victor Limay-Rios, Lana Reid, and Manish N. Raizada. 2023. "The Cultured Microbiome of Pollinated Maize Silks Shifts after Infection with Fusarium graminearum and Varies by Distance from the Site of Pathogen Inoculation" Pathogens 12, no. 11: 1322. https://doi.org/10.3390/pathogens12111322
APA StyleThompson, M. E. H., Shrestha, A., Rinne, J., Limay-Rios, V., Reid, L., & Raizada, M. N. (2023). The Cultured Microbiome of Pollinated Maize Silks Shifts after Infection with Fusarium graminearum and Varies by Distance from the Site of Pathogen Inoculation. Pathogens, 12(11), 1322. https://doi.org/10.3390/pathogens12111322