
Trichuris muris Model: Role in Understanding Intestinal Immune Response, Inflammation and Host Defense

 , , , and
, , , and 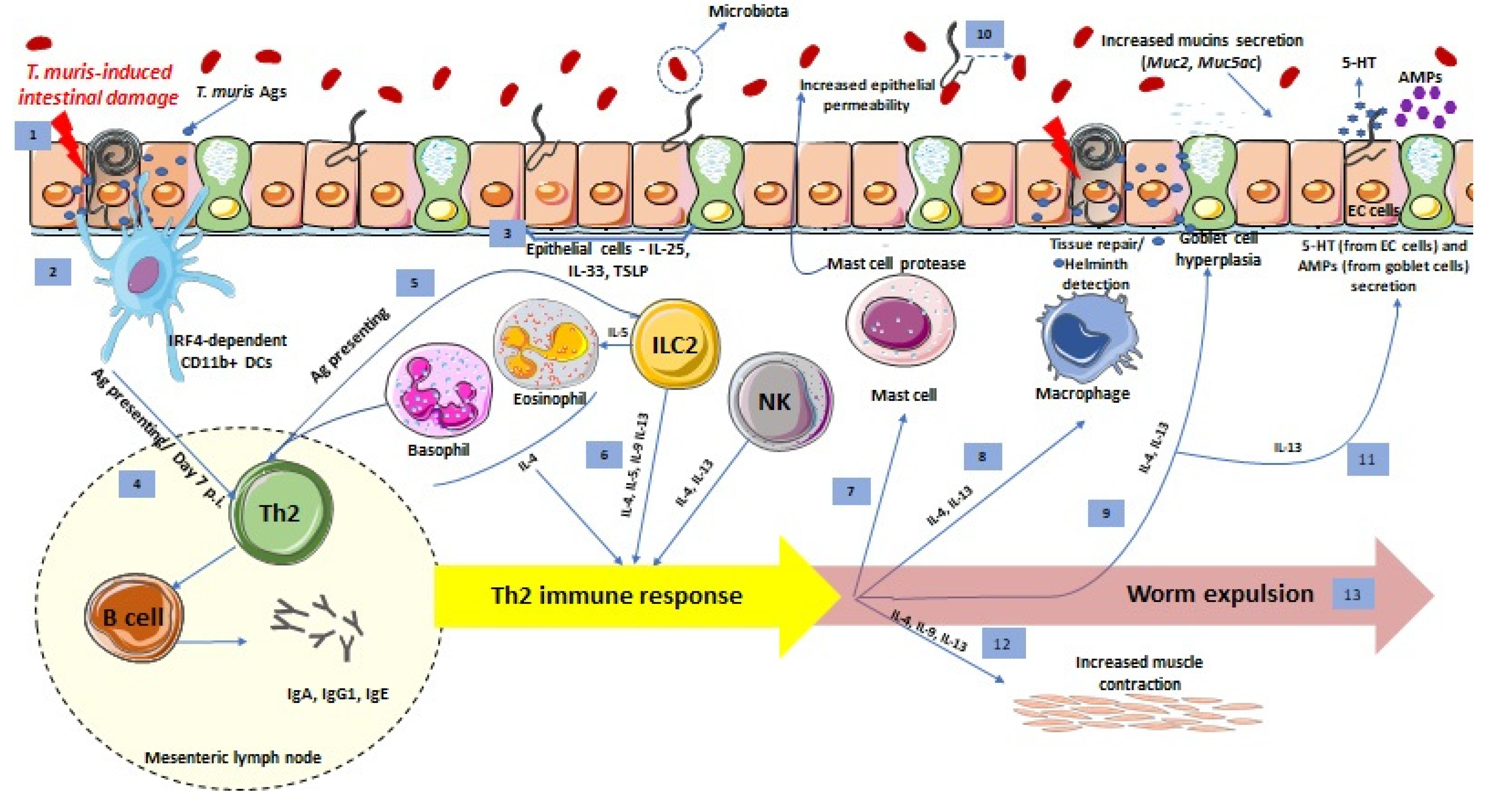{kind=link}
{kind=link}
Abstract
1. Introduction
2. T. muris Model
2.1. Life Cycle of T. muris
2.2. Immune Responses against T. muris Infection
2.2.1. Innate Immune Response
Macrophages
Dendritic Cells (DCs)
Basophils
Eosinophils
Mast Cells
Natural Killer (NK) Cells
Innate Lymphoid Cells (ILCs)
2.2.2. Adaptive Immunity
T Cells
B Cells
Regulatory T Cells (Tregs)
3. Effects of T. muris on the Cells in the Intestinal Epithelial Layer
3.1. Effects on Epithelial Cells
3.2. Effects on Goblet Cells and Mucus Layer
3.3. Effect on Enteroendocrine Cells
4. Effects of T. muris on Intestinal Muscle Function
5. Interaction of T. muris with Gut Microbiota
6. Role of T. muris in the Modulation of Immune and Inflammatory Disorders
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wright, J.E.; Werkman, M.; Dunn, J.C.; Anderson, R.M. Current epidemiological evidence for predisposition to high or low intensity human helminth infection: A systematic review. Parasit. Vectors 2018, 11, 1–12. [Google Scholar] [CrossRef]
- Dunn, J.C.; Turner, H.C.; Tun, A.; Anderson, R.M. Epidemiological surveys of, and research on, soil-transmitted helminths in Southeast Asia: A systematic review. Parasit. Vectors 2016, 9, 1–13. [Google Scholar] [CrossRef]
- Hotez, P.J.; Fenwick, A.; Savioli, L.; Molyneux, D.H. Rescuing the bottom billion through control of neglected tropical diseases. Lancet 2009, 373, 1570–1575. [Google Scholar] [CrossRef]
- Cruz, K.; Marcilla, A.; Kelly, P.; Vandenplas, M.; Osuna, A.; Trelis, M. Trichuris trichiura egg extract proteome reveals potential diagnostic targets and immunomodulators. PLoS Negl. Trop. Dis. 2021, 15, e0009221. [Google Scholar] [CrossRef] [PubMed]
- Gilman, R.H.; Chong, Y.H.; Davis, C.; Greenberg, B.; Virik, H.K.; Dixon, H.B. The adverse consequences of heavy Trichuris infection. Trans. R. Soc. Trop. Med. Hyg. 1983, 77, 432–438. [Google Scholar] [CrossRef]
- World Health Organization. Deworming for Health and Development: Report of the Third Global Meeting of the Partners for Parasite Control; World Health Organization: Geneva, Switzerland, 2005. [Google Scholar]
- Zaph, C.; Cooper, P.J.; Harris, N.L. Mucosal immune responses following intestinal nematode infection. Parasite Immunol. 2014, 36, 439–452. [Google Scholar] [CrossRef] [PubMed]
- Khan, W.I. Physiological changes in the gastrointestinal tract and host protective immunity: Learning from the mouse-Trichinella spiralis model. Parasitology 2008, 135, 671. [Google Scholar] [CrossRef] [PubMed]
- Klementowicz, J.E.; Travis, M.A.; Grencis, R.K. Trichuris muris: A model of gastrointestinal parasite infection. In Proceedings of the Seminars in immunopathology. Semin. Immunopathol. 2012, 34, 815–828. [Google Scholar] [CrossRef] [PubMed]
- Cliffe, L.J.; Grencis, R.K. The Trichuris muris system: A paradigm of resistance and susceptibility to intestinal nematode infection. Adv. Parasitol. 2004, 57, 255–307. [Google Scholar] [PubMed]
- Darlan, D.M.; Rozi, M.F.; Yulfi, H. Overview of Immunological Responses and Immunomodulation Properties of Trichuris sp.: Prospects for Better Understanding Human Trichuriasis. Life 2021, 11, 188. [Google Scholar] [CrossRef]
- Tilney, L.G.; Connelly, P.S.; Guild, G.M.; Vranich, K.A.; Artis, D. Adaptation of a nematode parasite to living within the mammalian epithelium. J. Exp. Zool. Part A Comp. Exp. Biol. 2005, 303, 927–945. [Google Scholar] [CrossRef]
- Hansen, T.V.A.; Hansen, M.; Nejsum, P.; Mejer, H.; Denwood, M.; Thamsborg, S.M. Glucose absorption by the bacillary band of Trichuris muris. PLoS Negl. Trop. Dis. 2016, 10, e0004971. [Google Scholar] [CrossRef] [PubMed]
- Else, K.J.; Wakelin, D.; Wassom, D.L.; Hauda, K.M. The influence of genes mapping within the major histocompatibility complex on resistance to Trichuris muris infections in mice. Parasitology 1990, 101, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Else, K.; Wakelin, D. The effects of H-2 and non-H-2 genes on the expulsion of the nematode Trichuris muris from inbred and congenic mice. Parasitology 1988, 96, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, R.H. T-lymphocyte recognition of antigen in association with gene products of the major histocompatibility complex. Annu. Rev. Immunol. 1985, 3, 237–261. [Google Scholar] [CrossRef]
- Hepworth, M.R.; Hardman, M.J.; Grencis, R.K. The role of sex hormones in the development of Th2 immunity in a gender-biased model of Trichuris muris infection. Eur. J. Immunol. 2010, 40, 406–416. [Google Scholar] [CrossRef]
- Bancroft, A.J.; Else, K.J.; Grencis, R.K. Low level infection with Trichuris muris significantly affects the polarization of the CD4 response. Eur. J. Immunol. 1994, 24, 3113–3118. [Google Scholar] [CrossRef] [PubMed]
- Bancroft, A.J.; Else, K.J.; Humphreys, N.E.; Grencis, R.K. The effect of challenge and trickle Trichuris muris infections on the polarisation of the immune response. Int. J. Parasitol. 2001, 31, 1627–1637. [Google Scholar] [CrossRef]
- Koyama, K.; Ito, Y. Comparative studies on immune responses to infection in susceptible B10. BR mice infected with different strains of the murine nematode parasite Trichuris muris. Parasite Immunol. 1996, 18, 257–263. [Google Scholar] [CrossRef]
- Bellaby, T.; Robinson, K.; Wakelin, D. Induction of differential T-helper-cell responses in mice infected with variants of the parasitic nematode Trichuris muris. Infect. Immun. 1996, 64, 791–795. [Google Scholar] [CrossRef]
- Harris, N.L. Recent advances in type-2-cell-mediated immunity: Insights from helminth infection. Immunity 2017, 47, 1024–1036. [Google Scholar] [CrossRef]
- Coakley, G.; Harris, N.L. Interactions between macrophages and helminths. Parasite Immunol. 2020, 42, e12717. [Google Scholar] [CrossRef] [PubMed]
- Little, M.C.; Hurst, R.J.M.; Else, K.J. Dynamic changes in macrophage activation and proliferation during the development and resolution of intestinal inflammation. J. Immunol. 2014, 193, 4684–4695. [Google Scholar] [CrossRef] [PubMed]
- Sorobetea, D.; Svensson-Frej, M.; Grencis, R. Immunity to gastrointestinal nematode infections. Mucosal Immunol. 2018, 11, 304–315. [Google Scholar] [CrossRef] [PubMed]
- De Schoolmeester, M.L.; Martinez Pomares, L.; Gordon, S.; Else, K.J. The mannose receptor binds Trichuris muris excretory/secretory proteins but is not essential for protective immunity. Immunology 2009, 126, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Cruickshank, S.M.; De Schoolmeester, M.L.; Svensson, M.; Howell, G.; Bazakou, A.; Logunova, L.; Little, M.C.; English, N.; Mack, M.; Grencis, R.K. Rapid dendritic cell mobilization to the large intestinal epithelium is associated with resistance to Trichuris muris infection. J. Immunol. 2009, 182, 3055–3062. [Google Scholar] [CrossRef] [PubMed]
- Mayer, J.U.; Demiri, M.; Agace, W.W.; MacDonald, A.S.; Svensson-Frej, M.; Milling, S.W. Different populations of CD11b+ dendritic cells drive Th2 responses in the small intestine and colon. Nat. Commun. 2017, 8, 1–12. [Google Scholar] [CrossRef]
- Joeris, T.; Müller-Luda, K.; Agace, W.W.; Mowat, A.M. Diversity and functions of intestinal mononuclear phagocytes. Mucosal Immunol. 2017, 10, 845–864. [Google Scholar] [CrossRef]
- Demiri, M.; Müller-Luda, K.; Agace, W.W.; Svensson-Frej, M. Distinct DC subsets regulate adaptive Th1 and 2 responses during Trichuris muris infection. Parasite Immunol. 2017, 39, e12458. [Google Scholar] [CrossRef]
- Little, M.C.; Bell, L.V.; Cliffe, L.J.; Else, K.J. The characterization of intraepithelial lymphocytes, lamina propria leukocytes, and isolated lymphoid follicles in the large intestine of mice infected with the intestinal nematode parasite Trichuris muris. J. Immunol. 2005, 175, 6713–6722. [Google Scholar] [CrossRef] [PubMed]
- Else, K.J.; Finkelman, F.D.; Maliszewski, C.R.; Grencis, R.K. Cytokine-mediated regulation of chronic intestinal helminth infection. J. Exp. Med. 1994, 179, 347–351. [Google Scholar] [CrossRef]
- Webb, L.M.; Wojno, E.D.T. The role of rare innate immune cells in Type 2 immune activation against parasitic helminths. Parasitology 2017, 144, 1288. [Google Scholar] [CrossRef]
- Siracusa, M.C.; Saenz, S.A.; Hill, D.A.; Kim, B.S.; Headley, M.B.; Doering, T.A.; Wherry, E.J.; Jessup, H.K.; Siegel, L.A.; Kambayashi, T. TSLP promotes interleukin-3-independent basophil haematopoiesis and type 2 inflammation. Nature 2011, 477, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Perrigoue, J.G.; Saenz, S.A.; Siracusa, M.C.; Allenspach, E.J.; Taylor, B.C.; Giacomin, P.R.; Nair, M.G.; Du, Y.; Zaph, C.; Van Rooijen, N. MHC class II–dependent basophil–CD4+ T cell interactions promote TH2 cytokine–dependent immunity. Nat. Immunol. 2009, 10, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Webb, L.M.; Oyesola, O.O.; Früh, S.P.; Kamynina, E.; Still, K.M.; Patel, R.K.; Peng, S.A.; Cubitt, R.L.; Grimson, A.; Grenier, J.K. The Notch signaling pathway promotes basophil responses during helminth-induced type 2 inflammation. J. Exp. Med. 2019, 216, 1268–1279. [Google Scholar] [CrossRef]
- Kim, S.; Prout, M.; Ramshaw, H.; Lopez, A.F.; LeGros, G.; Min, B. Cutting edge: Basophils are transiently recruited into the draining lymph nodes during helminth infection via IL-3, but infection-induced Th2 immunity can develop without basophil lymph node recruitment or IL-3. J. Immunol. 2010, 184, 1143–1147. [Google Scholar] [CrossRef]
- Phythian-Adams, A.T.; Cook, P.C.; Lundie, R.J.; Jones, L.H.; Smith, K.A.; Barr, T.A.; Hochweller, K.; Anderton, S.M.; Hämmerling, G.J.; Maizels, R.M. CD11c depletion severely disrupts Th2 induction and development in vivo. J. Exp. Med. 2010, 207, 2089–2096. [Google Scholar] [CrossRef]
- Sullivan, B.M.; Liang, H.-E.; Bando, J.K.; Wu, D.; Cheng, L.E.; McKerrow, J.K.; Allen, C.D.C.; Locksley, R.M. Genetic analysis of basophil function in vivo. Nat. Immunol. 2011, 12, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Dixon, H.; Blanchard, C.; Deschoolmeester, M.L.; Yuill, N.C.; Christie, J.W.; Rothenberg, M.E.; Else, K.J. The role of Th2 cytokines, chemokines and parasite products in eosinophil recruitment to the gastrointestinal mucosa during helminth infection. Eur. J. Immunol. 2006, 36, 1753–1763. [Google Scholar] [CrossRef]
- Svensson, M.; Bell, L.; Little, M.C.; De Schoolmeester, M.; Locksley, R.M.; Else, K.J. Accumulation of eosinophils in intestine draining mesenteric lymph nodes occurs after Trichuris muris infection. Parasite Immunol. 2011, 33, 1–11. [Google Scholar] [CrossRef]
- Sorobetea, D.; Holm, J.B.; Henningsson, H.; Kristiansen, K.; Svensson Frej, M. Acute infection with the intestinal parasite Trichuris muris has long term consequences on mucosal mast cell homeostasis and epithelial integrity. Eur. J. Immunol. 2017, 47, 257–268. [Google Scholar] [CrossRef]
- Betts, C.J.; Else, K.J. Mast cells, eosinophils and antibody mediated cellular cytotoxicity are not critical in resistance to Trichuris muris. Parasite Immunol. 1999, 21, 45–52. [Google Scholar] [CrossRef]
- Brillantes, M.; Beaulieu, A.M. Memory and memory-like NK cell responses to microbial pathogens. Front. Cell. Infect. Microbiol. 2020, 10, 102. [Google Scholar] [CrossRef]
- Krauss, M.E.Z. CD4+ T Cell Metabolism during Trichuris muris Infection; The University of Manchester: Manchester, UK, 2018; ISBN 1083519131. [Google Scholar]
- Hayes, K.S.; Bancroft, A.J.; Grencis, R.K. The role of TNF-α in Trichuris muris infection I: Influence of TNF-α receptor usage, gender and IL-13. Parasite Immunol. 2007, 29, 575–582. [Google Scholar] [CrossRef]
- Hepworth, M.R.; Grencis, R.K. Disruption of Th2 immunity results in a gender-specific expansion of IL-13 producing accessory NK cells during helminth infection. J. Immunol. 2009, 183, 3906–3914. [Google Scholar] [CrossRef]
- Martín-Fontecha, A.; Thomsen, L.L.; Brett, S.; Gerard, C.; Lipp, M.; Lanzavecchia, A.; Sallusto, F. Induced recruitment of NK cells to lymph nodes provides IFN-γ for TH 1 priming. Nature immunology. Nat. Immunol. 2004, 5, 1260–1265. [Google Scholar] [CrossRef] [PubMed]
- Wald, O.; Weiss, I.D.; Wald, H.; Shoham, H.; Bar-Shavit, Y.; Beider, K.; Galun, E.; Weiss, L.; Flaishon, L.; Shachar, I. IFN-γ acts on T cells to induce NK cell mobilization and accumulation in target organs. J. Immunol. 2006, 176, 4716–4729. [Google Scholar] [CrossRef] [PubMed]
- Artis, D.; Spits, H. The biology of innate lymphoid cells. Nature 2015, 517, 293–301. [Google Scholar] [CrossRef]
- Kumar, V. Innate lymphoid cells: New paradigm in immunology of inflammation. Immunol. Lett. 2014, 157, 23–37. [Google Scholar] [CrossRef]
- Oliphant, C.J.; Hwang, Y.Y.; Walker, J.A.; Salimi, M.; Wong, S.H.; Brewer, J.M.; Englezakis, A.; Barlow, J.L.; Hams, E.; Scanlon, S.T. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4+ T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity 2014, 41, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Glover, M.; Colombo, S.A.P.; Thornton, D.J.; Grencis, R.K. Trickle infection and immunity to Trichuris muris. PLoS Pathog. 2019, 15, e1007926. [Google Scholar] [CrossRef] [PubMed]
- Grencis, R.K.; Humphreys, N.E.; Bancroft, A.J. Immunity to gastrointestinal nematodes: Mechanisms and myths. Immunol. Rev. 2014, 260, 183–205. [Google Scholar] [CrossRef] [PubMed]
- Isah, A.U.J.; Ekwunife, O.I.; Ejie, I.L.; Mandrik, O. Effects of nutritional supplements on the re-infection rate of soil-transmitted helminths in school-age children: A systematic review and meta-analysis. PLoS ONE 2020, 15, e0237112. [Google Scholar] [CrossRef] [PubMed]
- Al-Mekhlafi, H.M.; Anuar, T.S.; Al-Zabedi, E.M.; Al-Maktari, M.T.; Mahdy, M.A.K.; Ahmed, A.; Sallam, A.A.; Abdullah, W.A.; Moktar, N.; Surin, J. Does vitamin A supplementation protect schoolchildren from acquiring soil-transmitted helminthiasis? A randomized controlled trial. Parasit. Vectors 2014, 7, 1–9. [Google Scholar] [CrossRef]
- Lee, T.D.G.; Wakelin, D.; Grencis, R.K. Cellular mechanisms of immunity to the nematode Trichuris muris. Int. J. Parasitol. 1983, 13, 349–353. [Google Scholar] [CrossRef]
- Yoichi, I. The absence of resistance in congenitally athymic nude mice toward infection with the intestinal nematode, Trichuris muris: Resistance restored by lymphoid cell transfer. Int. J. Parasitol. 1991, 21, 65–69. [Google Scholar] [CrossRef]
- Else, K.J.; Grencis, R.K. Antibody-independent effector mechanisms in resistance to the intestinal nematode parasite Trichuris muris. Infect. Immun. 1996, 64, 2950–2954. [Google Scholar] [CrossRef]
- Koyama, K.; Tamauchi, H.; Ito, Y. The role of CD4+ and CD8+ T cells in protective immunity to the murine nematode parasite Trichuris muris. Parasite Immunol. 1995, 17, 161–165. [Google Scholar] [CrossRef]
- Humphreys, N.E.; Worthington, J.J.; Little, M.C.; Rice, E.J.; Grencis, R.K. The role of CD8+ cells in the establishment and maintenance of a Trichuris muris infection. Parasite Immunol. 2004, 26, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Koyama, K. NK1. 1+ cell depletion in vivo fails to prevent protection against infection with the murine nematode parasite Trichuris muris. Parasite Immunol. 2002, 24, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, N.E.; Grencis, R.K. Effects of ageing on the immunoregulation of parasitic infection. Infect. Immun. 2002, 70, 5148–5157. [Google Scholar] [CrossRef]
- Bancroft, A.J.; McKenzie, A.N.J.; Grencis, R.K. A critical role for IL-13 in resistance to intestinal nematode infection. J. Immunol. 1998, 160, 3453–3461. [Google Scholar]
- Urban, J.F., Jr.; Madden, K.B.; Svetica, A.; Cheever, A.; Trotta, P.P.; Gause, W.C.; Katona, I.M.; Finkelman, F.D. The importance of Th2 cytokines in protective immunity to nematodes. Immunol. Rev. 1992, 127, 205–220. [Google Scholar] [CrossRef] [PubMed]
- Sharba, S.; Navabi, N.; Padra, M.; Persson, J.A.; Quintana-Hayashi, M.P.; Gustafsson, J.K.; Szeponik, L.; Venkatakrishnan, V.; Sjöling, Å.; Nilsson, S.; et al. Interleukin 4 induces rapid mucin transport, increases mucus thickness and quality and decreases colitis and Citrobacter rodentium in contact with epithelial cells. Virulence 2019, 10, 97–117. [Google Scholar] [CrossRef]
- Dardalhon, V.; Awasthi, A.; Kwon, H.; Galileos, G.; Gao, W.; Sobel, R.A.; Mitsdoerffer, M.; Strom, T.B.; Elyaman, W.; Ho, I.-C. IL-4 inhibits TGF-β-induced Foxp3+ T cells and, together with TGF-β, generates IL-9+ IL-10+ Foxp3− effector T cells. Nat. Immunol. 2008, 9, 1347–1355. [Google Scholar] [CrossRef]
- Veldhoen, M.; Uyttenhove, C.; Van Snick, J.; Helmby, H.; Westendorf, A.; Buer, J.; Martin, B.; Wilhelm, C.; Stockinger, B. Transforming growth factor-β’reprograms’ the differentiation of T helper 2 cells and promotes an interleukin 9–producing subset. Nat. Immunol. 2008, 9, 1341–1346. [Google Scholar] [CrossRef] [PubMed]
- Khan, W.I.; Richard, M.; Akiho, H.; Blennerhasset, P.A.; Humphreys, N.E.; Grencis, R.K.; Van Snick, J.; Collins, S.M. Modulation of intestinal muscle contraction by interleukin-9 (IL-9) or IL-9 neutralization: Correlation with worm expulsion in murine nematode infections. Infect. Immun. 2003, 71, 2430–2438. [Google Scholar] [CrossRef] [PubMed]
- Sahputra, R.; Ruckerl, D.; Couper, K.N.; Muller, W.; Else, K.J. The essential role played by B cells in supporting protective immunity against Trichuris muris infection is by controlling the Th1/Th2 balance in the mesenteric lymph nodes and depends on host genetic background. Front. Immunol. 2019, 10, 2842. [Google Scholar] [CrossRef]
- Blackwell, N.M.; Else, K.J. B cells and antibodies are required for resistance to the parasitic gastrointestinal nematode Trichuris muris. Infect. Immun. 2001, 69, 3860–3868. [Google Scholar] [CrossRef][Green Version]
- Makepeace, B.L.; Martin, C.; Turner, J.D.; Specht, S. Granulocytes in helminth infection-who is calling the shots? Curr. Med. Chem. 2012, 19, 1567–1586. [Google Scholar] [CrossRef] [PubMed]
- D’Elia, R.; Behnke, J.M.; Bradley, J.E.; Else, K.J. Regulatory T cells: A role in the control of helminth-driven intestinal pathology and worm survival. J. Immunol. 2009, 182, 2340–2348. [Google Scholar] [CrossRef]
- Rausch, S.; Huehn, J.; Loddenkemper, C.; Hepworth, M.R.; Klotz, C.; Sparwasser, T.; Hamann, A.; Lucius, R.; Hartmann, S. Establishment of nematode infection despite increased Th2 responses and immunopathology after selective depletion of Foxp3+ cells. Eur. J. Immunol. 2009, 39, 3066–3077. [Google Scholar] [CrossRef]
- Sawant, D.V.; Gravano, D.M.; Vogel, P.; Giacomin, P.; Artis, D.; Vignali, D.A.A. Regulatory T cells limit induction of protective immunity and promote immune pathology following intestinal helminth infection. J. Immunol. 2014, 192, 2904–2912. [Google Scholar] [CrossRef]
- Schopf, L.R.; Hoffmann, K.F.; Cheever, A.W.; Urban, J.F.; Wynn, T.A. IL-10 is critical for host resistance and survival during gastrointestinal helminth infection. J. Immunol. 2002, 168, 2383–2392. [Google Scholar] [CrossRef]
- Zaph, C.; Troy, A.E.; Taylor, B.C.; Berman-Booty, L.D.; Guild, K.J.; Du, Y.; Yost, E.A.; Gruber, A.D.; May, M.J.; Greten, F.R.; et al. Epithelial-cell-intrinsic IKK-β expression regulates intestinal immune homeostasis. Nature 2007, 446, 552–556. [Google Scholar] [CrossRef]
- Owyang, A.M.; Zaph, C.; Wilson, E.H.; Guild, K.J.; McClanahan, T.; Miller, H.R.P.; Cua, D.J.; Goldschmidt, M.; Hunter, C.A.; Kastelein, R.A.; et al. Interleukin 25 regulates type 2 cytokine-dependent immunity and limits chronic inflammation in the gastrointestinal tract. J. Exp. Med. 2006, 203, 843–849. [Google Scholar] [CrossRef]
- Fallon, P.G.; Ballantyne, S.J.; Mangan, N.E.; Barlow, J.L.; Dasvarma, A.; Hewett, D.R.; McIlgorm, A.; Jolin, H.E.; McKenzie, A.N.J. Identification of an interleukin (IL)-25–dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. J. Exp. Med. 2006, 203, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Saenz, S.A.; Siracusa, M.C.; Perrigoue, J.G.; Spencer, S.P.; Urban, J.F., Jr.; Tocker, J.E.; Budelsky, A.L.; Kleinschek, M.A.; Kastelein, R.A.; Kambayashi, T.; et al. IL25 elicits a multipotent progenitor cell population that promotes TH 2 cytokine responses. Nature 2010, 464, 1362–1366. [Google Scholar] [CrossRef]
- Schmitz, J.; Owyang, A.; Oldham, E.; Song, Y.; Murphy, E.; McClanahan, T.K.; Zurawski, G.; Moshrefi, M.; Qin, J.; Li, X.; et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 2005, 23, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, N.E.; Xu, D.; Hepworth, M.R.; Liew, F.Y.; Grencis, R.K. IL-33, a potent inducer of adaptive immunity to intestinal nematodes. J. Immunol. 2008, 180, 2443–2449. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.C.; Zaph, C.; Troy, A.E.; Du, Y.; Guild, K.J.; Comeau, M.R.; Artis, D. TSLP regulates intestinal immunity and inflammation in mouse models of helminth infection and colitis. J. Exp. Med. 2009, 206, 655–667. [Google Scholar] [CrossRef]
- Liu, Y.-J.; Soumelis, V.; Watanabe, N.; Ito, T.; Wang, Y.-H.; de Waal Malefyt, R.; Omori, M.; Zhou, B.; Ziegler, S.F. TSLP: An epithelial cell cytokine that regulates T cell differentiation by conditioning dendritic cell maturation. Annu. Rev. Immunol. 2007, 25, 193–219. [Google Scholar] [CrossRef]
- Soumelis, V.; Reche, P.A.; Kanzler, H.; Yuan, W.; Edward, G.; Homey, B.; Gilliet, M.; Ho, S.; Antonenko, S.; Lauerma, A.; et al. Human epithelial cells trigger dendritic cell–mediated allergic inflammation by producing TSLP. Nat. Immunol. 2002, 3, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Massacand, J.C.; Stettler, R.C.; Meier, R.; Humphreys, N.E.; Grencis, R.K.; Marsland, B.J.; Harris, N.L. Helminth products bypass the need for TSLP in Th2 immune responses by directly modulating dendritic cell function. Proc. Natl. Acad. Sci. USA 2009, 106, 13968–13973. [Google Scholar] [CrossRef] [PubMed]
- Dharmani, P.; Srivastava, V.; Kissoon-Singh, V.; Chadee, K. Role of intestinal mucins in innate host defense mechanisms against pathogens. J. Innate Immun. 2009, 1, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Hasnain, S.Z.; Thornton, D.J.; Grencis, R.K. Changes in the mucosal barrier during acute and chronic Trichuris muris infection. Parasite Immunol. 2011, 33, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Khan, W.I. Goblet cells and mucins: Role in innate defense in enteric infections. Pathogens 2013, 2, 55–70. [Google Scholar] [CrossRef]
- Hasnain, S.Z.; Wang, H.; Ghia, J.; Haq, N.; Deng, Y.; Velcich, A.; Grencis, R.K.; Thornton, D.J.; Khan, W.I. Mucin gene deficiency in mice impairs host resistance to an enteric parasitic infection. Gastroenterology 2010, 138, 1763–1771. [Google Scholar] [CrossRef] [PubMed]
- Else, K.J. Have gastrointestinal nematodes outwitted the immune system? Parasite Immunol. 2005, 27, 407–415. [Google Scholar] [CrossRef]
- Artis, D.; Potten, C.S.; Else, K.J.; Finkelman, F.D.; Grencis, R.K. Trichuris muris: Host intestinal epithelial cell hyperproliferation during chronic infection is regulated by interferon-γ. Exp. Parasitol. 1999, 92, 144–153. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, G.J.; Bancroft, A.; Grencis, R.K.; McKenzie, A.N.J. A distinct role for interleukin-13 in Th2-cell-mediated immune responses. Curr. Biol. 1998, 8, 339–342. [Google Scholar] [CrossRef]
- Khan, W.I.; Blennerhasset, P.; Ma, C.; Matthaei, K.I.; Collins, S.M. Stat6 dependent goblet cell hyperplasia during intestinal nematode infection. Parasite Immunol. 2001, 23, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Marillier, R.G.; Michels, C.; Smith, E.M.; Fick, L.C.E.; Leeto, M.; Dewals, B.; Horsnell, W.G.C.; Brombacher, F. IL-4/IL-13 independent goblet cell hyperplasia in experimental helminth infections. BMC Immunol. 2008, 9, 1–9. [Google Scholar] [CrossRef]
- Turner, J.-E.; Stockinger, B.; Helmby, H. IL-22 mediates goblet cell hyperplasia and worm expulsion in intestinal helminth infection. PLoS Pathog 2013, 9, e1003698. [Google Scholar] [CrossRef]
- Hasnain, S.Z.; Evans, C.M.; Roy, M.; Gallagher, A.L.; Kindrachuk, K.N.; Barron, L.; Dickey, B.F.; Wilson, M.S.; Wynn, T.A.; Grencis, R.K.; et al. Muc5ac: A critical component mediating the rejection of enteric nematodes. J. Exp. Med. 2011, 208, 893–900. [Google Scholar] [CrossRef] [PubMed]
- Hasnain, S.Z.; Dawson, P.A.; Lourie, R.; Hutson, P.; Tong, H.; Grencis, R.K.; McGuckin, M.A.; Thornton, D.J. Immune-driven alterations in mucin sulphation is an important mediator of Trichuris muris helminth expulsion. PLoS Pathog. 2017, 13, e1006218. [Google Scholar] [CrossRef] [PubMed]
- Artis, D.; Wang, M.L.; Keilbaugh, S.A.; He, W.; Brenes, M.; Swain, G.P.; Knight, P.A.; Donaldson, D.D.; Lazar, M.A.; Miller, H.R.P.; et al. RELM/FIZZ2 is a goblet cell-specific immune-effector molecule in the gastrointestinal tract. Proc. Natl. Acad. Sci. USA 2004, 101, 13596–13600. [Google Scholar] [CrossRef]
- Artis, D. New weapons in the war on worms: Identification of putative mechanisms of immune-mediated expulsion of gastrointestinal nematodes. Int. J. Parasitol. 2006, 36, 723–733. [Google Scholar] [CrossRef] [PubMed]
- Nair, M.G.; Guild, K.J.; Du, Y.; Zaph, C.; Yancopoulos, G.D.; Valenzuela, D.M.; Murphy, A.; Stevens, S.; Karow, M.; Artis, D. Goblet cell-derived resistin-like molecule β augments CD4+ T cell production of IFN-γ and infection-induced intestinal inflammation. J. Immunol. 2008, 181, 4709–4715. [Google Scholar] [CrossRef]
- Forman, R.A.; de Schoolmeester, M.L.; Hurst, R.J.M.; Wright, S.H.; Pemberton, A.D.; Else, K.J. The goblet cell is the cellular source of the anti-microbial angiogenin 4 in the large intestine post Trichuris muris infection. PLoS ONE 2012, 7, e42248. [Google Scholar] [CrossRef]
- Bell, L.V.; Else, K.J. Regulation of colonic epithelial cell turnover by IDO contributes to the innate susceptibility of SCID mice to Trichuris muris infection. Parasite Immunol. 2011, 33, 244–249. [Google Scholar] [CrossRef]
- Coakley, G.; Harris, N.L. The Intestinal Epithelium at the Forefront of Host–Helminth Interactions. Trends Parasitol. 2020, 36, 761–772. [Google Scholar] [CrossRef]
- Worthington, J.J.; Reimann, F.; Gribble, F.M. Enteroendocrine cells-sensory sentinels of the intestinal environment and orchestrators of mucosal immunity. Mucosal Immunol. 2018, 11, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Cliffe, L.J.; Humphreys, N.E.; Lane, T.E.; Potten, C.S.; Booth, C.; Grencis, R.K. Accelerated intestinal epithelial cell turnover: A new mechanism of parasite expulsion. Science 2005, 308, 1463–1465. [Google Scholar] [CrossRef] [PubMed]
- Motomura, Y.; Ghia, J.-E.; Wang, H.; Akiho, H.; El-Sharkawy, R.T.; Collins, M.; Wan, Y.; McLaughlin, J.T.; Khan, W.I. Enterochromaffin cell and 5-hydroxytryptamine responses to the same infectious agent differ in Th1 and Th2 dominant environments. Gut 2008, 57, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Steeds, J.; Motomura, Y.; Deng, Y.; Verma-Gandhu, M.; El-Sharkawy, R.T.; McLaughlin, J.T.; Grencis, R.K.; Khan, W.I. CD4+ T cell-mediated immunological control of enterochromaffin cell hyperplasia and 5-hydroxytryptamine production in enteric infection. Gut 2007, 56, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Manocha, M.; Shajib, M.S.; Rahman, M.M.; Wang, H.; Rengasamy, P.; Bogunovic, M.; Jordana, M.; Mayer, L.; Khan, W.I. IL-13-mediated immunological control of enterochromaffin cell hyperplasia and serotonin production in the gut. Mucosal Immunol. 2013, 6, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Kwon, Y.H.; Dewan, V.; Vahedi, F.; Syed, S.; Fontes, M.E.; Ashkar, A.A.; Surette, M.G.; Khan, W.I. TLR2 plays a pivotal role in mediating mucosal serotonin production in the gut. J. Immunol. 2019, 202, 3041–3052. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.J.; Wang, H.; Terc, J.D.; Zambrowicz, B.; Yang, Q.M.; Khan, W.I. Blocking peripheral serotonin synthesis by telotristat etiprate (LX1032/LX1606) reduces severity of both chemical-and infection-induced intestinal inflammation. Am. J. Physiol. Liver Physiol. 2015, 309, G455–G465. [Google Scholar] [CrossRef]
- Antignano, F.; Mullaly, S.C.; Burrows, K.; Zaph, C. Trichuris muris infection: A model of type 2 immunity and inflammation in the gut. JoVE 2011, 24, e2774. [Google Scholar] [CrossRef]
- Vallance, B.A.; Galeazzi, F.; Collins, S.M.; Snider, D.P. CD4 T Cells and Major Histocompatibility Complex Class II Expression Influence Worm Expulsion and Increased Intestinal Muscle Contraction during Trichinella spiralis Infection. Infect. Immun. 1999, 67, 6090–6097. [Google Scholar] [CrossRef] [PubMed]
- Khan, W.I.; Vallance, B.A.; Blennerhassett, P.A.; Deng, Y.; Verdu, E.F.; Matthaei, K.I.; Collins, S.M. Critical role for signal transducer and activator of transcription factor 6 in mediating intestinal muscle hypercontractility and worm expulsion in Trichinella spiralis-infected mice. Infect. Immun. 2001, 69, 838–844. [Google Scholar] [CrossRef] [PubMed]
- Vallance, B.A.; Blennerhassett, P.A.; Deng, Y.; Matthaei, K.I.; Young, I.G.; Collins, S.M. IL-5 contributes to worm expulsion and muscle hypercontractility in a primary T. spiralis infection. Am. J. Physiol. Liver Physiol. 1999, 277, G400–G408. [Google Scholar]
- Khan, W.I.; Blennerhassett, P.A.; Deng, Y.; Gauldie, J.; Vallance, B.A.; Collins, S.M. IL-12 gene transfer alters gut physiology and host immunity in nematode-infected mice. Am. J. Physiol. Liver Physiol. 2001, 281, G102–G110. [Google Scholar] [CrossRef]
- Motomura, Y.; Khan, W.I.; El-Sharkawy, R.T.; Verma-Gandhu, M.; Grencis, R.K.; Collins, S.M. Mechanisms underlying gut dysfunction in a murine model of chronic parasitic infection. Am. J. Physiol. Liver Physiol. 2010, 299, G1354–G1360. [Google Scholar] [CrossRef] [PubMed]
- Faulkner, H.; Renauld, J.-C.; Van Snick, J.; Grencis, R.K. Interleukin-9 enhances resistance to the intestinal nematode Trichuris muris. Infect. Immun. 1998, 66, 3832–3840. [Google Scholar] [CrossRef]
- Chen, Z.; Luo, J.; Li, J.; Kim, G.; Stewart, A.; Urban, J.F., Jr.; Huang, Y.; Chen, S.; Wu, L.-G.; Chesler, A.; et al. Interleukin-33 Promotes Serotonin Release from Enterochromaffin Cells for Intestinal Homeostasis. Immunity 2021, 54, 151–163. [Google Scholar] [CrossRef]
- Cortes, A.; Peachey, L.; Scotti, R.; Jenkins, T.P.; Cantacessi, C. Helminth-microbiota cross-talk–A journey through the vertebrate digestive system. Mol. Biochem. Parasitol. 2019, 233, 111222. [Google Scholar] [CrossRef]
- Leung, J.M.; Graham, A.L.; Knowles, S.C.L. Parasite-microbiota interactions with the vertebrate gut: Synthesis through an ecological lens. Front. Microbiol. 2018, 9, 843. [Google Scholar] [CrossRef]
- Hayes, K.S.; Bancroft, A.J.; Goldrick, M.; Portsmouth, C.; Roberts, I.S.; Grencis, R.K. Exploitation of the intestinal microflora by the parasitic nematode Trichuris muris. Science 2010, 328, 1391–1394. [Google Scholar] [CrossRef]
- White, E.C.; Houlden, A.; Bancroft, A.J.; Hayes, K.S.; Goldrick, M.; Grencis, R.K.; Roberts, I.S. Manipulation of host and parasite microbiotas: Survival strategies during chronic nematode infection. Sci. Adv. 2018, 4, eaap7399. [Google Scholar] [CrossRef]
- Leung, J.M.; Budischak, S.A.; Chung The, H.; Hansen, C.; Bowcutt, R.; Neill, R.; Shellman, M.; Loke, P.; Graham, A.L. Rapid environmental effects on gut nematode susceptibility in rewilded mice. PLoS Biol. 2018, 16, e2004108. [Google Scholar] [CrossRef]
- Holm, J.B.; Sorobetea, D.; Kiilerich, P.; Ramayo-Caldas, Y.; Estellé, J.; Ma, T.; Madsen, L.; Kristiansen, K.; Svensson-Frej, M. Chronic Trichuris muris infection decreases diversity of the intestinal microbiota and concomitantly increases the abundance of lactobacilli. PLoS ONE 2015, 10, e0125495. [Google Scholar]
- Houlden, A.; Hayes, K.S.; Bancroft, A.J.; Worthington, J.J.; Wang, P.; Grencis, R.K.; Roberts, I.S. Chronic Trichuris muris infection in C57BL/6 mice causes significant changes in host microbiota and metabolome: Effects reversed by pathogen clearance. PLoS ONE 2015, 10, e0125945. [Google Scholar]
- Ramanan, D.; Bowcutt, R.; Lee, S.C.; San Tang, M.; Kurtz, Z.D.; Ding, Y.; Honda, K.; Gause, W.C.; Blaser, M.J.; Bonneau, R.A.; et al. Helminth infection promotes colonization resistance via type 2 immunity. Science 2016, 352, 608–612. [Google Scholar] [CrossRef] [PubMed]
- Duque-Correa, M.A.; Karp, N.A.; McCarthy, C.; Forman, S.; Goulding, D.; Sankaranarayanan, G.; Jenkins, T.P.; Reid, A.J.; Cambridge, E.L.; Reviriego, C.B.; et al. Exclusive dependence of IL-10R signalling on intestinal microbiota homeostasis and control of whipworm infection. PLoS Pathog. 2019, 15, e1007265. [Google Scholar] [CrossRef] [PubMed]
- D’Elia, R.; Matthew, L.D.; Zeef, L.A.; Wright, S.H.; Pemberton, A.D.; Else, K.J. Expulsion of Trichuris muris is associated with increased expression of angiogenin 4 in the gut and increased acidity of mucins within the goblet cell. BMC Genomics 2009, 10, 1–17. [Google Scholar] [CrossRef]
- Hamann, K.J.; Barker, R.L.; Loegering, D.A.; Gleich, G.J. Comparative toxicity of purified human eosinophil granule proteins for newborn larvae of Trichinella spiralis. J. Parasitol. 1987, 73, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Bach, J.-F. The hygiene hypothesis in autoimmunity: The role of pathogens and commensals. Nat. Rev. Immunol. 2018, 18, 105. [Google Scholar] [CrossRef] [PubMed]
- McKay, D.M. The therapeutic helminth? Trends Parasitol. 2009, 25, 109–114. [Google Scholar] [CrossRef]
- Summers, R.W.; Elliott, D.E.; Urban, J.F., Jr.; Thompson, R.A.; Weinstock, J.V. Trichuris suis therapy for active ulcerative colitis: A randomized controlled trial. Gastroenterology 2005, 128, 825–832. [Google Scholar] [CrossRef]
- Reddy, A.; Fried, B. The use of Trichuris suis and other helminth therapies to treat Crohn’s disease. Parasitol. Res. 2007, 100, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Vegas-Sanchez, M.C.; Rollan-Landeras, E.; Garcia-Rodriguez, J.J.; Bolas-Fernandez, F. Induction of ulcerative colitis in mice influences the course of infection with the nematode Trichuris muris. J. Helminthol. 2015, 89, 593. [Google Scholar] [CrossRef] [PubMed]
- Elliott, D.E.; Setiawan, T.; Metwali, A.; Blum, A.; Urban, J.F., Jr.; Weinstock, J.V. Heligmosomoides polygyrus inhibits established colitis in IL 10 deficient mice. Eur. J. Immunol. 2004, 34, 2690–2698. [Google Scholar] [CrossRef] [PubMed]
- Elliott, D.E.; Li, J.; Blum, A.; Metwali, A.; Qadir, K.; Urban, J.F., Jr.; Weinstock, J.V. Exposure to schistosome eggs protects mice from TNBS-induced colitis. Am. J. Physiol. Liver Physiol. 2003, 284, G385–G391. [Google Scholar] [CrossRef] [PubMed]
- Hunter, M.M.; Wang, A.; Hirota, C.L.; McKay, D.M. Neutralizing anti-IL-10 antibody blocks the protective effect of tapeworm infection in a murine model of chemically induced colitis. J. Immunol. 2005, 174, 7368–7375. [Google Scholar] [CrossRef] [PubMed]
- Khan, W.I.; Blennerhasset, P.A.; Varghese, A.K.; Chowdhury, S.K.; Omsted, P.; Deng, Y.; Collins, S.M. Intestinal nematode infection ameliorates experimental colitis in mice. Infect. Immun. 2002, 70, 5931–5937. [Google Scholar] [CrossRef] [PubMed]
- Motomura, Y.; Wang, H.; Deng, Y.; El Sharkawy, R.T.; Verdu, E.F.; Khan, W.I. Helminth antigen based strategy to ameliorate inflammation in an experimental model of colitis. Clin. Exp. Immunol. 2009, 155, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Yamaguchi, T.; Nomura, T.; Ono, M. Regulatory T cells and immune tolerance. Cell 2008, 133, 775–787. [Google Scholar] [CrossRef]
- Bhardwaj, E.K.; Else, K.J.; Rogan, M.T.; Warhurst, G. Increased susceptibility to Trichuris muris infection and exacerbation of colitis in Mdr1a-/-mice. World J. Gastroenterol. WJG 2014, 20, 1797. [Google Scholar] [CrossRef]
- Wilson, M.S.; Ramalingam, T.R.; Rivollier, A.; Shenderov, K.; Mentink-Kane, M.M.; Madala, S.K.; Cheever, A.W.; Artis, D.; Kelsall, B.L.; Wynn, T.A. Colitis and Intestinal Inflammation in IL10−/− Mice Results from IL-13Rα2–Mediated Attenuation of IL-13 Activity. Gastroenterology 2011, 140, 254–264. [Google Scholar] [CrossRef]
- Levison, S.E.; McLaughlin, J.T.; Zeef, L.A.H.; Fisher, P.; Grencis, R.K.; Pennock, J.L. Colonic transcriptional profiling in resistance and susceptibility to trichuriasis: Phenotyping a chronic colitis and lessons for iatrogenic helminthosis. Inflamm. Bowel Dis. 2010, 16, 2065–2079. [Google Scholar] [CrossRef] [PubMed]
- Bramhall, M.; Rich, K.; Chakraborty, A.; Logunova, L.; Han, N.; Wilson, J.; McLaughlin, J.; Brass, A.; Cruickshank, S.M. Differential expression of soluble receptor for advanced glycation end-products in mice susceptible or resistant to chronic colitis. Inflamm. Bowel Dis. 2020, 26, 360–368. [Google Scholar] [CrossRef]
- Chenery, A.L.; Antignano, F.; Burrows, K.; Scheer, S.; Perona-Wright, G.; Zaph, C. Low-dose intestinal Trichuris muris infection alters the lung immune microenvironment and can suppress allergic airway inflammation. Infect. Immun. 2016, 84, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Yazdanbakhsh, M.; Kremsner, P.G.; Van Ree, R. Allergy, parasites, and the hygiene hypothesis. Science 2002, 296, 490–494. [Google Scholar] [CrossRef] [PubMed]
- Fleming, J.O.; Isaak, A.; Lee, J.E.; Luzzio, C.C.; Carrithers, M.D.; Cook, T.D.; Field, A.S.; Boland, J.; Fabry, Z. Probiotic helminth administration in relapsing-remitting multiple sclerosis: A phase 1 study. Mult. Scler. J. 2011, 17, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Osada, Y.; Shimizu, S.; Kumagai, T.; Yamada, S.; Kanazawa, T. Schistosoma mansoni infection reduces severity of collagen-induced arthritis via down-regulation of pro-inflammatory mediators. Int. J. Parasitol. 2009, 39, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.H.; Wang, H.; Denou, E.; Ghia, J.-E.; Rossi, L.; Fontes, M.E.; Bernier, S.P.; Shajib, M.S.; Banskota, S.; Collins, S.M.; et al. Modulation of gut microbiota composition by serotonin signaling influences intestinal immune response and susceptibility to colitis. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 709–728. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yousefi, Y.; Haq, S.; Banskota, S.; Kwon, Y.H.; Khan, W.I. Trichuris muris Model: Role in Understanding Intestinal Immune Response, Inflammation and Host Defense. Pathogens 2021, 10, 925. https://doi.org/10.3390/pathogens10080925
Yousefi Y, Haq S, Banskota S, Kwon YH, Khan WI. Trichuris muris Model: Role in Understanding Intestinal Immune Response, Inflammation and Host Defense. Pathogens. 2021; 10(8):925. https://doi.org/10.3390/pathogens10080925
Chicago/Turabian StyleYousefi, Yeganeh, Sabah Haq, Suhrid Banskota, Yun Han Kwon, and Waliul I. Khan. 2021. "Trichuris muris Model: Role in Understanding Intestinal Immune Response, Inflammation and Host Defense" Pathogens 10, no. 8: 925. https://doi.org/10.3390/pathogens10080925
APA StyleYousefi, Y., Haq, S., Banskota, S., Kwon, Y. H., & Khan, W. I. (2021). Trichuris muris Model: Role in Understanding Intestinal Immune Response, Inflammation and Host Defense. Pathogens, 10(8), 925. https://doi.org/10.3390/pathogens10080925