
Prevalence and Evolution of Noroviruses between 1966 and 2019, Implications for Vaccine Design

 , ,
, ,
Abstract
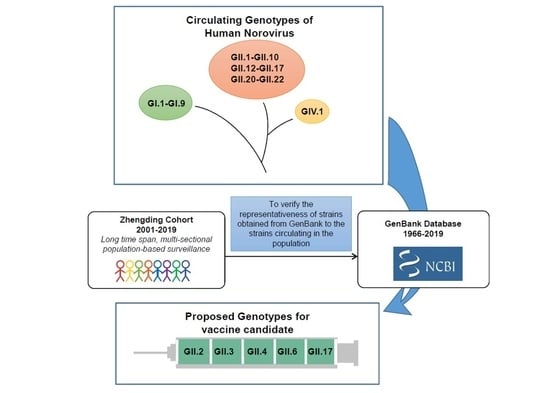
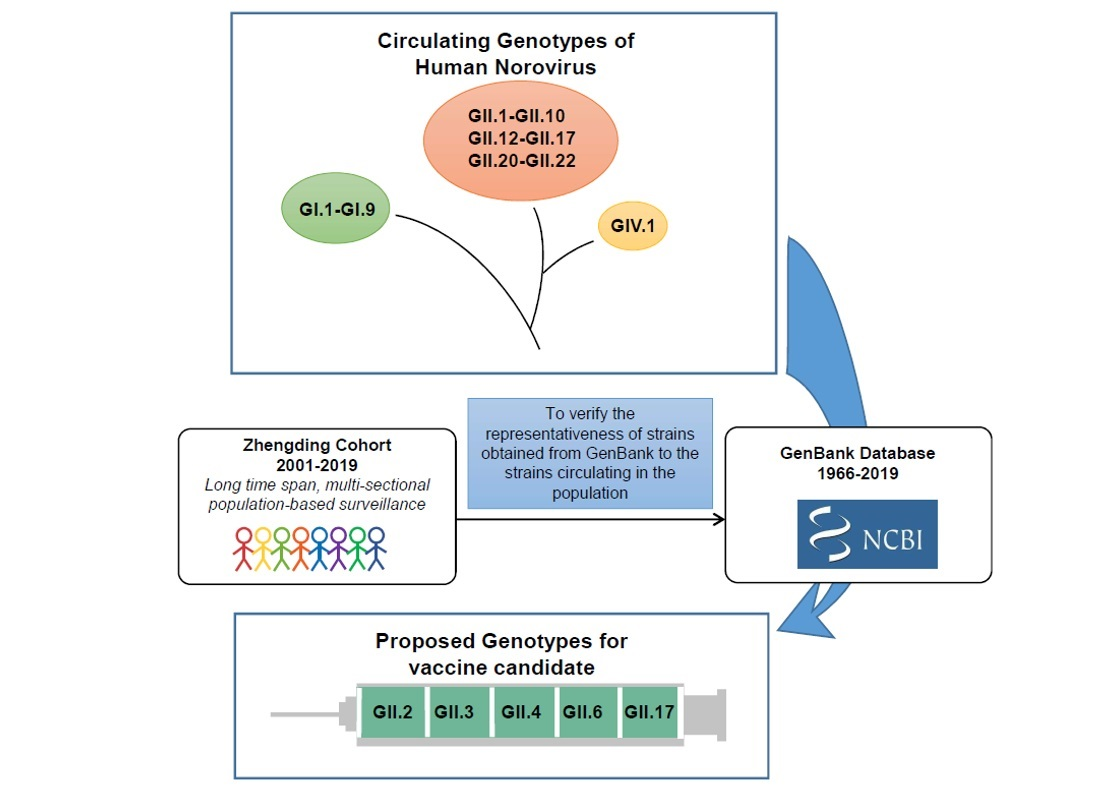
1. Introduction
2. Results
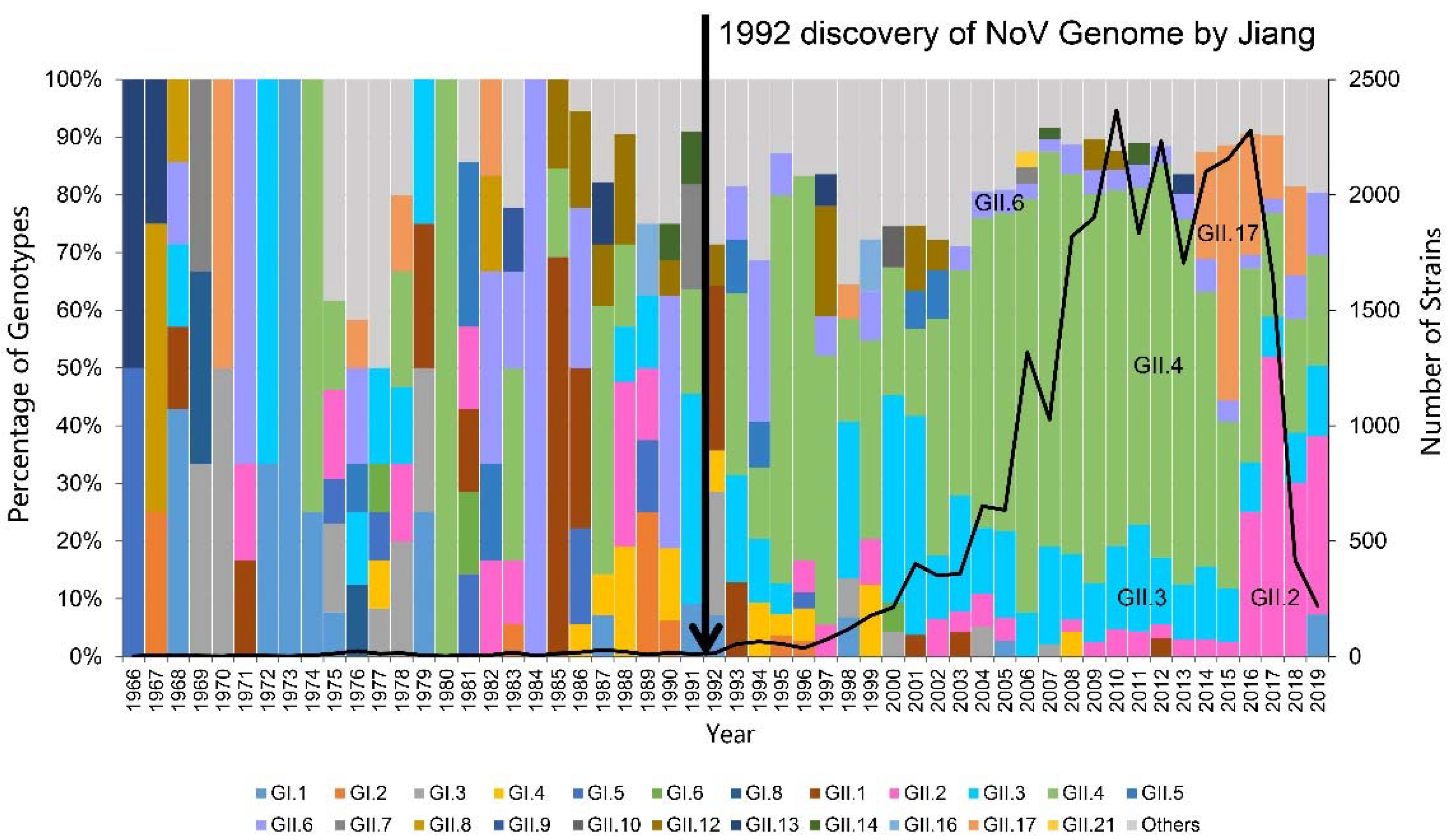
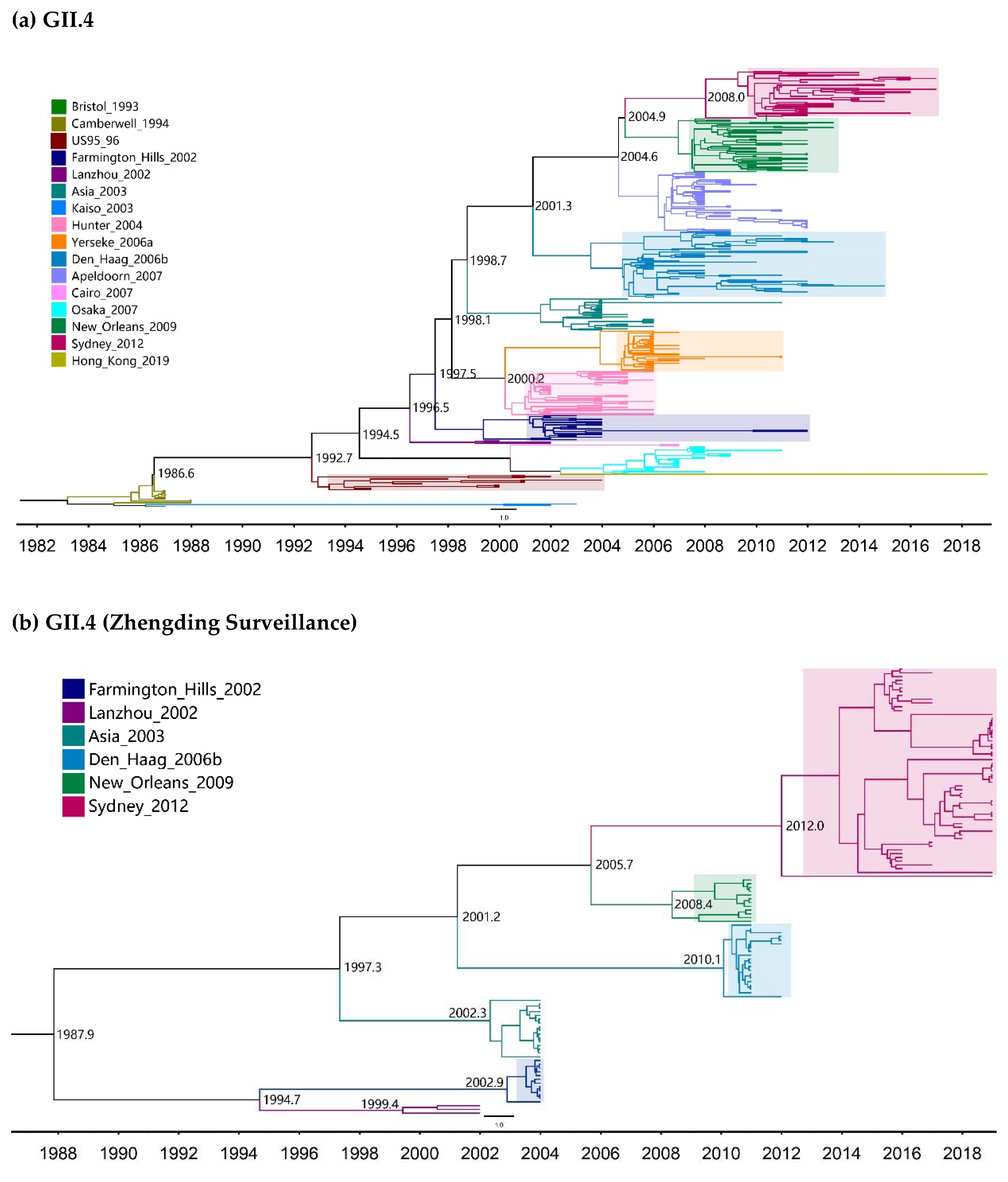
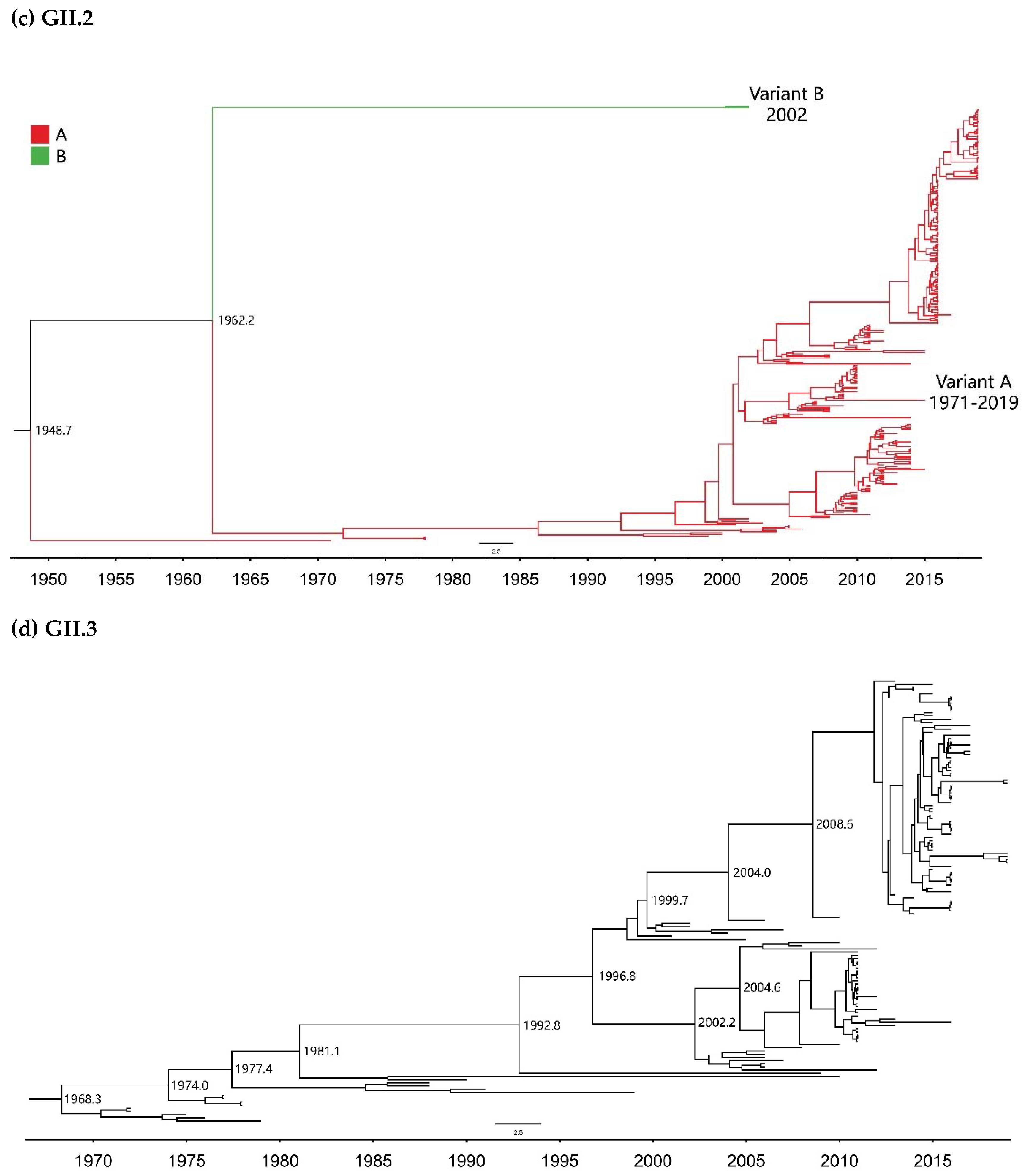
2.1. Global Temporal Dynamics of NoVs
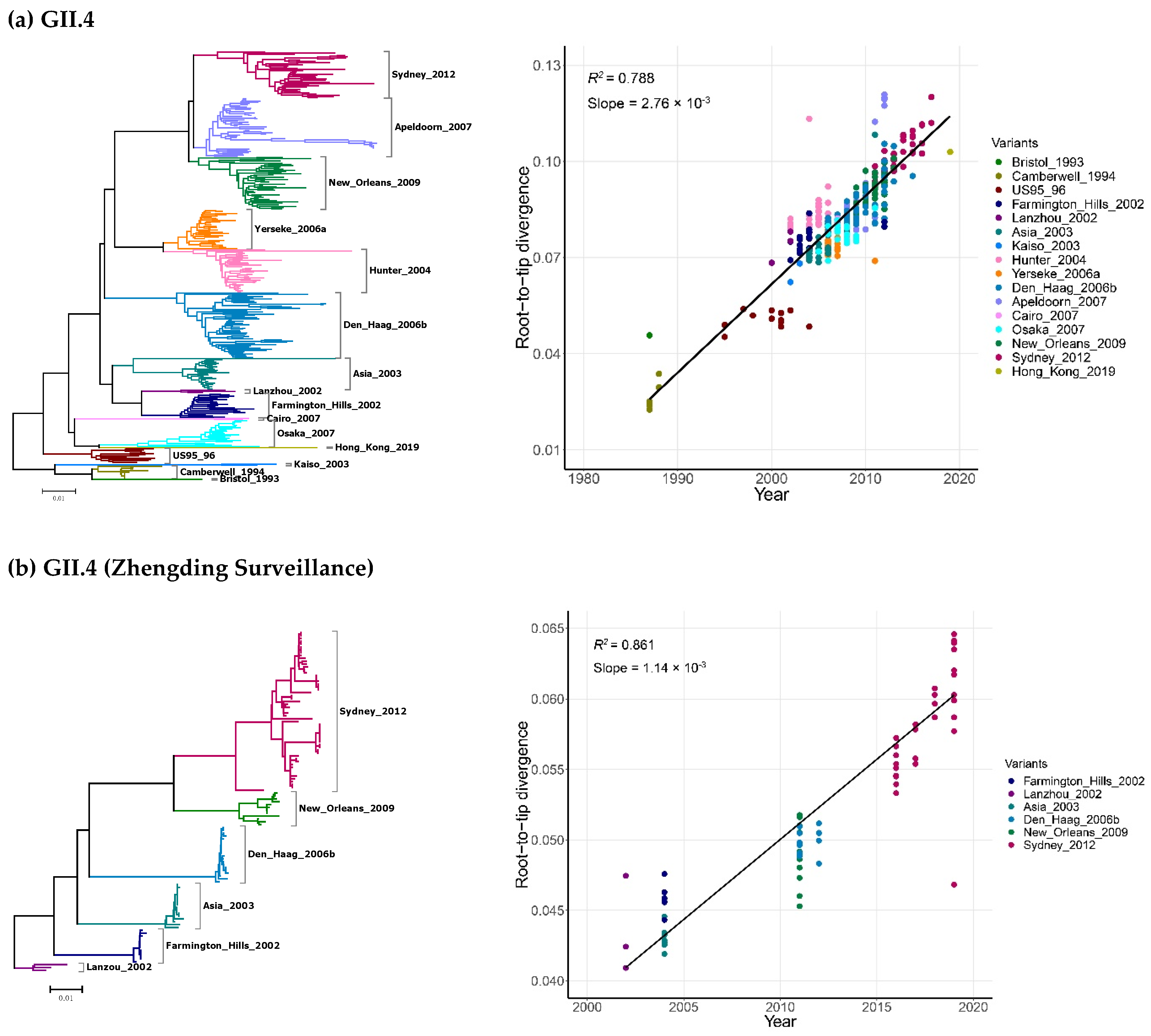
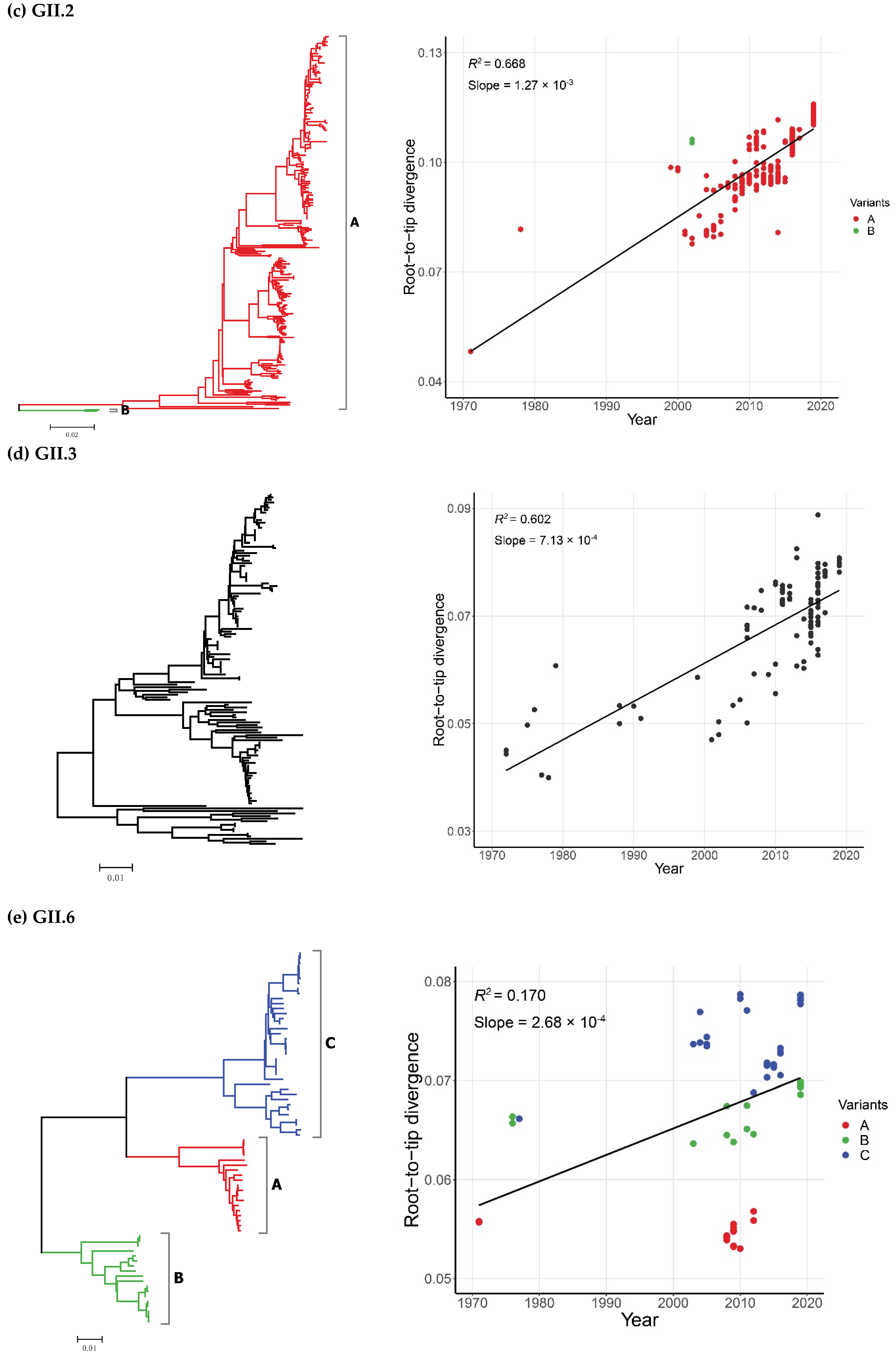
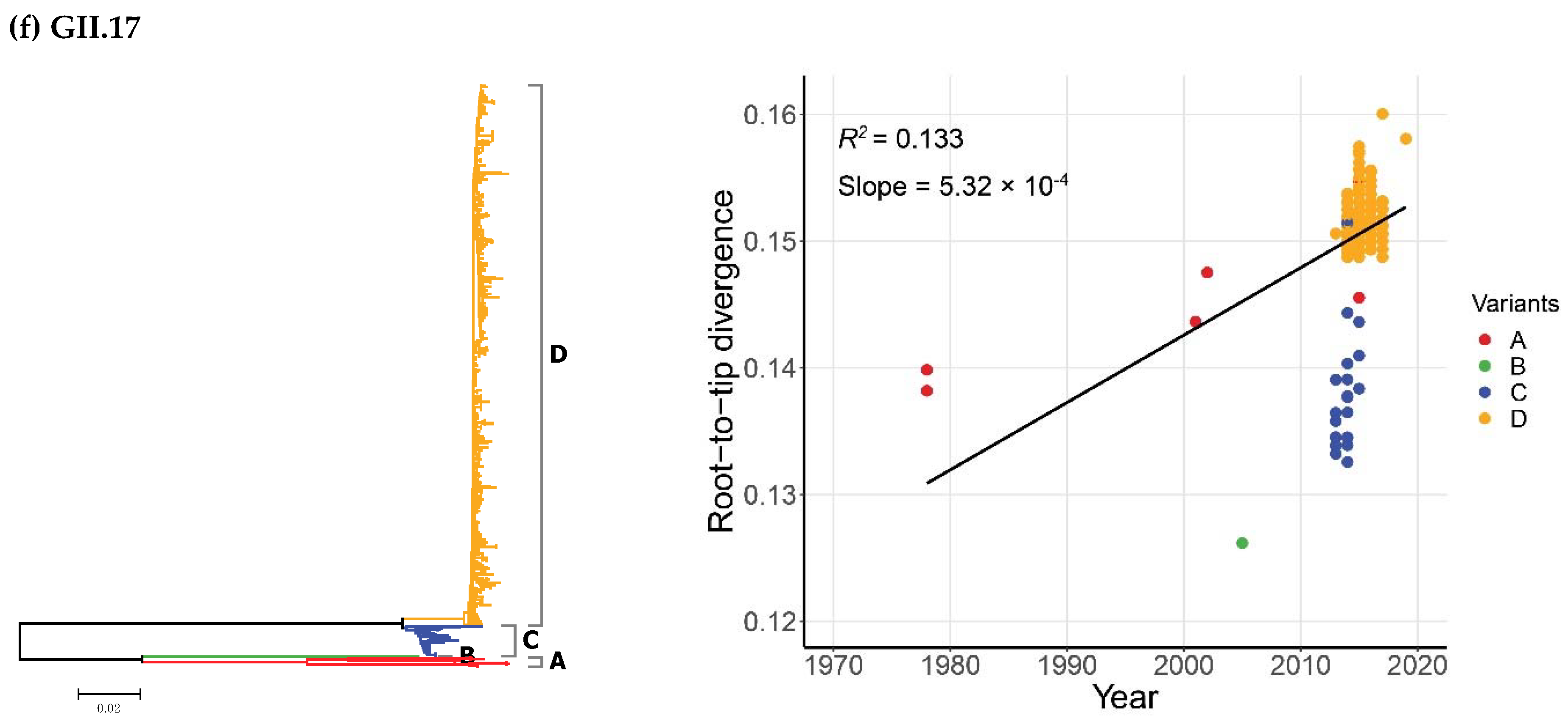
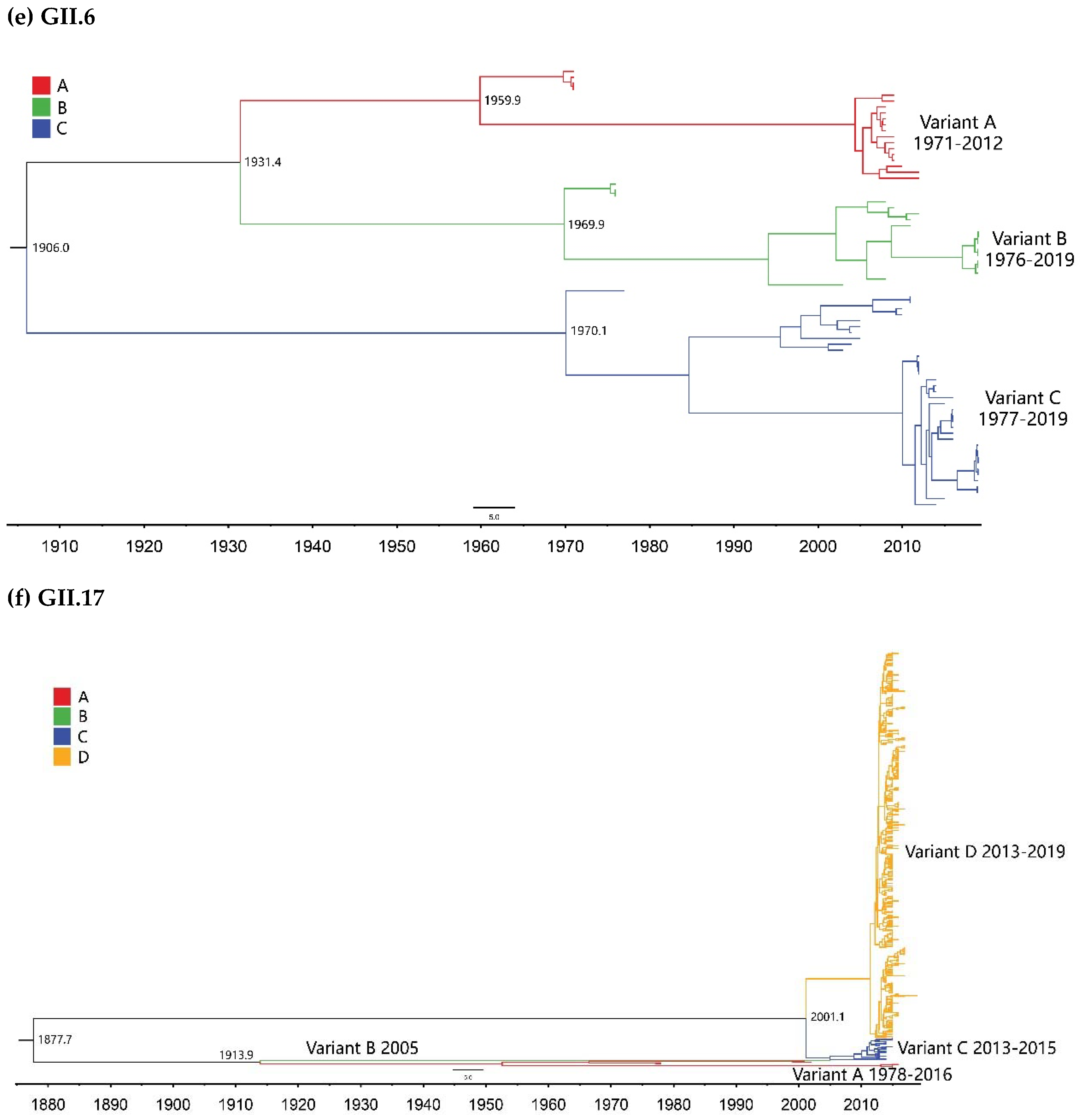
2.2. Time-Scale Evolution of the Globally Collected NoV Strains
2.3. Genetic Distances Based on Amino Acid of Complete VP1s
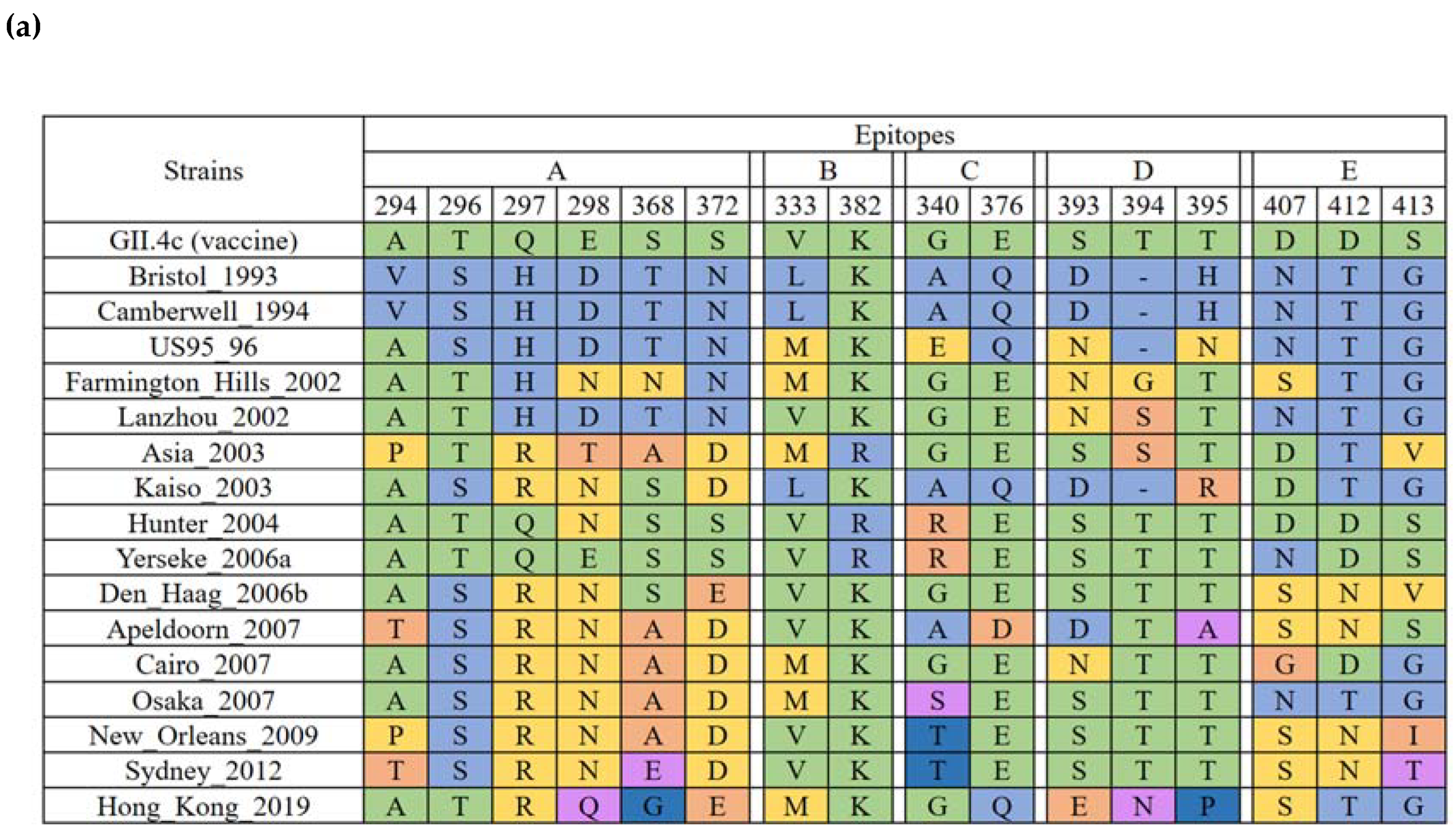
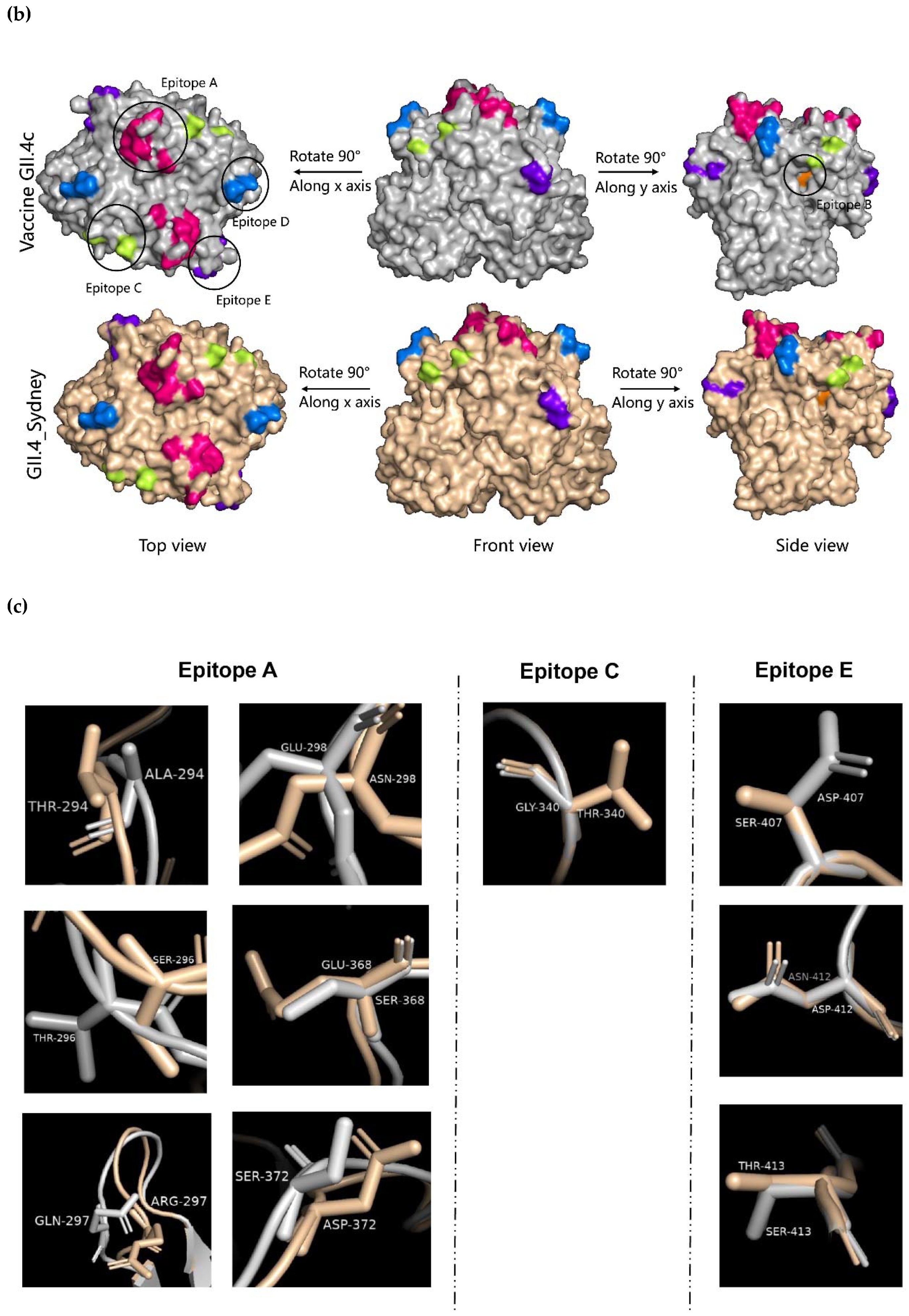
2.4. Evolution of the GII.4 Blockade Epitopes
3. Discussion
4. Materials and Methods
4.1. Dataset from Population-Based Diarrhea Surveillance
4.1.1. Sample Collection
4.1.2. Complete VP1 Sequencing
4.2. Dataset from GenBank
4.3. Multiple Alignments and Phylogenetic Analyses
4.4. Evolutionary Dynamic Analyses
4.5. Homologous Modeling
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Troeger, C.; Blacker, B.F.; Khalil, I.A.; Rao, P.C.; Cao, S.; Zimsen, S.R.M.; Albertson, S.B.; Stanaway, J.D.; Deshpande, A.; Abebe, Z.; et al. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of diarrhoea in 195 countries: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Infect. Dis. 2018, 18, 1211–1228. [Google Scholar] [CrossRef]
- Robilotti, E.; Deresinski, S.; Pinsky, B.A. Norovirus. Clin. Microbiol. Rev. 2015, 28, 134–164. [Google Scholar] [CrossRef]
- Prasad, B.V.; Hardy, M.E.; Dokland, T.; Bella, J.; Rossmann, M.G.; Estes, M.K. X-ray crystallographic structure of the Norwalk virus capsid. Science 1999, 286, 287–290. [Google Scholar] [CrossRef]
- Vinje, J. Advances in laboratory methods for detection and typing of norovirus. J. Clin. Microbiol. 2015, 53, 373–381. [Google Scholar] [CrossRef]
- De Graaf, M.; van Beek, J.; Koopmans, M.P. Human norovirus transmission and evolution in a changing world. Nat. Rev. Microbiol. 2016, 14, 421–433. [Google Scholar] [CrossRef]
- De Graaf, M.; van Beek, J.; Vennema, H.; Podkolzin, A.T.; Hewitt, J.; Bucardo, F.; Templeton, K.; Mans, J.; Nordgren, J.; Reuter, G.; et al. Emergence of a novel GII.17 norovirus—End of the GII.4 era. Eurosurveillance 2015, 20, 21178. [Google Scholar] [CrossRef]
- Supadej, K.; Khamrin, P.; Kumthip, K.; Malasao, R.; Chaimongkol, N.; Saito, M.; Oshitani, H.; Ushijima, H.; Maneekarn, N. Distribution of norovirus and sapovirus genotypes with emergence of NoV GII.P16/GII.2 recombinant strains in Chiang Mai, Thailand. J. Med. Virol. 2019, 91, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, K.; Matsushima, Y.; Motoya, T.; Mizukoshi, F.; Ueki, Y.; Sakon, N.; Murakami, K.; Shimizu, T.; Okabe, N.; Nagata, N.; et al. Phylogeny and Immunoreactivity of Norovirus GII.P16-GII.2, Japan, Winter 2016–17. Emerg. Infect. Dis. 2018, 24, 144–148. [Google Scholar] [CrossRef] [PubMed]
- Niendorf, S.; Jacobsen, S.; Faber, M.; Eis-Hübinger, A.M.; Hofmann, J.; Zimmermann, O.; Höhne, M.; Bock, C.T. Steep rise in norovirus cases and emergence of a new recombinant strain GII.P16-GII.2, Germany, winter 2016. Eurosurveillance 2017, 22, 30447. [Google Scholar] [CrossRef] [PubMed]
- Ao, Y.Y.; Cong, X.; Jin, M.; Sun, X.M.; Wei, X.M.; Wang, J.J.; Zhang, Q.; Song, J.; Yu, J.M.; Cui, J.; et al. Genetic Analysis of Reemerging GII.P16-GII.2 Noroviruses in 2016–2017 in China. J. Infect. Dis. 2018, 218, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Clinical Trial of Quadrivalent Recombinant Norovirus Vaccine. High Tech Ind. 2019, 71.
- Esposito, S.; Principi, N. Norovirus Vaccine: Priorities for Future Research and Development. Front. Immunol. 2020, 11, 1383. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Penfield, N.W.; Ramani, S.; Estes, M.K.; Atmar, R.L. Prospects and Challenges in the Development of a Norovirus Vaccine. Clin. Ther. 2017, 39, 1537–1549. [Google Scholar] [CrossRef] [PubMed]
- Parra, G.I.; Squires, R.B.; Karangwa, C.K.; Johnson, J.A.; Lepore, C.J.; Sosnovtsev, S.V.; Green, K.Y. Static and Evolving Norovirus Genotypes: Implications for Epidemiology and Immunity. PLoS Pathog. 2017, 13, e1006136. [Google Scholar] [CrossRef] [PubMed]
- Tohma, K.; Lepore, C.J.; Ford-Siltz, L.A.; Parra, G.I. Evolutionary dynamics of non-GII genotype 4 (GII.4) noroviruses reveal limited and independent diversification of variants. J. Gen. Virol. 2018, 99, 1027–1035. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, J.A.M.; Bandeira, R.D.S.; Oliveira, D.S.; Dos Santos, L.F.P.; Gabbay, Y.B. Genotype diversity and molecular evolution of noroviruses: A 30-year (1982-2011) comprehensive study with children from Northern Brazil. PLoS ONE 2017, 12, e0178909. [Google Scholar] [CrossRef]
- Sherwood, J.; Mendelman, P.M.; Lloyd, E.; Liu, M.; Boslego, J.; Borkowski, A.; Jackson, A.; Faix, D. Efficacy of an intramuscular bivalent norovirus GI.1/GII.4 virus-like particle vaccine candidate in healthy US adults. Vaccine 2020, 38, 6442–6449. [Google Scholar] [CrossRef]
- Lindesmith, L.C.; Beltramello, M.; Donaldson, E.F.; Corti, D.; Swanstrom, J.; Debbink, K.; Lanzavecchia, A.; Baric, R.S. Immunogenetic mechanisms driving norovirus GII.4 antigenic variation. PLoS Pathog. 2012, 8, e1002705. [Google Scholar] [CrossRef]
- Ruis, C.; Lindesmith, L.C.; Mallory, M.L.; Brewer-Jensen, P.D.; Bryant, J.M.; Costantini, V.; Monit, C.; Vinje, J.; Baric, R.S.; Goldstein, R.A.; et al. Preadaptation of pandemic GII.4 noroviruses in unsampled virus reservoirs years before emergence. Virus Evol. 2020, 6, veaa067. [Google Scholar] [CrossRef]
- Chan, M.C.; Leung, T.F.; Chung, T.W.; Kwok, A.K.; Nelson, E.A.; Lee, N.; Chan, P.K. Virus Genotype Distribution and Virus Burden in Children and Adults Hospitalized for Norovirus Gastroenteritis, 2012-2014, Hong Kong. Sci. Rep. 2015, 5, 11507. [Google Scholar] [CrossRef]
- Wu, X.; Han, J.; Chen, L.; Xu, D.; Shen, Y.; Zha, Y.; Zhu, X.; Ji, L. Prevalence and genetic diversity of noroviruses in adults with acute gastroenteritis in Huzhou, China, 2013–2014. Arch. Virol. 2015, 160, 1705–1713. [Google Scholar] [CrossRef]
- Zakikhany, K.; Allen, D.J.; Brown, D.; Iturriza-Gomara, M. Molecular evolution of GII-4 Norovirus strains. PLoS ONE 2012, 7, e41625. [Google Scholar] [CrossRef] [PubMed]
- Debbink, K.; Lindesmith, L.C.; Donaldson, E.F.; Costantini, V.; Beltramello, M.; Corti, D.; Swanstrom, J.; Lanzavecchia, A.; Vinje, J.; Baric, R.S. Emergence of new pandemic GII.4 Sydney norovirus strain correlates with escape from herd immunity. J. Infect. Dis. 2013, 208, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Villabruna, N.; Koopmans, M.P.G.; de Graaf, M. Animals as Reservoir for Human Norovirus. Viruses 2019, 11, 478. [Google Scholar] [CrossRef] [PubMed]
- Karst, S.M.; Baric, R.S. What is the reservoir of emergent human norovirus strains? J. Virol. 2015, 89, 5756–5759. [Google Scholar] [CrossRef]
- Chen, H.; Qian, F.; Xu, J.; Chan, M.; Shen, Z.; Zai, S.; Shan, M.; Cai, J.; Zhang, W.; He, J.; et al. A novel norovirus GII.17 lineage contributed to adult gastroenteritis in Shanghai, China, during the winter of 2014–2015. Emerg. Microbes Infect. 2015, 4, e67. [Google Scholar] [CrossRef] [PubMed]
- Chan, M.C.; Lee, N.; Hung, T.N.; Kwok, K.; Cheung, K.; Tin, E.K.; Lai, R.W.; Nelson, E.A.; Leung, T.F.; Chan, P.K. Rapid emergence and predominance of a broadly recognizing and fast-evolving norovirus GII.17 variant in late 2014. Nat. Commun. 2015, 6, 10061. [Google Scholar] [CrossRef] [PubMed]
- Debbink, K.; Donaldson, E.F.; Lindesmith, L.C.; Baric, R.S. Genetic mapping of a highly variable norovirus GII.4 blockade epitope: Potential role in escape from human herd immunity. J. Virol. 2012, 86, 1214–1226. [Google Scholar] [CrossRef]
- Lindesmith, L.C.; Ferris, M.T.; Mullan, C.W.; Ferreira, J.; Debbink, K.; Swanstrom, J.; Richardson, C.; Goodwin, R.R.; Baehner, F.; Mendelman, P.M.; et al. Broad blockade antibody responses in human volunteers after immunization with a multivalent norovirus VLP candidate vaccine: Immunological analyses from a phase I clinical trial. PLoS Med. 2015, 12, e1001807. [Google Scholar] [CrossRef] [PubMed]
- Motoya, T.; Nagasawa, K.; Matsushima, Y.; Nagata, N.; Ryo, A.; Sekizuka, T.; Yamashita, A.; Kuroda, M.; Morita, Y.; Suzuki, Y.; et al. Molecular Evolution of the VP1 Gene in Human Norovirus GII.4 Variants in 1974–2015. Front. Microbiol. 2017, 8, 2399. [Google Scholar] [CrossRef] [PubMed]
- Bok, K.; Abente, E.J.; Realpe-Quintero, M.; Mitra, T.; Sosnovtsev, S.V.; Kapikian, A.Z.; Green, K.Y. Evolutionary dynamics of GII.4 noroviruses over a 34-year period. J. Virol. 2009, 83, 11890–11901. [Google Scholar] [CrossRef] [PubMed]
- Qiao, N.; Wang, X.Y.; Liu, L. Temporal Evolutionary Dynamics of Norovirus GII.4 Variants in China between 2004 and 2015. PLoS ONE 2016, 11, e0163166. [Google Scholar] [CrossRef]
- Siebenga, J.J.; Lemey, P.; Kosakovsky Pond, S.L.; Rambaut, A.; Vennema, H.; Koopmans, M. Phylodynamic reconstruction reveals norovirus GII.4 epidemic expansions and their molecular determinants. PLoS Pathog. 2010, 6, e1000884. [Google Scholar] [CrossRef] [PubMed]
- Tse, E.H.Y.; Zhang, L.-Y.; Lau, S.-L.; Chan, M.C.-W. Genome Sequence of a Human Norovirus GII.4 Hong Kong [P31] Variant in Hong Kong, China. Microbiol. Resour. Announc. 2020, 9, e01391-19. [Google Scholar] [CrossRef] [PubMed]
- Iritani, N.; Kaida, A.; Abe, N.; Sekiguchi, J.; Kubo, H.; Takakura, K.; Goto, K.; Ogura, H.; Seto, Y. Increase of GII.2 norovirus infections during the 2009-2010 season in Osaka City, Japan. J. Med. Virol. 2012, 84, 517–525. [Google Scholar] [CrossRef]
- Iritani, N.; Kaida, A.; Kubo, H.; Abe, N.; Murakami, T.; Vennema, H.; Koopmans, M.; Takeda, N.; Ogura, H.; Seto, Y. Epidemic of genotype GII.2 noroviruses during spring 2004 in Osaka City, Japan. J. Clin. Microbiol. 2008, 46, 2406–2409. [Google Scholar] [CrossRef][Green Version]
- Tohma, K.; Lepore, C.J.; Ford-Siltz, L.A.; Parra, G.I. Phylogenetic Analyses Suggest that Factors Other Than the Capsid Protein Play a Role in the Epidemic Potential of GII.2 Norovirus. mSphere 2017, 2, e00187-17. [Google Scholar] [CrossRef] [PubMed]
- Ao, Y.; Xie, X.; Dong, X.; Jin, M.; Duan, Z. Genetic Analysis of an Emerging GII.P2-GII.2 Norovirus Associated with a 2016 Outbreak of Acute Gastroenteritis in China. Virol. Sin. 2019, 34, 111–114. [Google Scholar] [CrossRef]
- Jin, M.; Wu, S.; Kong, X.; Xie, H.; Fu, J.; He, Y.; Feng, W.; Liu, N.; Li, J.; Rainey, J.; et al. Norovirus Outbreak Surveillance, China, 2016–2018. Emerg. Infect. Dis. J. 2020, 26, 437. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, S.; Wang, C.; Zheng, L.; Ma, J.; Li, C.; Huo, Y.; Wang, Y. Genomic characterization of GII.3 noroviruses isolated from children in Zhengzhou city, China, 2015/16. Arch. Virol. 2018, 163, 2737–2742. [Google Scholar] [CrossRef]
- Boonchan, M.; Guntapong, R.; Sripirom, N.; Ruchusatsawat, K.; Singchai, P.; Rungnobhakhun, P.; Tacharoenmuang, R.; Mizushima, H.; Tatsumi, M.; Takeda, N.; et al. The dynamics of norovirus genotypes and genetic analysis of a novel recombinant GII.P12-GII.3 among infants and children in Bangkok, Thailand between 2014 and 2016. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2018, 60, 133–139. [Google Scholar] [CrossRef]
- Karangwa, C.K.; Parra, G.I.; Bok, K.; Johnson, J.A.; Levenson, E.A.; Green, K.Y. Sequential Gastroenteritis Outbreaks in a Single Year Caused by Norovirus Genotypes GII.2 and GII.6 in an Institutional Setting. Open Forum Infect. Dis. 2017, 4, ofx236. [Google Scholar] [CrossRef]
- Epifanova, N.V. Genetic variants of norovirus of GII.6 genotype. Mol. Genet. Microbiol. Virol. 2016, 30, 192–200. [Google Scholar] [CrossRef]
- Chan-It, W.; Thongprachum, A.; Khamrin, P.; Kobayashi, M.; Okitsu, S.; Mizuguchi, M.; Ushijima, H. Emergence of a new norovirus GII.6 variant in Japan, 2008–2009. J. Med. Virol. 2012, 84, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.F.; Qiao, K.; Wang, X.G.; Ding, K.Y.; Su, H.L.; Li, C.Z.; Yan, H.J. Acute gastroenteritis outbreak caused by a GII.6 norovirus. World J. Gastroenterol. 2015, 21, 5295–5302. [Google Scholar] [CrossRef]
- Cannon, J.L.; Barclay, L.; Collins, N.R.; Wikswo, M.E.; Castro, C.J.; Magana, L.C.; Gregoricus, N.; Marine, R.L.; Chhabra, P.; Vinje, J. Genetic and Epidemiologic Trends of Norovirus Outbreaks in the United States from 2013 to 2016 Demonstrated Emergence of Novel GII.4 Recombinant Viruses. J. Clin. Microbiol. 2017, 55, 2208–2221. [Google Scholar] [CrossRef] [PubMed]
- Van Beek, J.; de Graaf, M.; Al-Hello, H.; Allen, D.J.; Ambert-Balay, K.; Botteldoorn, N.; Brytting, M.; Buesa, J.; Cabrerizo, M.; Chan, M.; et al. Molecular surveillance of norovirus, 2005–2016: An epidemiological analysis of data collected from the NoroNet network. Lancet Infect. Dis. 2018, 18, 545–553. [Google Scholar] [CrossRef]
- Rackoff, L.A.; Bok, K.; Green, K.Y.; Kapikian, A.Z. Epidemiology and evolution of rotaviruses and noroviruses from an archival WHO Global Study in Children (1976–1979) with implications for vaccine design. PLoS ONE 2013, 8, e59394. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.F.; Jin, M.; Li, Y.N.; Kang, H.H.; Duan, Z.J. The research progress on epidemic and evolutionary characteristics of norovirus GII.17. Chin. J. Front. Health Quar. 2017, 40, 136–140. [Google Scholar]
- Sang, S.; Yang, X. Evolutionary dynamics of GII.17 norovirus. PeerJ 2018, 6, e4333. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.X.; Zhou, H.L.; Mo, Z.J.; Wang, S.M.; Hao, Z.Y.; Li, Y.; Zhen, S.S.; Zhang, C.J.; Zhang, X.J.; Ma, J.C.; et al. Burden of viral gastroenteritis in children living in rural China: Population-based surveillance. Int. J. Infect. Dis. IJID Off. Publ. Int. Soc. Infect. Dis. 2019, 90, 151–160. [Google Scholar] [CrossRef]
- Motomura, K.; Oka, T.; Yokoyama, M.; Nakamura, H.; Mori, H.; Ode, H.; Hansman, G.S.; Katayama, K.; Kanda, T.; Tanaka, T.; et al. Identification of monomorphic and divergent haplotypes in the 2006–2007 norovirus GII/4 epidemic population by genomewide tracing of evolutionary history. J. Virol. 2008, 82, 11247–11262. [Google Scholar] [CrossRef] [PubMed]
- Hansman, G.S.; Natori, K.; Shirato-Horikoshi, H.; Ogawa, S.; Oka, T.; Katayama, K.; Tanaka, T.; Miyoshi, T.; Sakae, K.; Kobayashi, S.; et al. Genetic and antigenic diversity among noroviruses. J. Gen. Virol. 2006, 87, 909–919. [Google Scholar] [CrossRef]
- NCBI. Open Reading Frame Finder. Available online: https://www.ncbi.nlm.nih.gov/orffinder/ (accessed on 31 January 2020).
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef]
- Kroneman, A.; Vennema, H.; Deforche, K.; Avoort, H.V.D.; Penaranda, S.; Oberste, M.S.; Vinje, J.; Koopmans, M. An automated genotyping tool for enteroviruses and noroviruses. J. Clin. Virol. Off. Publ. Pan Am. Soc. Clin. Virol. 2011, 51, 121–125. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef]
- Ho, S.Y.; Shapiro, B. Skyline-plot methods for estimating demographic history from nucleotide sequences. Mol. Ecol. Resour. 2011, 11, 423–434. [Google Scholar] [CrossRef]
- Lindesmith, L.C.; McDaniel, J.R.; Changela, A.; Verardi, R.; Kerr, S.A.; Costantini, V.; Brewer-Jensen, P.D.; Mallory, M.L.; Voss, W.N.; Boutz, D.R.; et al. Sera Antibody Repertoire Analyses Reveal Mechanisms of Broad and Pandemic Strain Neutralizing Responses after Human Norovirus Vaccination. Immunity 2019, 50, 1530–1541.e8. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Lou, Z.; Tan, M.; Chen, Y.; Liu, Y.; Zhang, Z.; Zhang, X.C.; Jiang, X.; Li, X.; Rao, Z. Structural basis for the recognition of blood group trisaccharides by norovirus. J. Virol. 2007, 81, 5949–5957. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Genotypes | Variants | Sequences | Years | Time Span (Years) |
|---|---|---|---|---|
| GII.4 | Bristol_1993 | 1 | 1987 | 1 |
| Camberwell_1994 | 11 | 1987–1988 | 2 | |
| US95_96 | 13 | 1995–2004 | 10 | |
| Farmington_Hills_2002 | 21 | 2002–2012 | 11 | |
| Lanzhou_2002 | 3 | 2000–2002 | 3 | |
| Asia_2003 | 27 | 2004–2011 | 8 | |
| Kaiso_2003 | 2 | 2002–2003 | 2 | |
| Hunter_2004 | 37 | 2002–2006 | 5 | |
| Yerseke_2006a | 34 | 2006–2015 | 10 | |
| Den_Haag_2006b | 56 | 2006–2015 | 10 | |
| Apeldoorn_2007 | 50 | 2007–2009 | 3 | |
| Cairo_2007 | 2 | 2007 | 1 | |
| Osaka_2007 | 23 | 2005–2011 | 6 | |
| New_Orleans_2009 | 45 | 2008–2013 | 6 | |
| Sydney_2012 | 40 | 2010–2019 | 10 | |
| Hong_Kong_2019 | 1 | 2019 | 1 | |
| GII.2 | A | 299 | 1971–2019 | 49 |
| B | 2 | 2002 | 1 | |
| GII.3 | - | 139 | 1972–2019 | 48 |
| GII.6 | A | 19 | 1971–2012 | 49 |
| B | 18 | 1976–2019 | 44 | |
| C | 37 | 1977–2019 | 43 | |
| GII.17 | A | 8 | 1978–2016 | 39 |
| B | 1 | 2005 | 1 | |
| C | 26 | 2013–2015 | 3 | |
| D | 460 | 2013–2019 | 7 |
| Genotypes | Years | No. of Sequences | Molecular Clock | Nucleotide Substitution Rate 10−3 Substitution/Site/Year (95% HPD) | TMRCA | |
|---|---|---|---|---|---|---|
| No. of Years | Dates (Ranges) | |||||
| GII.4 | 1987–2019 | 366 | Strict Clock | 4.95 (4.53–5.39) | 38.5 (37.0–40.1) | 1980.5 (1978.9–1982.0) |
| UCLN | 5.37 (4.84–5.88) | 38.4 (34.7–43.5) | 1980.6 (1975.5–1984.3) | |||
| UCED | 5.91 (5.33–6.52) | 37.6 (33.7–43.2) | 1981.4 (1975.8–1985.3) | |||
| GII.4 (Zhengding) | 2002–2019 | 119 | Strict Clock | 1.55 (1.17–2.01) | 51.2 (38.5–63.7) | 1967.8 (1955.3–1980.5) |
| UCLN | 1.67 (1.10–2.52) | 50.3 (29.5–70.4) | 1968.7 (1948.6–1989.5) | |||
| UCED | 2.97 (1.48–4.43) | 32.3 (17.9–56.1) | 1986.7 (1962.9–2001.1) | |||
| GII.2 | 1971–2019 | 301 | Strict Clock | 2.30 (2.01–2.60) | 88.3 (79.1–97.4) | 1930.7 (1921.6–1939.9) |
| UCLN | 2.68 (2.19–3.20) | 81.5 (60.5–105.6) | 1937.5 (1913.4–1958.5) | |||
| UCED | 3.16 (2.52–3.76) | 73.5 (55.0–99.3) | 1945.5 (1919.7–1964.0) | |||
| GII.3 | 1972–2019 | 139 | Strict Clock | 3.40 (3.00–3.80) | 51.0 (49.3–52.9) | 1968.0 (1966.1–1969.7) |
| UCLN | 3.51 (2.99–4.04) | 50.9 (47.3–55.1) | 1968.1 (1963.9–1971.7) | |||
| UCED | 3.59 (2.99–4.21) | 51.9 (47.0–60.1) | 1967.1 (1958.9–1972.0) | |||
| GII.6 | 1971–2019 | 74 | Strict Clock | 2.39 (1.98–2.83) | 163.4 (136.0–192.1) | 1855.6 (1826.9–1883.0) |
| UCLN | 2.64 (1.96–3.34) | 142.3 (96.9–189.6) | 1876.7 (1829.4–1922.1) | |||
| UCED | 3.07 (1.86–4.28) | 122.0 (68.1–193.7) | 1897.0 (1825.3–1950.9) | |||
| GII.17 | 1978–2019 | 495 | Strict Clock | 1.54 (1.29–1.79) | 227.1 (183.1–274.2) | 1791.9 (1744.8–1835.9) |
| UCLN | 1.94 (1.26–2.65) | 157.5 (67.5–276.0) | 1861.5 (1743.0–1951.5) | |||
| UCED | 1.98 (1.37–2.60) | 150.4 (74.5–253.4) | 1868.6 (1765.6–1944.5) | |||
| Variants | Vaccine GII.4c | GII.4 Sydney_2012 | GII.2 Variant A | GII.3 Genotype | GII.6 Variant B | GII.6 Variant C |
|---|---|---|---|---|---|---|
| GII.4 Sydney_2012 | 0.053 | |||||
| GII.2 Variant A | 0.354 | 0.355 | ||||
| GII.3 Genotype | 0.319 | 0.338 | 0.299 | |||
| GII.6 Variant B | 0.348 | 0.357 | 0.284 | 0.265 | ||
| GII.6 Variant C | 0.342 | 0.352 | 0.301 | 0.261 | 0.076 | |
| GII.17 Variant D | 0.335 | 0.335 | 0.278 | 0.278 | 0.280 | 0.272 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, H.-L.; Chen, L.-N.; Wang, S.-M.; Tan, M.; Qiu, C.; Qiu, T.-Y.; Wang, X.-Y. Prevalence and Evolution of Noroviruses between 1966 and 2019, Implications for Vaccine Design. Pathogens 2021, 10, 1012. https://doi.org/10.3390/pathogens10081012
Zhou H-L, Chen L-N, Wang S-M, Tan M, Qiu C, Qiu T-Y, Wang X-Y. Prevalence and Evolution of Noroviruses between 1966 and 2019, Implications for Vaccine Design. Pathogens. 2021; 10(8):1012. https://doi.org/10.3390/pathogens10081012
Chicago/Turabian StyleZhou, Hong-Lu, Li-Na Chen, Song-Mei Wang, Ming Tan, Chao Qiu, Tian-Yi Qiu, and Xuan-Yi Wang. 2021. "Prevalence and Evolution of Noroviruses between 1966 and 2019, Implications for Vaccine Design" Pathogens 10, no. 8: 1012. https://doi.org/10.3390/pathogens10081012
APA StyleZhou, H.-L., Chen, L.-N., Wang, S.-M., Tan, M., Qiu, C., Qiu, T.-Y., & Wang, X.-Y. (2021). Prevalence and Evolution of Noroviruses between 1966 and 2019, Implications for Vaccine Design. Pathogens, 10(8), 1012. https://doi.org/10.3390/pathogens10081012