
Antistaphylococcal Activity of the FtsZ Inhibitor C109

 , ,
, ,  , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
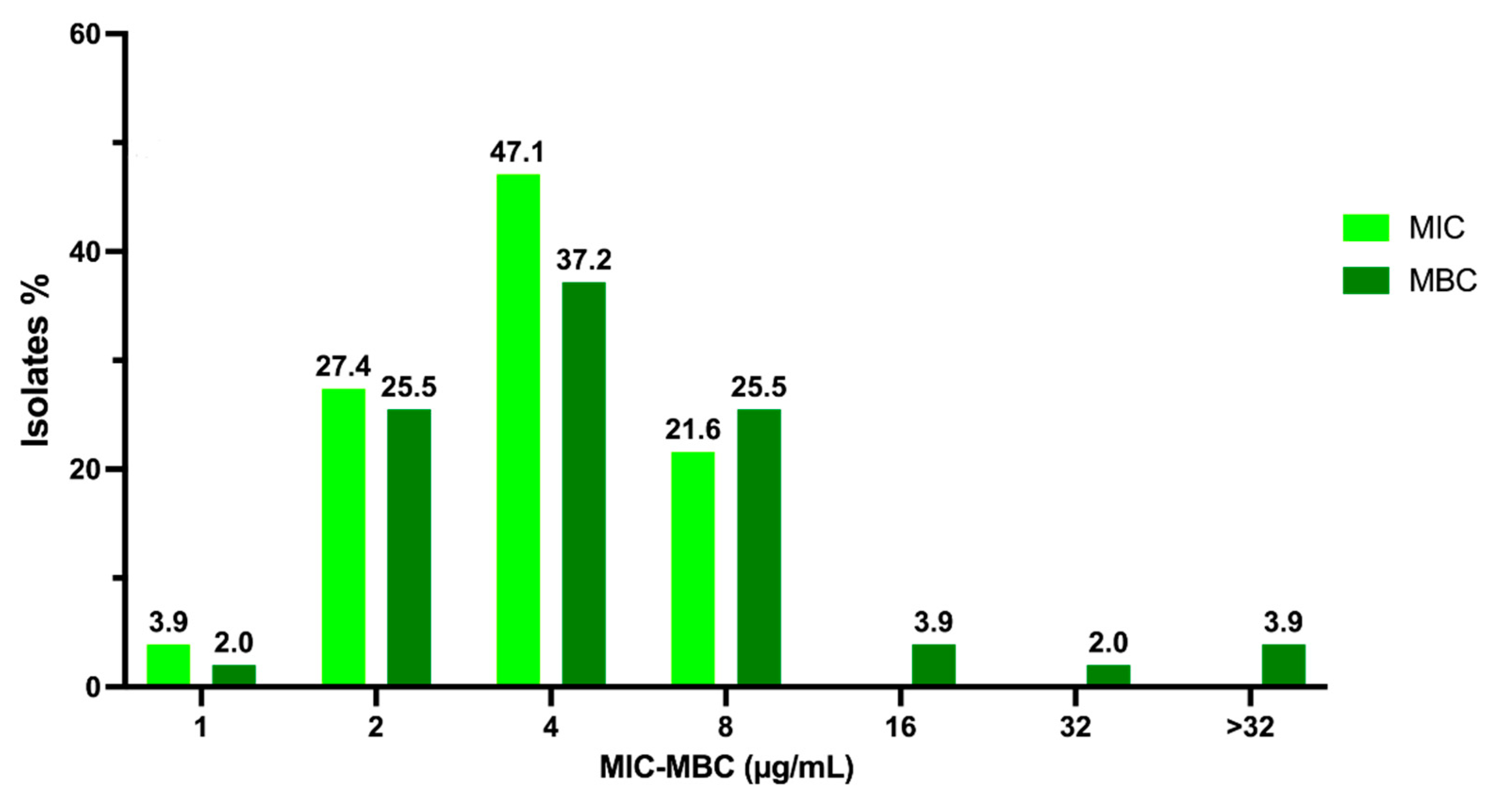
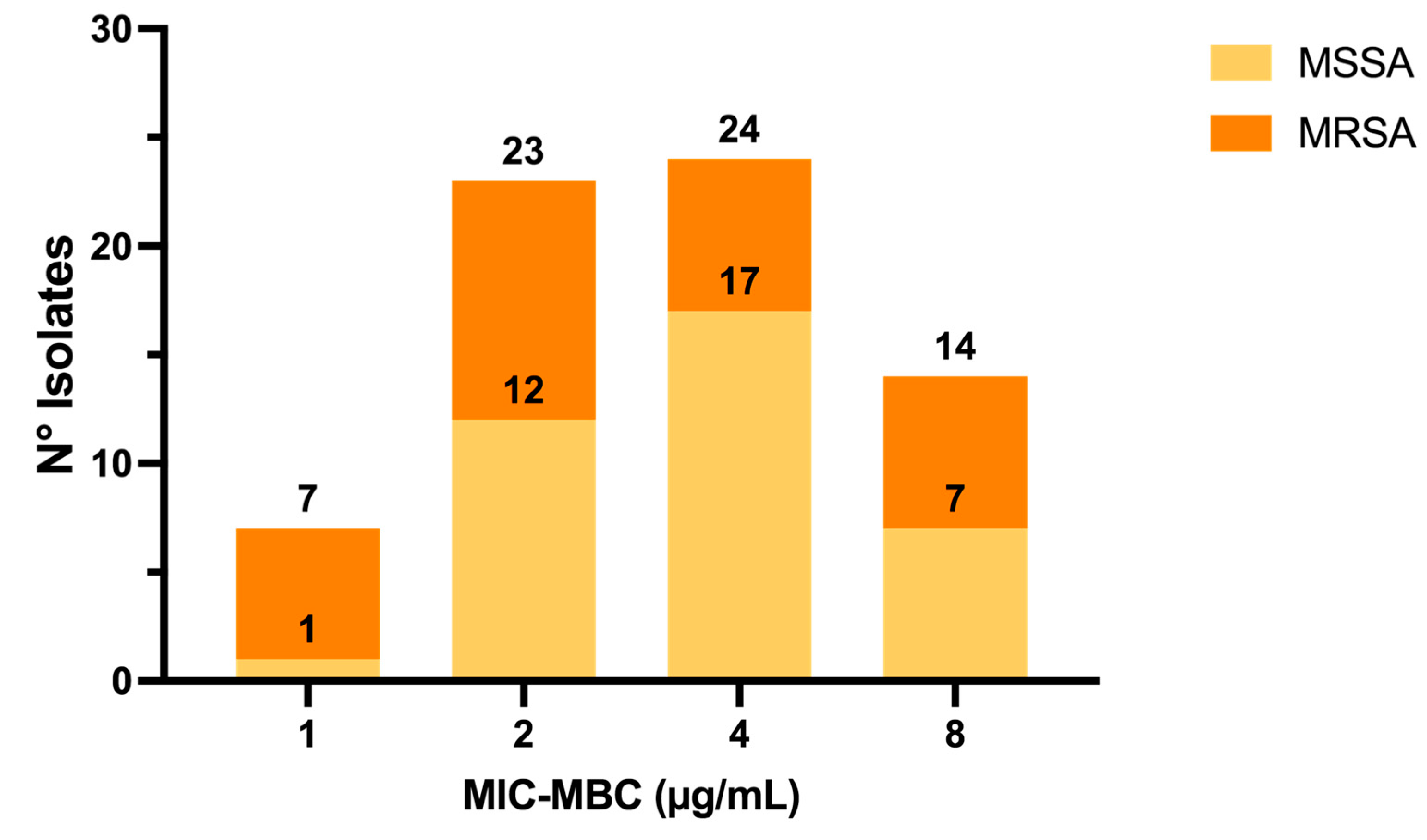
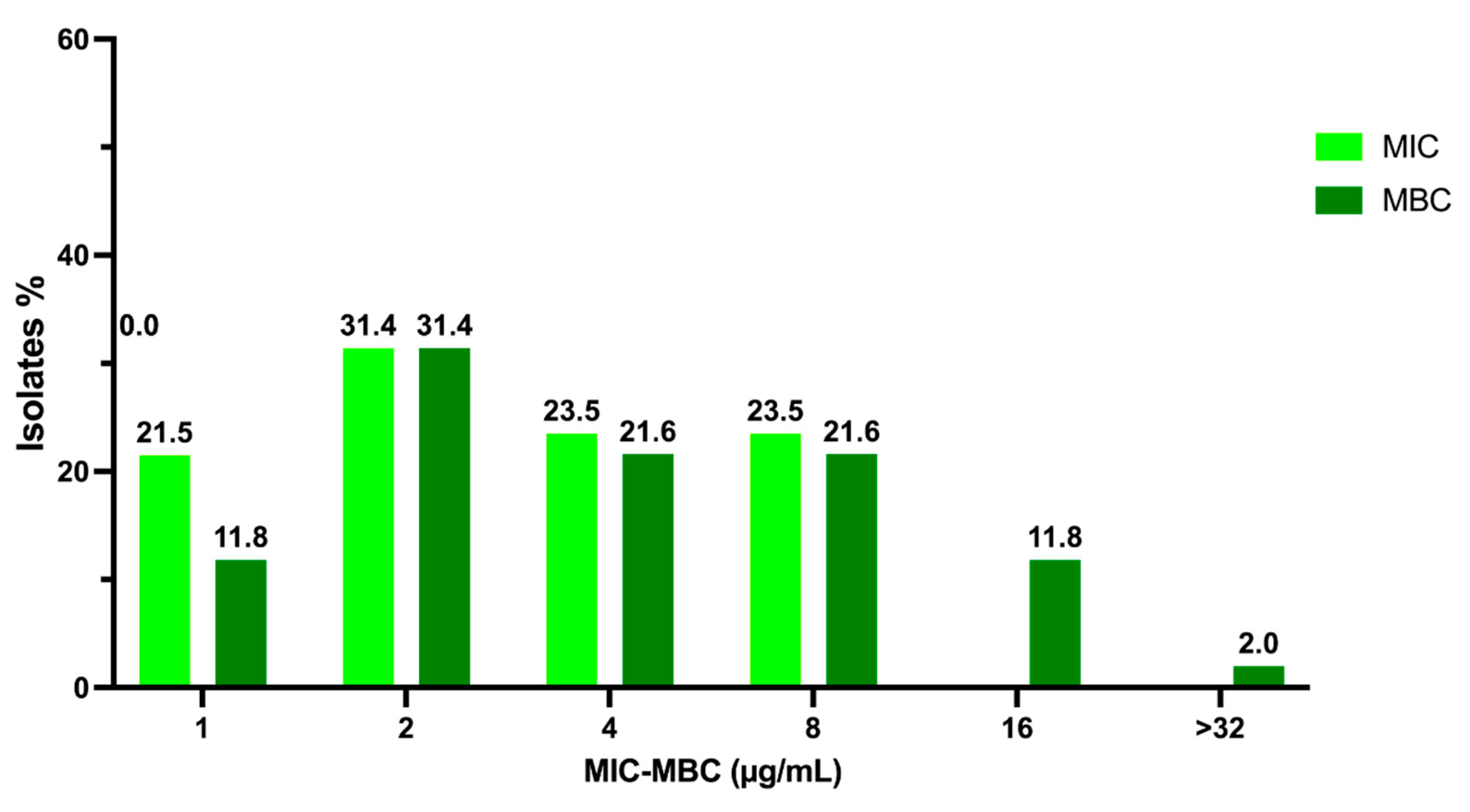
2.1. Activity of C109 against Sensitive (MSSA) and Resistant (MRSA) S. aureus Strains
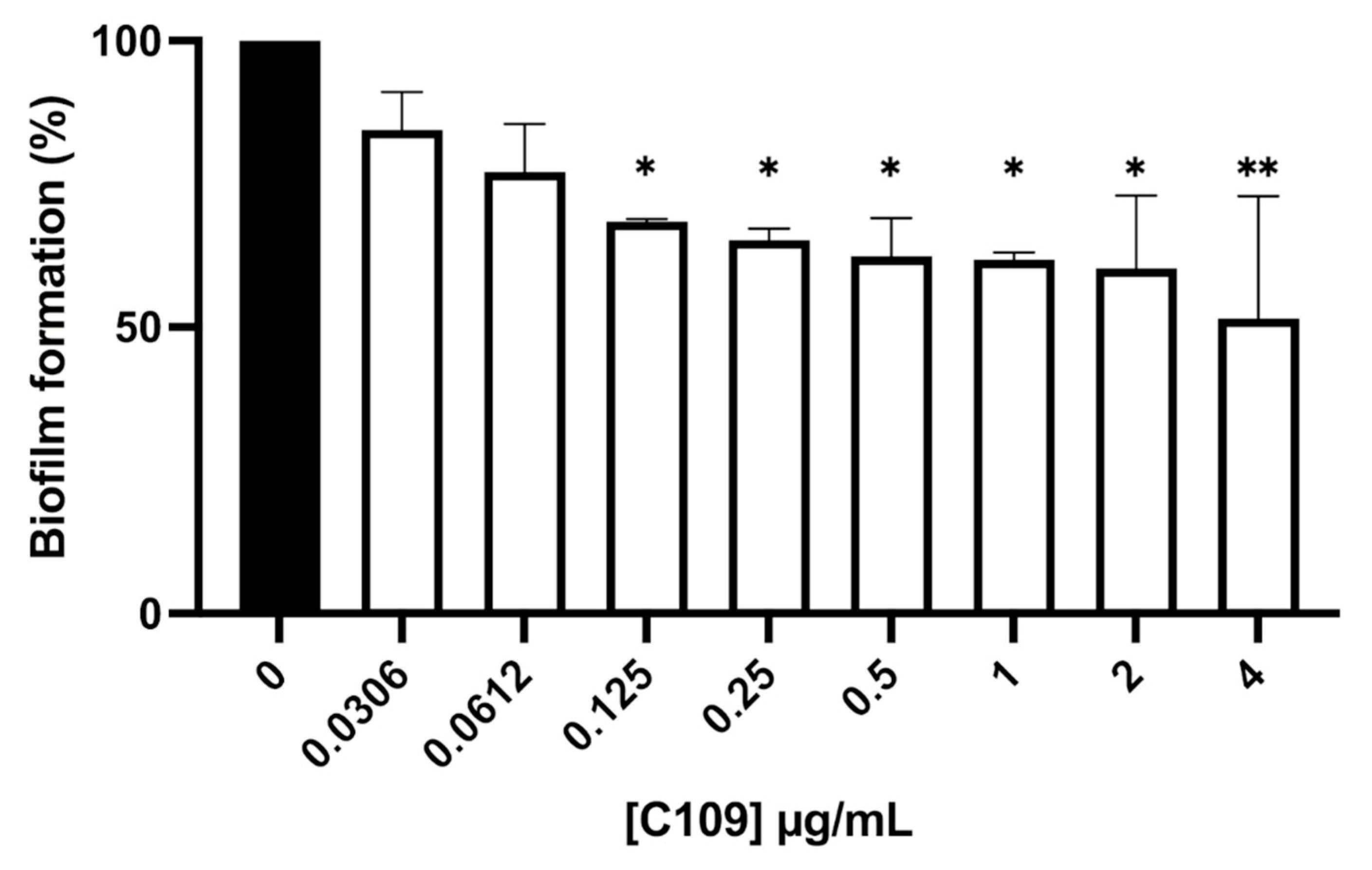
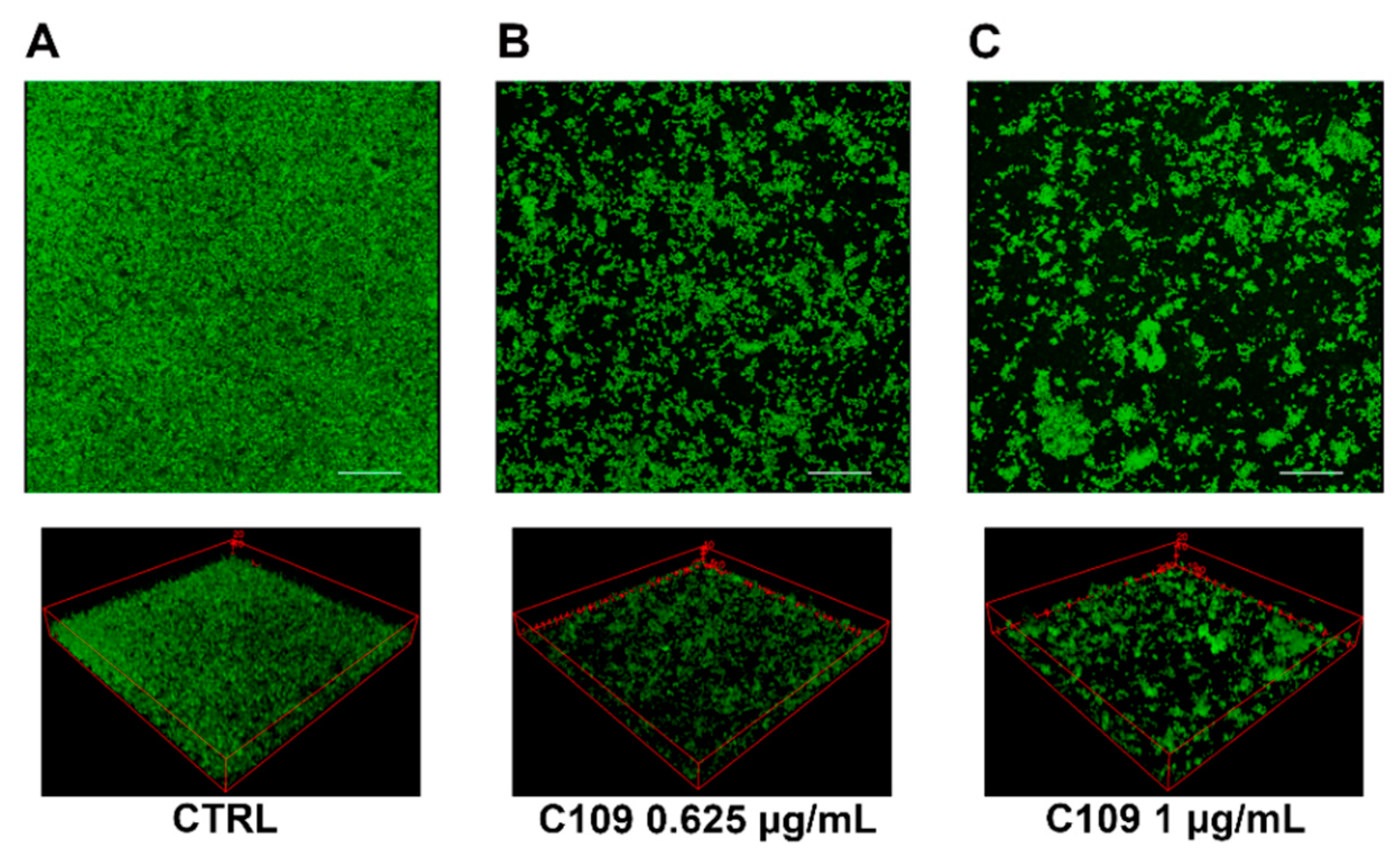
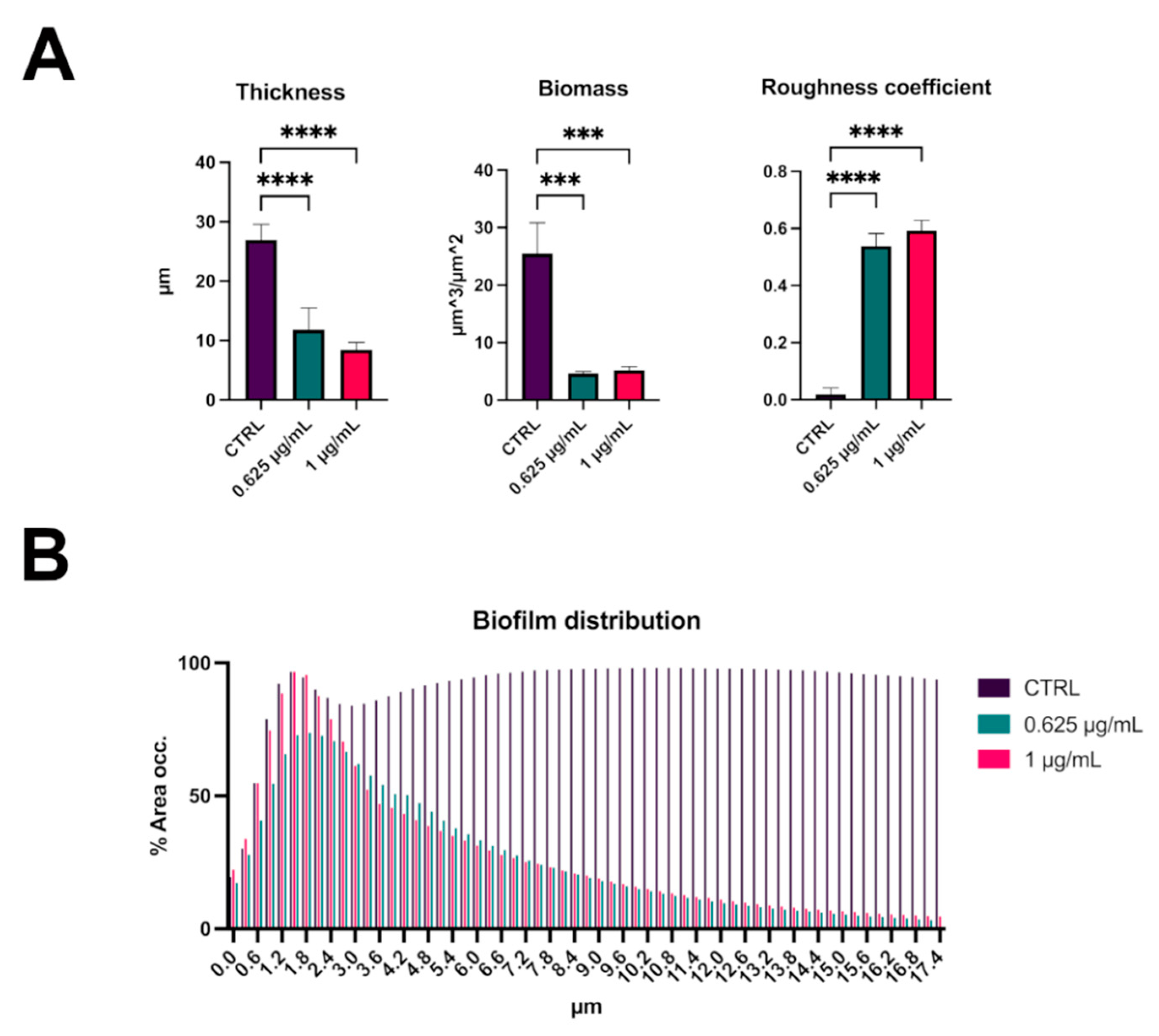
2.2. C109 Biofilm Inhibitory Activity against S. aureus ATCC 25923
2.3. C109 Biofilm Inhibitory Activity against S. aureus Mu50
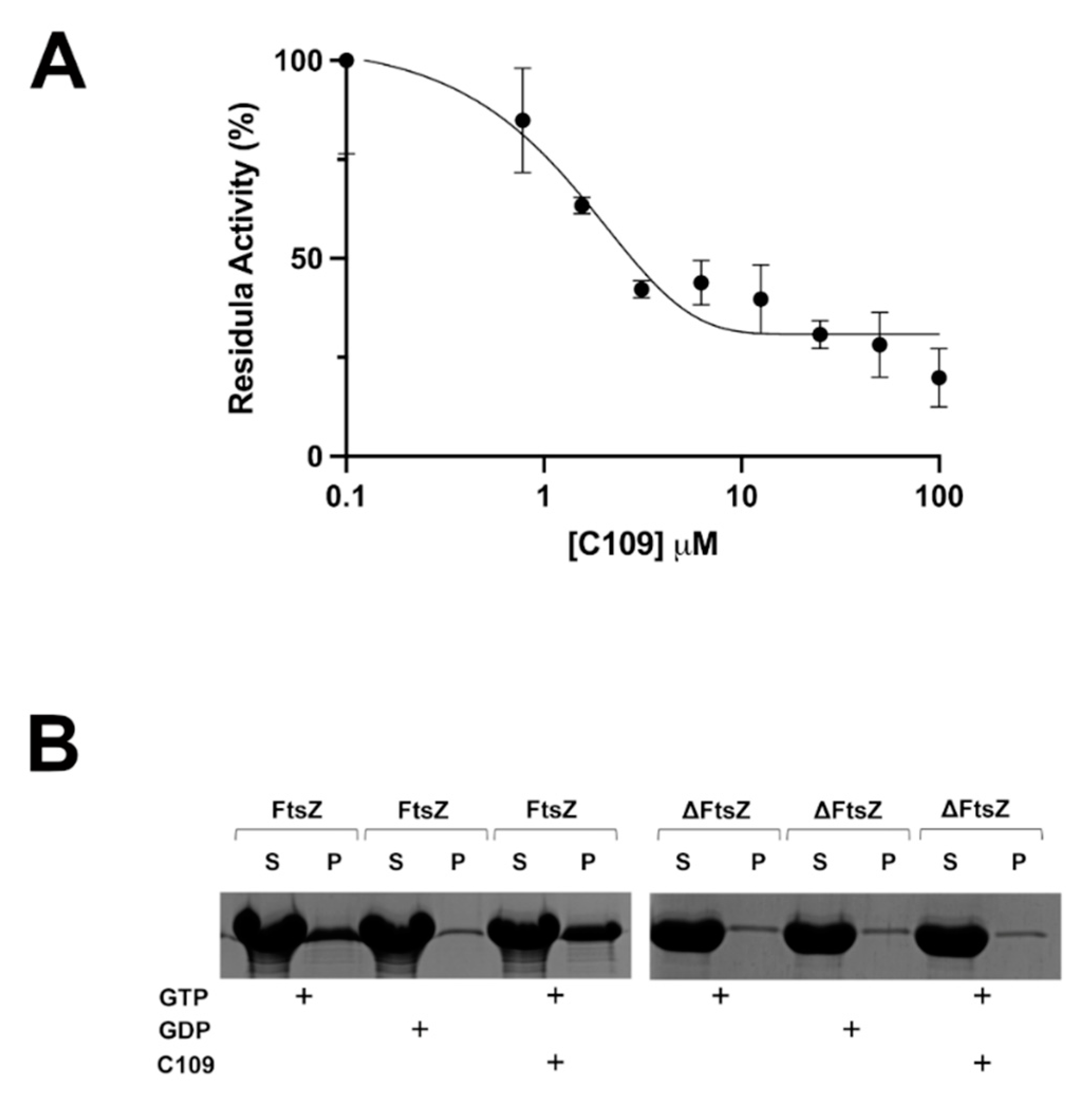
2.4. C109 Activity on Purified S. aureus FtsZ
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Culture Media
4.2. Minimum Inhibitory Concentration (MIC) and Minimum Bactericidal Concentration (MBC) Determination for S. aureus Clinical Isolates
4.3. In Vitro Biofilm Inhibition Test in 96-Well Microtiter Plates
4.4. Biofilm Eradication Assay in 96-Well Microtiter Plates
4.5. Biofilm Evaluation by Confocal Laser Scanning Microscopy
4.6. Preparation of the Artificial Dermis for the Biofilm Wound Model
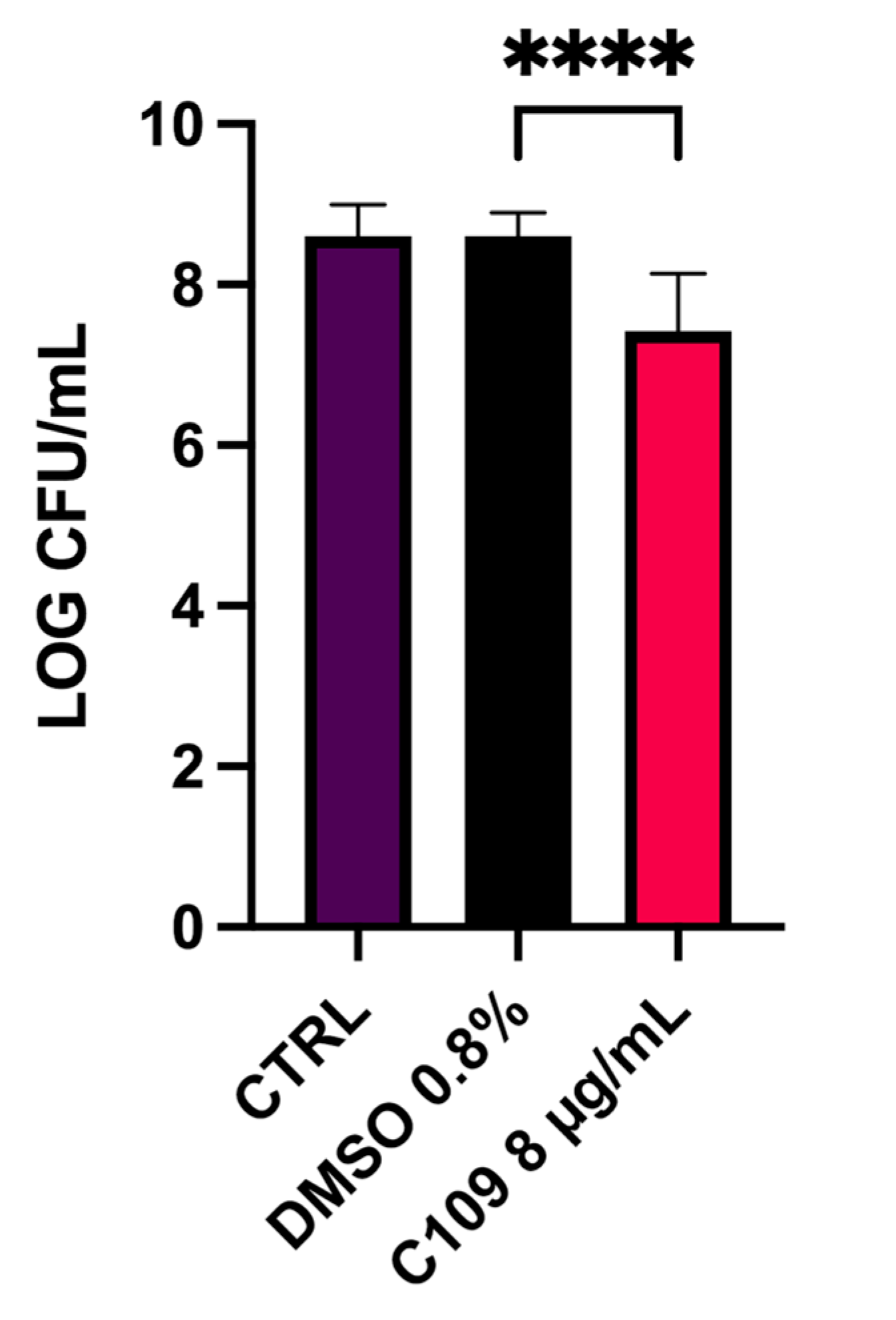
4.7. S. aureus Mu50 Biofilm Inhibition in a Wound Model
4.8. Cloning, Expression, and Purification of the S. aureus 25923 FtsZ Proteins
4.9. In Vitro FtsZ GTPase Activity
4.10. In Vitro FtsZ Polymerization Assay
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dayan, G.H.; Mohamed, N.; Scully, I.L.; Cooper, D.; Begier, E.; Eiden, J.; Jansen, K.U.; Gurtman, A.; Anderson, A.S. Staphylococcus aureus: The current state of disease, pathophysiology and strategies for prevention. Expert Rev. Vaccines 2016, 15, 1373–1392. [Google Scholar] [CrossRef] [PubMed]
- Gangell, C.; Gard, S.; Douglas, T.; Park, J.; de Klerk, N.; Keil, T.; Brennan, S.; Ranganathan, S.; Robins-Browne, R.; Sly, P.D.; et al. Inflammatory responses to individual microorganisms in the lungs of children with cystic fibrosis. Clin. Infect. Dis. 2011, 53, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Pillarisetti, N.; Williamson, E.; Linnane, B.; Skoric, B.; Robertson, C.F.; Robinson, P.; Massie, J.; Hall, G.L.; Sly, P.; Stick, S.; et al. Infection, inflammation, and lung function decline in infants with cystic fibrosis. Am. J. Respir. Crit. Care Med. 2011, 184, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Cystic Fibrosis Foundation Patient Registry. 2018 Annual Report. 2019. Available online: https://www.cff.org/Research/Researcher-Resources/Patient-Registry/2019-Patient-Registry-Annual-Data-Report.pdf (accessed on 18 June 2021).
- European Cystic Fibrosis Society Patient Registry. Annual Data Report 2018. 2020. Available online: https://www.ecfs.eu/sites/default/files/general-content-files/working-groups/ecfs-patient-registry/ECFSPR_Report_2018_v1.4.pdf (accessed on 18 June 2021).
- Prunier, A.L.; Malbruny, B.; Laurans, M.; Brouard, J.; Duhamel, J.F.; Leclercq, R. High rate of macrolide resistance in Staphylococcus aureus strains from patients with cystic fibrosis reveals high proportions of hypermutable strains. J. Infect. Dis. 2003, 187, 1709–1716. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Wu, Y.; Riehle, A.; Ma, J.; Kamler, M.; Gulbins, E.; Grassmé, H. Staphylococcus aureus Survives in Cystic Fibrosis Macrophages, Forming a Reservoir for Chronic Pneumonia. Infect. Immun. 2017, 85, e00883-16. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Coureuil, M.; Ramond, E.; Euphrasie, D.; Dupuis, M.; Tros, F.; Meyer, J.; Nemazanyy, I.; Chhuon, C.; Guerrera, I.C.; et al. Chronic Staphylococcus aureus Lung Infection Correlates with Proteogenomic and Metabolic Adaptations Leading to an Increased Intracellular Persistence. Clin. Infect. Dis. 2019, 69, 1937–1945. [Google Scholar] [CrossRef] [PubMed]
- Kriegeskorte, A.; Lorè, N.I.; Bragonzi, A.; Riva, C.; Kelkenberg, M.; Becker, K.; Proctor, R.A.; Peters, G.; Kahl, B.C. Thymidine-Dependent Staphylococcus aureus Small-Colony Variants Are Induced by Trimethoprim-Sulfamethoxazole (SXT) and Have Increased Fitness during SXT Challenge. Antimicrob. Agents Chemother. 2015, 59, 7265–7272. [Google Scholar] [CrossRef]
- Archer, N.K.; Mazaitis, M.J.; Costerton, J.W.; Leid, J.G.; Powers, M.E.; Shirtliff, M.E. Staphylococcus aureus biofilms: Properties, regulation, and roles in human disease. Virulence 2011, 2, 445–459. [Google Scholar] [CrossRef]
- McKenney, D.; Pouliot, K.L.; Wang, Y.; Murthy, V.; Ulrich, M.; Döring, G.; Lee, J.C.; Goldmann, D.A.; Pier, G.B. Broadly protective vaccine for Staphylococcus aureus based on an in vivo-expressed antigen. Science 1999, 284, 1523–1527. [Google Scholar] [CrossRef]
- Ren, C.L.; Morgan, W.J.; Konstan, M.W.; Schechter, M.S.; Wagener, J.S.; Fisher, K.A.; Regelmann, W.E. Investigators and Coordinators of the Epidemiologic Study of Cystic Fibrosis. Presence of methicillin resistant Staphylococcus aureus in respiratory cultures from cystic fibrosis patients is associated with lower lung function. Pediatric Pulmonol. 2007, 42, 513–518. [Google Scholar] [CrossRef]
- Dasenbrook, E.C.; Checkley, W.; Merlo, C.A.; Konstan, M.W.; Lechtzin, N.; Boyle, M.P. Association between respiratory tract methicillin-resistant Staphylococcus aureus and survival in cystic fibrosis. JAMA 2010, 303, 2386–2392. [Google Scholar] [CrossRef]
- Akil, N.; Muhlebach, M.S. Biology and management of methicillin resistant Staphylococcus aureus in cystic fibrosis. Pediatric Pulmonol. 2018, 53, S64–S74. [Google Scholar] [CrossRef]
- Cafiso, V.; Bertuccio, T.; Spina, D.; Campanile, F.; Bongiorno, D.; Santagati, M.; Sciacca, A.; Sciuto, C.; Stefani, S. Methicillin resistance and vancomycin heteroresistance in Staphylococcus aureus in cystic fibrosis patients. Eur. J. Clin. Microbiol. Infect. Dis. 2010, 29, 1277–1285. [Google Scholar] [CrossRef]
- Sen, C.K.; Gordillo, G.M.; Roy, S.; Kirsner, R.; Lambert, L.; Hunt, T.K.; Gottrup, F.; Gurtner, G.C.; Longaker, M.T. Human skin wounds: A major and snowballing threat to public health and the economy. Wound Repair Regen. 2009, 17, 763–771. [Google Scholar] [CrossRef]
- Gottrup, F. A specialized wound-healing center concept: Importance of a multidisciplinary department structure and surgical treatment facilities in the treatment of chronic wounds. Am. J. Surg. 2004, 187, 38S–43S. [Google Scholar] [CrossRef]
- Schultz, G.S.; Sibbald, R.G.; Falanga, V.; Ayello, E.A.; Dowsett, C.; Harding, K.; Romanelli, M.; Stacey, M.C.; Teot, L.; Vanscheidt, W. Wound bed preparation: A systematic approach to wound management. Wound Repair Regen. 2003, 11, S1–S28. [Google Scholar] [CrossRef]
- Gjødsbøl, K.; Christensen, J.J.; Karlsmark, T.; Jørgensen, B.; Klein, B.M.; Krogfelt, K.A. Multiple bacterial species reside in chronic wounds: A longitudinal study. Int. Wound J. 2006, 3, 225–231. [Google Scholar] [CrossRef]
- Silva, V.; Almeida, F.; Carvalho, J.A.; Castro, A.P.; Ferreira, E.; Manageiro, V.; Tejedor-Junco, M.T.; Caniça, M.; Igrejas, G.; Poeta, P. Emergence of community-acquired methicillin-resistant Staphylococcus aureus EMRSA-15 clone as the predominant cause of diabetic foot ulcer infections in Portugal. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 179–186. [Google Scholar] [CrossRef]
- Tentolouris, N.; Jude, E.B.; Smirnof, I.; Knowles, E.A.; Boulton, A.J. Methicillin-resistant Staphylococcus aureus: An increasing problem in a diabetic foot clinic. Diabet. Med. 1999, 16, 767–771. [Google Scholar] [CrossRef]
- Silva, V.; Peirone, C.; Amaral, J.S.; Capita, R.; Alonso-Calleja, C.; Marques-Magallanes, J.A.; Martins, Â.; Carvalho, Á.; Maltez, L.; Pereira, J.E.; et al. High Efficacy of Ozonated Oils on the Removal of Biofilms Produced by Methicillin-Resistant Staphylococcus aureus (MRSA) from Infected Diabetic Foot Ulcers. Molecules 2020, 25, 3601. [Google Scholar] [CrossRef]
- Scoffone, V.C.; Ryabova, O.; Makarov, V.; Iadarola, P.; Fumagalli, M.; Fondi, M.; Fani, R.; De Rossi, E.; Riccardi, G.; Buroni, S. Efflux-mediated resistance to a benzothiadiazol derivative effective against Burkholderia cenocepacia. Front. Microbiol. 2015, 6, 815. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hogan, A.M.; Scoffone, V.C.; Makarov, V.; Gislason, A.S.; Tesfu, H.; Stietz, M.S.; Brassinga, A.K.C.; Domaratzki, M.; Li, X.; Azzalin, A.; et al. Competitive Fitness of Essential Gene Knockdowns Reveals a Broad-Spectrum Antibacterial Inhibitor of the Cell Division Protein FtsZ. Antimicrob. Agents Chemother. 2018, 62, e01231-18. [Google Scholar] [CrossRef] [PubMed]
- Costabile, G.; Provenzano, R.; Azzalin, A.; Scoffone, V.C.; Chiarelli, L.R.; Rondelli, V.; Grillo, I.; Zinn, T.; Lepioshkin, A.; Savina, S.; et al. PEGylated mucus-penetrating nanocrystals for lung delivery of a new FtsZ inhibitor against Burkholderia cenocepacia infection. Nanomedicine 2020, 23, 102113. [Google Scholar] [CrossRef]
- Chiarelli, L.R.; Scoffone, V.C.; Trespidi, G.; Barbieri, G.; Riabova, O.; Monakhova, N.; Porta, A.; Manina, G.; Riccardi, G.; Makarov, V.; et al. Chemical, Metabolic, and Cellular Characterization of a FtsZ Inhibitor Effective Against Burkholderia cenocepacia. Front. Microbiol. 2020, 11, 562. [Google Scholar] [CrossRef] [PubMed]
- Buroni, S.; Makarov, V.; Scoffone, V.C.; Trespidi, G.; Riccardi, G.; Chiarelli, L.R. The cell division protein FtsZ as a cellular target to hit cystic fibrosis pathogens. Eur. J. Med. Chem. 2020, 190, 112132. [Google Scholar] [CrossRef] [PubMed]
- Trespidi, G.; Scoffone, V.C.; Barbieri, G.; Riccardi, G.; De Rossi, E.; Buroni, S. Molecular Characterization of the Burkholderia cenocepacia dcw Operon and FtsZ Interactors as New Targets for Novel Antimicrobial Design. Antibiotics 2020, 9, 841. [Google Scholar] [CrossRef] [PubMed]
- Zoraghi, R.; Reiner, N.E. Protein interaction networks as starting points to identify novel antimicrobial drug targets. Curr. Opin. Microbiol. 2013, 16, 566–572. [Google Scholar] [CrossRef]
- Brackman, G.; Garcia-Fernandez, M.J.; Lenoir, J.; De Meyer, L.; Remon, J.P.; De Beer, T.; Concheiro, A.; Alvarez-Lorenzo, C.; Coenye, T. Dressings Loaded with Cyclodextrin-Hamamelitannin Complexes Increase Staphylococcus aureus Susceptibility Toward Antibiotics Both in Single as well as in Mixed Biofilm Communities. Macromol. Biosci. 2016, 16, 859–869. [Google Scholar] [CrossRef]
- Wagstaff, J.M.; Tsim, M.; Oliva, M.A.; García-Sanchez, A.; Kureisaite-Ciziene, D.; Andreu, J.M.; Löwe, J. A Polymerization-Associated Structural Switch in FtsZ That Enables Treadmilling of Model Filaments. MBio 2017, 8, e00254-17. [Google Scholar] [CrossRef]
- Delaney, J.A.; Schneider-Lindner, V.; Brassard, P.; Suissa, S. Mortality after infection with methicillin-resistant Staphylococcus aureus (MRSA) diagnosed in the community. BMC Med. 2008, 6, 2. [Google Scholar] [CrossRef]
- Mottola, C.; Matias, C.S.; Mendes, J.J.; Melo-Cristino, J.; Tavares, L.; Cavaco-Silva, P.; Oliveira, M. Susceptibility patterns of Staphylococcus aureus biofilms in diabetic foot infections. BMC Microbiol. 2016, 16, 119. [Google Scholar] [CrossRef] [PubMed]
- Romeo, L.; Lanza Cariccio, V.; Iori, R.; Rollin, P.; Bramanti, P.; Mazzon, E. The α-Cyclodextrin/Moringin Complex: A New Promising Antimicrobial Agent against Staphylococcus aureus. Molecules 2018, 23, 2097. [Google Scholar] [CrossRef] [PubMed]
- Bocquet, L.; Sahpaz, S.; Bonneau, N.; Beaufay, C.; Mahieux, S.; Samaillie, J.; Roumy, V.; Jacquin, J.; Bordage, S.; Hennebelle, T.; et al. Phenolic Compounds from Humulus lupulus as Natural Antimicrobial Products: New Weapons in the Fight against Methicillin Resistant Staphylococcus aureus, Leishmania Mexicana and Trypanosoma brucei Strains. Molecules 2019, 24, 1024. [Google Scholar] [CrossRef] [PubMed]
- Li, C.L.; Xu, T.T.; Chen, R.B.; Huang, X.X.; Zhao, Y.C.; Bao, Y.Y.; Zhao, W.D.; Zheng, Z.Y. Cloning, expression and characterization of antimicrobial porcine β defensin 1 in Escherichia coli. Protein Expr. Purif. 2013, 88, 47–53. [Google Scholar] [CrossRef]
- Choi, M.; Hasan, N.; Cao, J.; Lee, J.; Hlaing, S.P.; Yoo, J.W. Chitosan-based nitric oxide-releasing dressing for anti-biofilm and in vivo healing activities in MRSA biofilm-infected wounds. Int. J. Biol. Macromol. 2020, 142, 680–692. [Google Scholar] [CrossRef]
- Di Domenico, E.G.; Rimoldi, S.G.; Cavallo, I.; D’Agosto, G.; Trento, E.; Cagnoni, G.; Palazzin, A.; Pagani, C.; Romeri, F.; De Vecchi, E.; et al. Microbial biofilm correlates with an increased antibiotic tolerance and poor therapeutic outcome in infective endocarditis. BMC Microbiol. 2019, 19, 228. [Google Scholar] [CrossRef]
- Van den Driessche, F.; Brackman, G.; Swimberghe, R.; Rigole, P.; Coenye, T. Screening a repurposing library for potentiators of antibiotics against Staphylococcus aureus biofilms. Int. J. Antimicrob. Agents 2017, 49, 315–320. [Google Scholar] [CrossRef]
- Cohan, M.C.; Eddelbuettel, A.M.P.; Levin, P.A.; Pappu, R.V. Dissecting the Functional Contributions of the Intrinsically Disordered C-terminal Tail of Bacillus subtilis FtsZ. J. Mol. Biol. 2020, 432, 3205–3221. [Google Scholar] [CrossRef]
- Elsen, N.L.; Lu, J.; Parthasarathy, G.; Reid, J.C.; Sharma, S.; Soisson, S.M.; Lumb, K.J. Mechanism of action of the cell-division inhibitor PC190723: Modulation of FtsZ assembly cooperativity. J. Am. Chem. Soc. 2012, 134, 12342–12345. [Google Scholar] [CrossRef]
- Huecas, S.; Canosa-Valls, A.J.; Araújo-Bazán, L.; Ruiz, F.M.; Laurents, D.V.; Fernández-Tornero, C.; Andreu, J.M. Nucleotide-induced folding of cell division protein FtsZ from Staphylococcus aureus. FEBS J. 2020, 287, 4048–4067. [Google Scholar] [CrossRef]
- Fujita, J.; Sugiyama, S.; Terakado, H.; Miyazaki, M.; Ozawa, M.; Ueda, N.; Kuroda, N.; Tanaka, S.I.; Yoshizawa, T.; Uchihashi, T.; et al. Dynamic Assembly/Disassembly of Staphylococcus aureus FtsZ Visualized by High-Speed Atomic Force Microscopy. Int. J. Mol. Sci. 2021, 22, 1697. [Google Scholar] [CrossRef]
- Hiramatsu, K.; Hanaki, H.; Ino, T.; Yabuta, K.; Oguri, T.; Tenover, F.C. Methicillin-resistant Staphylococcus aureus clinical strain with reduced vancomycin susceptibility. J. Antimicrob. Chemother. 1997, 40, 135–136. [Google Scholar] [CrossRef]
- EUCAST. The European Committee on Antimicrobial Susceptibility Testing. EUCAST Reading Guide for Broth Microdilution. Version 2.0. 2020. Available online: https://www.eucast.org/fileadmin/src/media/PDFs/EUCAST_files/Disk_test_documents/2020_manuals/Reading_guide_BMD_v_2.0_2020.pdf (accessed on 9 June 2021).
- Elshikh, M.; Ahmed, S.; Funston, S.; Dunlop, P.; McGaw, M.; Marchant, R.; Banat, I.M. Resazurin-based 96-well plate microdilution method for the determination of minimum inhibitory concentration of biosurfactants. Biotechnol. Lett. 2016, 38, 1015–1019. [Google Scholar] [CrossRef]
- Vandecandelaere, I.; Van Acker, H.; Coenye, T. A Microplate-Based System as In Vitro Model of Biofilm Growth and Quantification. Methods Mol. Biol. 2016, 1333, 53–66. [Google Scholar]
- Heydorn, A.; Nielsen, A.T.; Hentzer, M.; Sternberg, C.; Givskov, M.; Ersbøll, B.K.; Molin, S. Quantification of biofilm structures by the novel computer program COMSTAT. Microbiology 2000, 146, 2395–2407. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trespidi, G.; Scoffone, V.C.; Barbieri, G.; Marchesini, F.; Abualshaar, A.; Coenye, T.; Ungaro, F.; Makarov, V.; Migliavacca, R.; De Rossi, E.; et al. Antistaphylococcal Activity of the FtsZ Inhibitor C109. Pathogens 2021, 10, 886. https://doi.org/10.3390/pathogens10070886
Trespidi G, Scoffone VC, Barbieri G, Marchesini F, Abualshaar A, Coenye T, Ungaro F, Makarov V, Migliavacca R, De Rossi E, et al. Antistaphylococcal Activity of the FtsZ Inhibitor C109. Pathogens. 2021; 10(7):886. https://doi.org/10.3390/pathogens10070886
Chicago/Turabian StyleTrespidi, Gabriele, Viola Camilla Scoffone, Giulia Barbieri, Federica Marchesini, Aseel Abualshaar, Tom Coenye, Francesca Ungaro, Vadim Makarov, Roberta Migliavacca, Edda De Rossi, and et al. 2021. "Antistaphylococcal Activity of the FtsZ Inhibitor C109" Pathogens 10, no. 7: 886. https://doi.org/10.3390/pathogens10070886
APA StyleTrespidi, G., Scoffone, V. C., Barbieri, G., Marchesini, F., Abualshaar, A., Coenye, T., Ungaro, F., Makarov, V., Migliavacca, R., De Rossi, E., & Buroni, S. (2021). Antistaphylococcal Activity of the FtsZ Inhibitor C109. Pathogens, 10(7), 886. https://doi.org/10.3390/pathogens10070886